Emerging Roles of BRD7 in Pathophysiology



Abstract
:1. Introduction
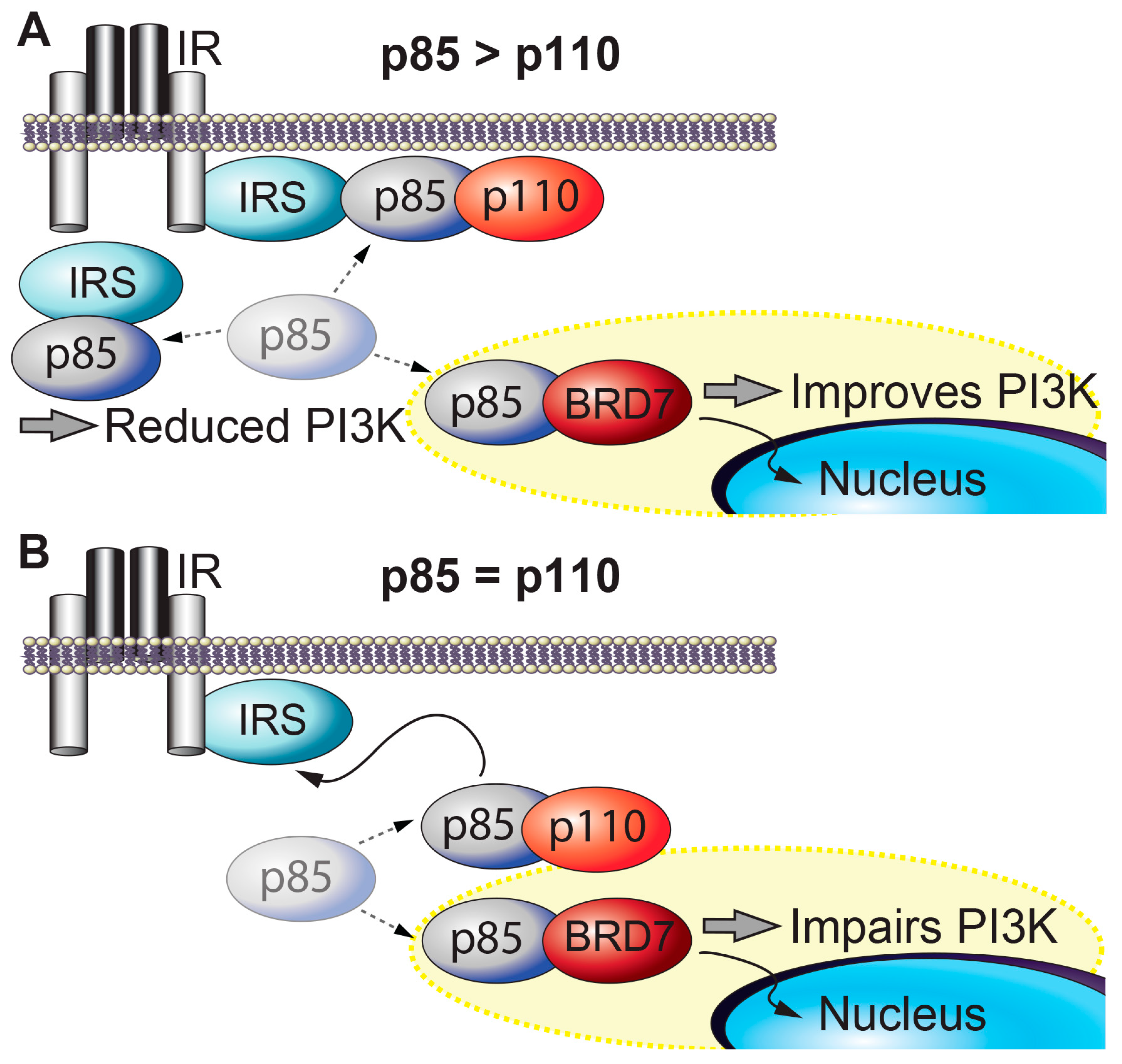
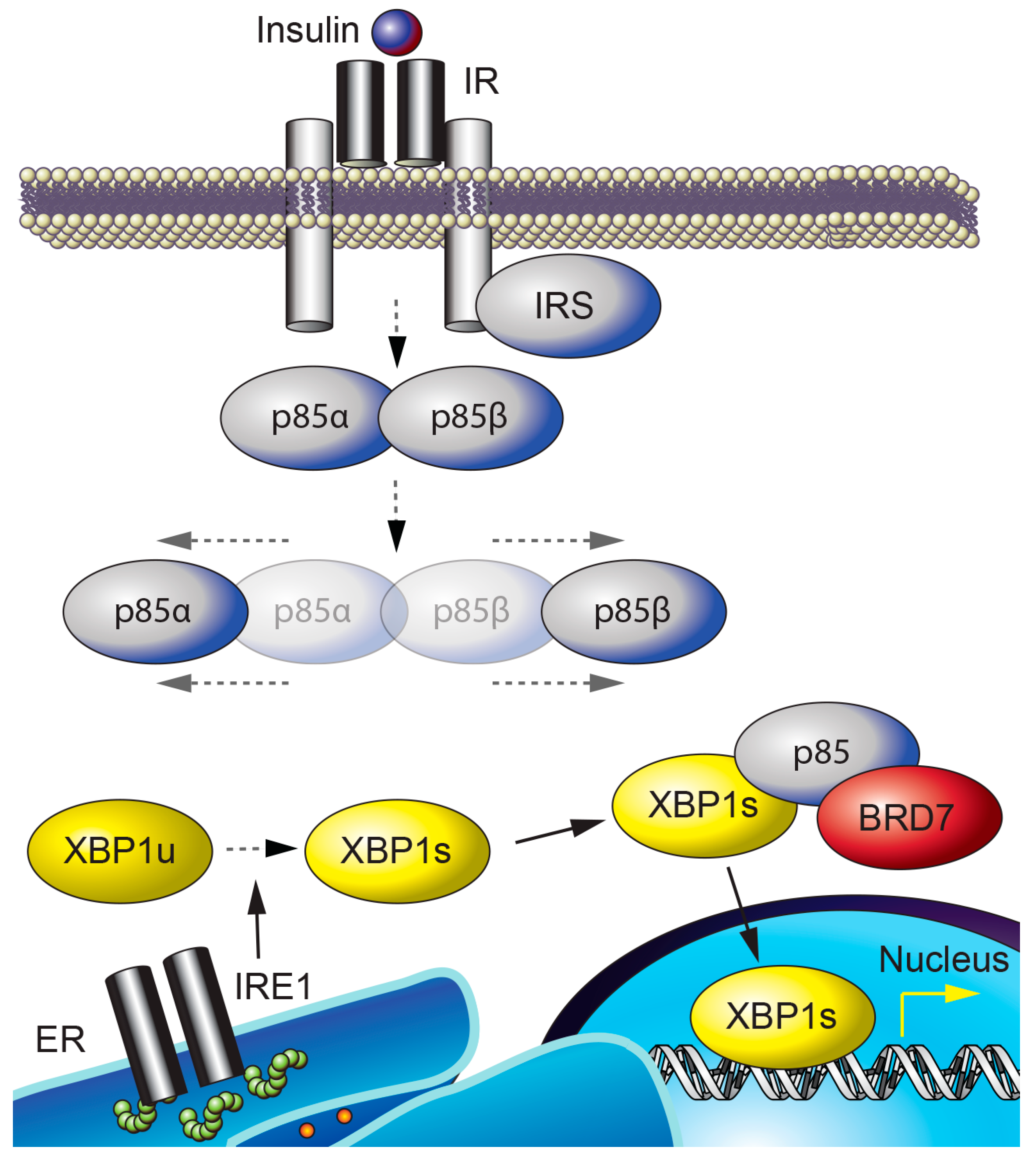
2. BRD7 in Metabolism
3. BRD7 in Cancer
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BRD7 | Bromodomain-containing protein 7 |
| PTP-BL | Protein tyrosine phosphatase BAS-like protein |
| PBAF | Polybromo-associated BRG1-associated factor |
| SWI/SNF | SWItch/Sucrose Non-Fermentable complex |
| PI3K | Phosphatidylinositol 3-kinase |
| GSK3β | Glycogen synthase kinase 3β |
| MEF | Mouse embryonic fibroblast |
| GS | Glycogen synthase |
| ER | Endoplasmic reticulum |
| UPR | Unfolded protein response |
| PERK | PKR-like ER kinase |
| ATF6 | Activating transcription factor 6 |
| IRE1 | Inositol-requiring enzyme 1 |
| XBP1 | X-Box Binding Protein-1 |
| XBP1s | The spliced form of XBP1 |
| NPC | Nasopharyngeal carcinoma |
| NLS | Nuclear localization signal |
| Dvl-1 | Disheveled 1 |
| ERα | Estrogen receptor α |
| TRIM24 | Tripartite motif-containing 24 |
| TIF1 | Transcriptional intermediary family 1 |
| APC/C | Anaphase-promoting complex/cyclosome |
| NSCLC | Non-small cell lung carcinomas |
| XAF1 | X-linked inhibitor of apoptosis associated factor 1 |
| HMVEC | Human lung microvascular endothelial cell |
References
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar] [CrossRef] [Green Version]
- Jeanmougin, F.; Wurtz, J.M.; Le Douarin, B.; Chambon, P.; Losson, R. The bromodomain revisited. Trends Biochem. Sci 1997, 22, 151–153. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Winston, F.; Allis, C.D. The bromodomain: A chromatin-targeting module? Nat. Struct. Biol. 1999, 6, 601–604. [Google Scholar] [CrossRef]
- Jacobson, R.H.; Ladurner, A.G.; King, D.S.; Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 2000, 288, 1422–1425. [Google Scholar] [CrossRef]
- Tamkun, J.W.; Deuring, R.; Scott, M.P.; Kissinger, M.; Pattatucci, A.M.; Kaufman, T.C.; Kennison, J.A. brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 1992, 68, 561–572. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Owen, D.J.; Ornaghi, P.; Yang, J.C.; Lowe, N.; Evans, P.R.; Ballario, P.; Neuhaus, D.; Filetici, P.; Travers, A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000, 19, 6141–6149. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, R.; Zhou, M.M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Devel. 2009, 12, 659–665. [Google Scholar]
- Cuppen, E.; van Ham, M.; Pepers, B.; Wieringa, B.; Hendriks, W. Identification and molecular characterization of BP75, a novel bromodomain-containing protein. FEBS Lett. 1999, 459, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Andres Salazar Hernandez, M.; Herrema, H.; Delibasi, T.; Park, S.W. The role of BRD7 in embryo development and glucose metabolism. J. Cell Mol. Med. 2016, 20, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhou, J.; Liu, H.Y.; Zhou, M.; Wang, L.L.; Zhang, Q.H.; Yang, Y.X.; Xiong, W.; Shen, S.R.; Li, X.L.; et al. The transcriptional regulation role of BRD7 by binding to acetylated histone through bromodomain. J. Cell Biochem. 2006, 97, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Kzhyshkowska, J.; Rusch, A.; Wolf, H.; Dobner, T. Regulation of transcription by the heterogeneous nuclear ribonucleoprotein E1B-AP5 is mediated by complex formation with the novel bromodomain-containing protein BRD7. Biochem. J. 2003, 371, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, M.D.; Aslanian, A.; Dong, M.Q.; Yates, J.R., 3rd; Emerson, B.M. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 2008, 283, 32254–32263. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Liu, J.; Zhang, J.; Shen, W.; Huang, H.; Xu, C.; Dai, H.; Wu, J.; Shi, Y. Solution structure of BRD7 bromodomain and its interaction with acetylated peptides from histone H3 and H4. Biochem. Biophys. Res. Commun. 2007, 358, 435–441. [Google Scholar] [CrossRef]
