The Emerging Role of the RNA-Binding Protein SFPQ in Neuronal Function and Neurodegeneration


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The RNA-Binding Protein SFPQ