- Staal, A.; Enserink, J.M.; Stein, J.L.; Stein, G.S.; van Wijnen, A.J. Molecular characterization of celtix-1, a bromodomain protein interacting with the transcription factor interferon regulatory factor 2. J. Cell Physiol. 2000, 185, 269–279. [Google Scholar] [CrossRef]
- Liu, Z.; Yan, M.; Liang, Y.; Liu, M.; Zhang, K.; Shao, D.; Jiang, R.; Li, L.; Wang, C.; Nussenzveig, D.R.; et al. Nucleoporin Seh1 Interacts with Olig2/Brd7 to Promote Oligodendrocyte Differentiation and Myelination. Neuron 2019, 102, 587–601.e587. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, M.; Liu, J.; He, Z.; Zhang, Y.; You, H.; Huang, J.; Lin, X.; Feng, X.H. Tumor suppressor bromodomain-containing protein 7 cooperates with Smads to promote transforming growth factor-beta responses. Oncogene 2017, 36, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Drost, J.; Mantovani, F.; Tocco, F.; Elkon, R.; Comel, A.; Holstege, H.; Kerkhoven, R.; Jonkers, J.; Voorhoeve, P.M.; Agami, R.; et al. BRD7 is a candidate tumour suppressor gene required for p53 function. Nat. Cell Biol. 2010, 12, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Burrows, A.E.; Smogorzewska, A.; Elledge, S.J. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc. Natl. Acad. Sci. USA 2010, 107, 14280–14285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harte, M.T.; O’Brien, G.J.; Ryan, N.M.; Gorski, J.J.; Savage, K.I.; Crawford, N.T.; Mullan, P.B.; Harkin, D.P. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res. 2010, 70, 2538–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Yoshihara, E.; He, N.; Hah, N.; Fan, W.; Pinto, A.F.M.; Huddy, T.; Wang, Y.; Ross, B.; Estepa, G.; et al. Vitamin D Switches BAF Complexes to Protect beta Cells. Cell 2018, 173, 1135–1149.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, M.; Okumura, F.; Tsukiyama, T.; Watanabe, M.; Miyajima, N.; Tanaka, J.; Imamura, M.; Hatakeyama, S. TRIM24 mediates ligand-dependent activation of androgen receptor and is repressed by a bromodomain-containing protein, BRD7, in prostate cancer cells. Biochim. Biophys. Acta 2009, 1793, 1828–1836. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.I.; Kim, W.; Choi, K.J.; Bae, S.; Jeong, J.H.; Kim, K.S. XIAP-associating factor 1, a transcriptional target of BRD7, contributes to endothelial cell senescence. Oncotarget 2016, 7, 5118–5130. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ma, J.; Zhang, B.C.; Li, X.L.; Shen, S.R.; Zhu, S.G.; Xiong, W.; Liu, H.Y.; Huang, H.; Zhou, M.; et al. BRD7, a novel bromodomain gene, inhibits G1-S progression by transcriptionally regulating some important molecules involved in ras/MEK/ERK and Rb/E2F pathways. J. Cell Physiol. 2004, 200, 89–98. [Google Scholar] [CrossRef]
- Zhang, Q.; Wei, L.; Yang, H.; Yang, W.; Yang, Q.; Zhang, Z.; Wu, K.; Wu, J. Bromodomain containing protein represses the Ras/Raf/MEK/ERK pathway to attenuate human hepatoma cell proliferation during HCV infection. Cancer Lett. 2016, 371, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, J.; Park, J.; Chung, J. BP75, bromodomain-containing M(r) 75,000 protein, binds dishevelled-1 and enhances Wnt signaling by inactivating glycogen synthase kinase-3 beta. Cancer Res. 2003, 63, 4792–4795. [Google Scholar]
- Peng, C.; Liu, H.Y.; Zhou, M.; Zhang, L.M.; Li, X.L.; Shen, S.R.; Li, G.Y. BRD7 suppresses the growth of Nasopharyngeal Carcinoma cells (HNE1) through negatively regulating beta-catenin and ERK pathways. Mol. Cell Biochem. 2007, 303, 141–149. [Google Scholar] [CrossRef]
- Park, Y.A.; Lee, J.W.; Kim, H.S.; Lee, Y.Y.; Kim, T.J.; Choi, C.H.; Choi, J.J.; Jeon, H.K.; Cho, Y.J.; Ryu, J.Y.; et al. Tumor suppressive effects of bromodomain-containing protein 7 (BRD7) in epithelial ovarian carcinoma. Clin. Cancer Res. 2014, 20, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.A.; Lee, J.W.; Choi, J.J.; Jeon, H.K.; Cho, Y.; Choi, C.; Kim, T.J.; Lee, N.W.; Kim, B.G.; Bae, D.S. The interactions between MicroRNA-200c and BRD7 in endometrial carcinoma. Gynecol. Oncol. 2012, 124, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xie, Y. BRD7-Mediated miR-3148 Inhibits Progression of Cervical Cancer by Targeting Wnt3a/beta-Catenin Pathway. Reprod. Sci. 2020, 27, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Lee, J.Y.; Cantley, L.C. BRD7, a tumor suppressor, interacts with p85alpha and regulates PI3K activity. Mol. Cell 2014, 54, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.W.; Herrema, H.; Salazar, M.; Cakir, I.; Cabi, S.; Basibuyuk Sahin, F.; Chiu, Y.H.; Cantley, L.C.; Ozcan, U. BRD7 regulates XBP1s’ activity and glucose homeostasis through its interaction with the regulatory subunits of PI3K. Cell Metab. 2014, 20, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golick, L.; Han, Y.; Kim, Y.; Park, S.W. BRD7 regulates the insulin-signaling pathway by increasing phosphorylation of GSK3beta. Cell Mol. Life Sci. 2018, 75, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Wang, Y.C.; Liu, X.J.; Wang, Q.; Zhang, C.M.; Zhang, L.P.; Liu, H.; Zhang, X.Y.; Mao, Y.; Ge, Z.M. BRD7 mediates hyperglycaemia-induced myocardial apoptosis via endoplasmic reticulum stress signalling pathway. J. Cell Mol. Med. 2017, 21, 1094–1105. [Google Scholar] [CrossRef]
- Zhao, R.; Liu, Y.; Wang, H.; Yang, J.; Niu, W.; Fan, S.; Xiong, W.; Ma, J.; Li, X.; Phillips, J.B.; et al. BRD7 plays an anti-inflammatory role during early acute inflammation by inhibiting activation of the NF-small ka, CyrillicB signaling pathway. Cell Mol. Immunol. 2017, 14, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Yan, A.; Yue, T.; Li, L.; Li, W.; Li, Q.; Li, J. Bromodomain-containing protein 7 deficiency augments atherosclerotic lesions in ApoE(-/-) mice. Biochem. Biophys. Res. Commun. 2018, 495, 2202–2208. [Google Scholar] [CrossRef]
- Niu, W.; Luo, Y.; Zhou, Y.; Li, M.; Wu, C.; Duan, Y.; Wang, H.; Fan, S.; Li, Z.; Xiong, W.; et al. BRD7 suppresses invasion and metastasis in breast cancer by negatively regulating YB1-induced epithelial-mesenchymal transition. J. Exp. Clin. Cancer Res. 2020, 39, 30. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.J.; Hu, K.S.; Chen, D.L.; Zeng, Z.L.; Luo, H.Y.; Wang, F.; Wang, D.S.; Wang, Z.Q.; He, F.; Xu, R.H. Prognostic relevance of BRD7 expression in colorectal carcinoma. Eur. J. Clin. Investig. 2013, 43, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Kim, Y.; Salazar Hernandez, M.A.; Han, Y.; Liu, R.; Park, S.W. BRD7 deficiency leads to the development of obesity and hyperglycemia. Sci. Rep. 2019, 9, 5327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Wang, Y.; Pan, Q.Z.; Tang, Y.; Wang, Q.J.; Pan, K.; Huang, L.X.; He, J.; Zhao, J.J.; Jiang, S.S.; et al. Bromodomain-containing protein 7 (BRD7) as a potential tumor suppressor in hepatocellular carcinoma. Oncotarget 2016, 7, 16248–16261. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, Y.; Zhu, G.; Liu, X.; Zhao, M.; Li, X.; Yang, Q. MicroRNA-410 promotes cell proliferation by targeting BRD7 in non-small cell lung cancer. FEBS Lett. 2015, 589, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, L.; Niu, Z.; Zhou, M.; Peng, C.; Li, X.; Deng, T.; Shi, L.; Tan, Y.; Li, G. Promoter methylation inhibits BRD7 expression in human nasopharyngeal carcinoma cells. BMC Cancer 2008, 8, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, K.; Liao, D.; Wu, W.; Han, A.J.; Shi, H.J.; Wang, F.; Wang, X.; Zhong, L.; Duan, T.; Wu, Y.; et al. Targeting the anaphase-promoting complex/cyclosome (APC/C)- bromodomain containing 7 (BRD7) pathway for human osteosarcoma. Oncotarget 2014, 5, 3088–3100. [Google Scholar] [CrossRef]
- Luo, J.; Field, S.J.; Lee, J.Y.; Engelman, J.A.; Cantley, L.C. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J. Cell Biol. 2005, 170, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Ueki, K.; Yballe, C.M.; Brachmann, S.M.; Vicent, D.; Watt, J.M.; Kahn, C.R.; Cantley, L.C. Increased insulin sensitivity in mice lacking p85beta subunit of phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Mauvais-Jarvis, F.; Ueki, K.; Fruman, D.A.; Hirshman, M.F.; Sakamoto, K.; Goodyear, L.J.; Iannacone, M.; Accili, D.; Cantley, L.C.; Kahn, C.R. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J. Clin. Investig. 2002, 109, 141–149. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Kondo, T.; Sajan, M.; Luo, J.; Bronson, R.; Asano, T.; Farese, R.; Cantley, L.C.; Kahn, C.R. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006, 3, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Dalle Pezze, P.; Sonntag, A.G.; Thien, A.; Prentzell, M.T.; Godel, M.; Fischer, S.; Neumann-Haefelin, E.; Huber, T.B.; Baumeister, R.; Shanley, D.P.; et al. A dynamic network model of mTOR signaling reveals TSC-independent mTORC2 regulation. Sci. Signal. 2012, 5, ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolensek, J.; Rupnik, M.S.; Stozer, A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015, 7, e1024405. [Google Scholar] [CrossRef] [Green Version]
- Stoffers, D.A. The development of beta-cell mass: Recent progress and potential role of GLP-1. Horm. Metab. Res. 2004, 36, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takiishi, T.; Gysemans, C.; Bouillon, R.; Mathieu, C. Vitamin D and diabetes. Endocrinol. Metab. Clin. 2010, 39, 419–446, table of contents. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Dan, H.C.; Sun, M.; Yang, L.; Feldman, R.I.; Sui, X.M.; Ou, C.C.; Nellist, M.; Yeung, R.S.; Halley, D.J.; Nicosia, S.V.; et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J. Biol. Chem. 2002, 277, 35364–35370. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriburi, R.; Bommiasamy, H.; Buldak, G.L.; Robbins, G.R.; Frank, M.; Jackowski, S.; Brewer, J.W. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J. Biol. Chem. 2007, 282, 7024–7034. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Lee, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B.; et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003, 289, 76–79. [Google Scholar] [CrossRef]
- Van Gaal, L.F.; Mertens, I.L.; De Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.B.; Lavie, C.J.; Blair, S.N. Obesity and Cardiovascular Disease. Circ. Res. 2016, 118, 1752–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pischon, T.; Nothlings, U.; Boeing, H. Obesity and cancer. Proc. Nutr. Soc. 2008, 67, 128–145. [Google Scholar] [CrossRef] [Green Version]
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Tian, J.; Zhong, R.; Tian, Y.; Chen, W.; Qian, J.; Zou, L.; Xiao, M.; Shen, N.; Yang, H.; et al. Genetic variants in the SWI/SNF complex and smoking collaborate to modify the risk of pancreatic cancer in a Chinese population. Mol. Carcinog. 2015, 54, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, B.; Gao, S. BRD7 Acts as a Tumor Suppressor Gene in Lung Adenocarcinoma. PLoS ONE 2016, 11, e0156701. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, A.; Subramaniam, R.; Narayanan, V.; Annamalai, T.; Ramanathan, A. BRD7 promoter hypermethylation as an indicator of well differentiated oral squamous cell carcinomas. Asian Pac. J. Cancer Prev. 2015, 16, 1615–1619. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Zhao, J.; Niu, L.; An, G.; Guo, Y.; Ni, L. Up-Regulation of MiR-300 Promotes Proliferation and Invasion of Osteosarcoma by Targeting BRD7. PLoS ONE 2015, 10, e0127682. [Google Scholar] [CrossRef]
- Hu, K.; Wu, W.; Li, Y.; Lin, L.; Chen, D.; Yan, H.; Xiao, X.; Chen, H.; Chen, Z.; Zhang, Y.; et al. Poly(ADP-ribosyl)ation of BRD7 by PARP1 confers resistance to DNA-damaging chemotherapeutic agents. EMBO Rep. 2019, 20, e46166. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, H.; Xu, X.; Zhou, H.; Li, X.; Peng, C.; Shen, S.; Xiong, W.; Ma, J.; Zeng, Z.; et al. Identification of nuclear localization signal that governs nuclear import of BRD7 and its essential roles in inhibiting cell cycle progression. J. Cell Biochem. 2006, 98, 920–930. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Deiry, W.S. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016, 76, 5189–5191. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Drost, J.; Voorhoeve, P.M.; Del Sal, G.; Agami, R. Gene regulation and tumor suppression by the bromodomain-containing protein BRD7. Cell Cycle 2010, 9, 2777–2781. [Google Scholar] [CrossRef] [Green Version]
- Kohno, K.; Izumi, H.; Uchiumi, T.; Ashizuka, M.; Kuwano, M. The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays 2003, 25, 691–698. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef]
- Liang, Y.; Dong, B.; Shen, J.; Ma, C.; Ma, Z. Clinical significance of bromodomain-containing protein 7 and its association with tumor progression in prostate cancer. Oncol. Lett. 2019, 17, 849–856. [Google Scholar] [CrossRef] [Green Version]
- Torres-Padilla, M.E.; Zernicka-Goetz, M. Role of TIF1alpha as a modulator of embryonic transcription in the mouse zygote. J. Cell Biol. 2006, 174, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Pines, J. The APC/C: A smorgasbord for proteolysis. Mol. Cell 2009, 34, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, M.; Shen, M.; Kong, D.; Zhang, F.; Shao, J.; Tan, S.; Wang, S.; Chen, A.; Cao, P.; et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 2020, 36, 101619. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Edelman, S.V. Type II diabetes mellitus. Adv. Intern. Med. 1998, 43, 449–500. [Google Scholar]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, T.A.; Grant, P. Drug therapies in type 2 diabetes: An era of personalised medicine. Clin. Med. 2016, 16, 441–447. [Google Scholar] [CrossRef]
- Park, S.W.; Ozcan, U. Potential for therapeutic manipulation of the UPR in disease. Semin. Immunopathol. 2013, 35, 351–373. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Zhou, Y.; Lee, J.; Ozcan, U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 19320–19325. [Google Scholar] [CrossRef] [Green Version]
- Winnay, J.N.; Boucher, J.; Mori, M.A.; Ueki, K.; Kahn, C.R. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 2010, 16, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Bavelloni, A.; Santi, S.; Sirri, A.; Riccio, M.; Faenza, I.; Zini, N.; Cecchi, S.; Ferri, A.; Auron, P.; Maraldi, N.M.; et al. Phosphatidylinositol 3-kinase translocation to the nucleus is induced by interleukin 1 and prevented by mutation of interleukin 1 receptor in human osteosarcoma Saos-2 cells. J. Cell Sci. 1999, 112 Pt 5, 631–640. [Google Scholar]
- Martelli, A.M.; Borgatti, P.; Bortul, R.; Manfredini, M.; Massari, L.; Capitani, S.; Neri, L.M. Phosphatidylinositol 3-kinase translocates to the nucleus of osteoblast-like MC3T3-E1 cells in response to insulin-like growth factor I and platelet-derived growth factor but not to the proapoptotic cytokine tumor necrosis factor alpha. J. Bone Miner. Res. 2000, 15, 1716–1730. [Google Scholar] [CrossRef] [PubMed]
- Neri, L.M.; Milani, D.; Bertolaso, L.; Stroscio, M.; Bertagnolo, V.; Capitani, S. Nuclear translocation of phosphatidylinositol 3-kinase in rat pheochromocytoma PC 12 cells after treatment with nerve growth factor. Cell Mol. Biol. (Noisy-le-grand) 1994, 40, 619–626. [Google Scholar]
- Taniguchi, C.M.; Aleman, J.O.; Ueki, K.; Luo, J.; Asano, T.; Kaneto, H.; Stephanopoulos, G.; Cantley, L.C.; Kahn, C.R. The p85alpha regulatory subunit of phosphoinositide 3-kinase potentiates c-Jun N-terminal kinase-mediated insulin resistance. Mol. Cell Biol. 2007, 27, 2830–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, C.M.; Tran, T.T.; Kondo, T.; Luo, J.; Ueki, K.; Cantley, L.C.; Kahn, C.R. Phosphoinositide 3-kinase regulatory subunit p85alpha suppresses insulin action via positive regulation of PTEN. Proc. Natl. Acad. Sci. USA 2006, 103, 12093–12097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, C.M.; Winnay, J.; Kondo, T.; Bronson, R.T.; Guimaraes, A.R.; Aleman, J.O.; Luo, J.; Stephanopoulos, G.; Weissleder, R.; Cantley, L.C.; et al. The phosphoinositide 3-kinase regulatory subunit p85alpha can exert tumor suppressor properties through negative regulation of growth factor signaling. Cancer Res. 2010, 70, 5305–5315. [Google Scholar] [CrossRef] [Green Version]
- Ueki, K.; Fruman, D.A.; Yballe, C.M.; Fasshauer, M.; Klein, J.; Asano, T.; Cantley, L.C.; Kahn, C.R. Positive and negative roles of p85 alpha and p85 beta regulatory subunits of phosphoinositide 3-kinase in insulin signaling. J. Biol. Chem. 2003, 278, 48453–48466. [Google Scholar] [CrossRef] [Green Version]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Chagpar, R.B.; Links, P.H.; Pastor, M.C.; Furber, L.A.; Hawrysh, A.D.; Chamberlain, M.D.; Anderson, D.H. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 5471–5476. [Google Scholar] [CrossRef] [Green Version]
- Rabinovsky, R.; Pochanard, P.; McNear, C.; Brachmann, S.M.; Duke-Cohan, J.S.; Garraway, L.A.; Sellers, W.R. p85 Associates with unphosphorylated PTEN and the PTEN-associated complex. Mol. Cell Biol. 2009, 29, 5377–5388. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, B.; Kaidanovich, O.; Talior, I.; Eldar-Finkelman, H. Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J. Pharmacol. Exp. Ther. 2003, 305, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochemical. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [Green Version]
- Rayasam, G.V.; Tulasi, V.K.; Sodhi, R.; Davis, J.A.; Ray, A. Glycogen synthase kinase 3: More than a namesake. Br. J. Pharmacol. 2009, 156, 885–898. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Foster, D.A. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2015, 2, 807–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, R.; Guo, C.; Jiang, S.; Yang, J.; Xu, Y.; Liu, Y.; Fan, L.; Xiong, W.; Ma, J.; et al. Knockout of BRD7 results in impaired spermatogenesis and male infertility. Sci. Rep. 2016, 6, 21776. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Cao, W.; Zhou, M.; Li, C.; Luo, Y.; Wang, H.; Zhao, R.; Jiang, S.; Yang, J.; Liu, Y.; et al. Inactivation of BRD7 results in impaired cognitive behavior and reduced synaptic plasticity of the medial prefrontal cortex. Behav. Brain Res. 2015, 286, 1–10. [Google Scholar] [CrossRef]
- Heffner, C.S.; Herbert Pratt, C.; Babiuk, R.P.; Sharma, Y.; Rockwood, S.F.; Donahue, L.R.; Eppig, J.T.; Murray, S.A. Supporting conditional mouse mutagenesis with a comprehensive cre characterization resource. Nat. Commun. 2012, 3, 1218. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Function | Mechanism | Ref |
|---|---|---|
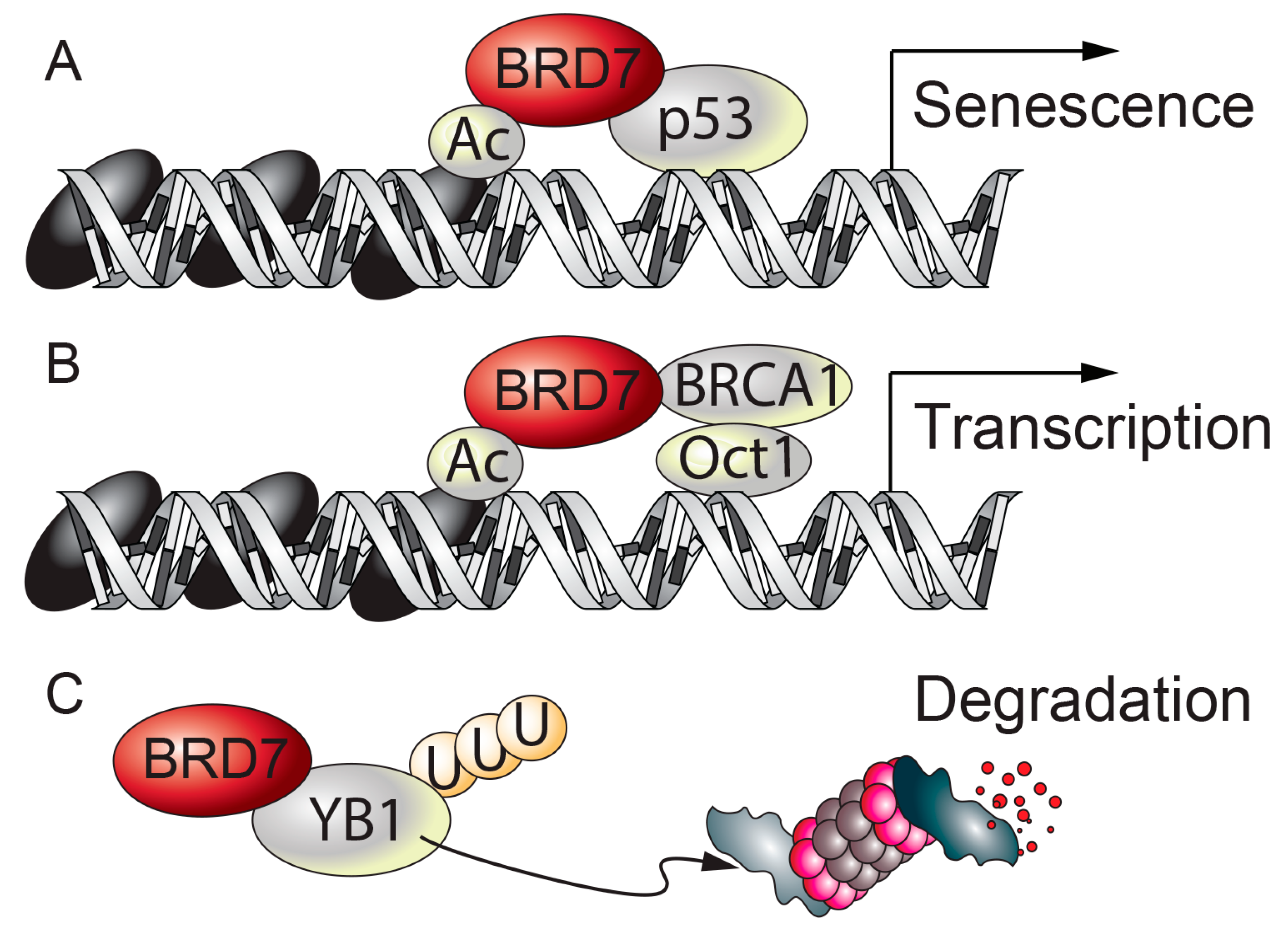
| Chromatin Remodeling | BRD7 binds to acetylated histone H3 through its bromodomain and BRD7 is involved in chromatin remodeling. | [14] |
| The bromodomain of BRD7 contains the left-handed four-helix bundle topology, which binds to acetylated lysine on histone H3 or H4. | [17] | |
| BRD7 is a component of the PBAF chromatin remodeling complex. | [16] | |
| Transcriptional Regulation | BRD7 interacts with interferon regulatory factor 2 (IRF2). | [18] |
| Seh1 recruits Olig2 and BRD7 to form a complex and increases the transcription of myelination-associated genes and chromatin modification. | [19] | |
| BRD7 interacts with E1B-AP5, which is involved in mRNA processing and transport. The complex associates with histones. | [15] | |
| BRD7 binds to Smad proteins and increases TGFβ-Smad-dependent transcriptional activity. | [20] | |
| BRD7 interacts with p53 and regulates the transcriptional activity of a subset of p53 target genes involved in induction of replicative and oncogenic stress senescence. | [21,22] | |
| BRD7 facilitates the recruitment of BRCA1 and Oct-1 to the ESR1 promoter and regulates the transcription of ERα. | [23] | |
| The association between vitamin D receptor and PBAF is increased by BRD7 in β cells, leading to transcriptional activation of genes involved in anti-inflammatory responses and maintenance of β cell function. | [24] | |
| BRD7 negatively regulates the transcriptional activity of androgen receptor in the CWR22Rv1 prostate cancer cell line by binding to TRIM24, an activator of androgen receptor. | [25] | |
| BRD7 is required for the expression of the tumor suppressor XIAP-associating factor 1 in human pulmonary microvascular endothelial cells. | [26] | |
| Cell Cycle Progression | Overexpression of BRD7 in HNE2 cells inhibits the G1-S phase transition and downregulates expression of proteins in the ras/MEK/ER and E2F/Rb pathways. | [27] |
| BRD7 attenuates ras/raf/MEK/ERK signaling and represses cell proliferation. | [28] | |
| Wnt Signaling | BRD7 interacts with DVL1 and promotes Wnt signaling in HEK293T cells in a DVL1-dependent manner by inhibiting the activity of GSK3β and increasing the nuclear translocation of β-catenin. | [29] |
| BRD7 overexpression in HNE1 nasopharyngeal carcinoma cells inhibits nuclear accumulation of β-catenin. | [30] | |
| BRD7 negatively regulates the β-catenin pathway in A2780 and SKOV3 ovarian cancer cell lines by inhibiting nuclear translocation of β-catenin. | [31] | |
| BRD7 expression is inhibited by microRNA-200c in HEC-1A and Ishikawa endometrial carcinoma cells, which leads to increased nuclear translocation of β-catenin and consequent increased transcription of cyclin D1 and c-myc. | [32] | |
| BRD7 transcriptionally upregulates miR-3148 in C33A cells, which reduces Wnt3a expression, and thus inhibits oncogenic Wnt3a/β-catenin signaling. | [33] | |
| Insulin Signaling | BRD7 interacts with p85α/β, the regulatory subunits of PI3K, and increases their nuclear translocation. This increases PI3K-Akt signaling in the liver, but attenuates Akt activity in HeLa cervical cancer cells. | [34,35] |
| BRD7 increases phosphorylation of glycogen synthase kinase 3β (GSK3β) at residue Ser9, which leads to inactivation of GSK3β. | [36] | |
| Unfolded Protein Response | BRD7 increases the nuclear translocation of the spliced form of X-box binding protein 1 (XBP1s), upregulates the transcription of XBP1-target genes, and relieves ER stress. | [35] |
| BRD7 is required for hyperglycemia-induced apoptosis in H9c2 cardiomyoblasts. Reduction of BRD7 in the heart of diabetic rats alleviates ER stress-induced myocardial apoptosis. | [37] | |
| Inflammation | MEFs that lack BRD7 and EIIα-Cre-derived BRD7-deficient mice show increased nuclear translocation of p65 and NF-κB transcriptional activity. | [38] |
| BRD7 knockdown in ApoE-knockout mice promotes atherosclerotic lesion formation and vascular inflammation by promoting the transcriptional activity of NF-κB. | [39] |
| Tissue Type | Disease | Mechanism of Progression | Ref. |
|---|---|---|---|
| Breast | Cancer | The BRD7 locus is frequently found deleted in human breast tumors. BRD7 is required for p53-mediated transcription of a subset of p53 target genes. | [21] |
| Cancer | BRD7 overexpression suppresses the epithelial-mesenchymal transition and metastasis of breast cancer cells through increasing degradation of the oncogenic protein YB1. | [40] | |
| Colon | Cancer | BRD7 is downregulated in colorectal cancer tissues. BRD7 expression level is correlated with survival time in colorectal cancer patients. | [41] |
| Liver | Obesity | BRD7 levels are decreased in the liver of genetically obese ob/ob and high-fat diet-induced obese mice. | [35] |
| Obesity | Heterozygous whole-body and liver-specific knockout of BRD7 leads to increased weight gain in mice, exacerbated by high-fat diet feeding. Long-term upregulation of hepatic BRD7 reduces weight gain in mice challenged with a high-fat diet. | [42] | |
| Type 2 diabetes | Upregulation of BRD7 in the liver of obese and type 2 diabetic mice decreases blood glucose levels and improves glucose homeostasis. | [35,42] | |
| Cancer | BRD7 is downregulated in hepatocellular carcinoma (HCC), and higher BRD7 levels are correlated with improved outcomes in HCC patients. BRD7 inhibits HCC tumor growth in a xenograft mouse model. | [43] | |
| Lung | Cancer | BRD7 expression levels are downregulated in non-small cell lung cancer (NSCLC). Upregulated expression of microRNA-410 in NSCLC leads to decreased BRD7 expression and increased Akt phosphorylation. | [44] |
| Nasopharynx | Cancer | BRD7 expression levels are downregulated in nasopharyngeal carcinoma. | [27] |
| Cancer | High methylation frequency of the BRD7 promoter is found in tumor and blood samples of patients with nasopharyngeal carcinoma. | [45] | |
| Bone | Cancer | BRD7 is degraded by anaphase promoting complex in U2OS osteosarcoma cells. Upregulation of degradation-resistant BRD7 reduces cell growth and tumorigenesis. | [46] |
| Ovary | Cancer | The expression of BRD7 is decreased in high-grade serous ovarian cancer tissues. Overexpression of BRD7 in A2780 and SKOV3 ovarian cancer cell lines increases apoptosis and inhibits cell migration. | [31] |
| Pancreas | Diabetes | BRD7 increases the association between vitamin D receptor and PBAF in β cells. This complex maintains β cell function and reduces glucose levels in db/db and streptozotocin-treated mice. | [24] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.W.; Lee, J.M. Emerging Roles of BRD7 in Pathophysiology. Int. J. Mol. Sci. 2020, 21, 7127. https://doi.org/10.3390/ijms21197127
Park SW, Lee JM. Emerging Roles of BRD7 in Pathophysiology. International Journal of Molecular Sciences. 2020; 21(19):7127. https://doi.org/10.3390/ijms21197127
Chicago/Turabian StylePark, Sang Won, and Junsik M. Lee. 2020. "Emerging Roles of BRD7 in Pathophysiology" International Journal of Molecular Sciences 21, no. 19: 7127. https://doi.org/10.3390/ijms21197127
APA StylePark, S. W., & Lee, J. M. (2020). Emerging Roles of BRD7 in Pathophysiology. International Journal of Molecular Sciences, 21(19), 7127. https://doi.org/10.3390/ijms21197127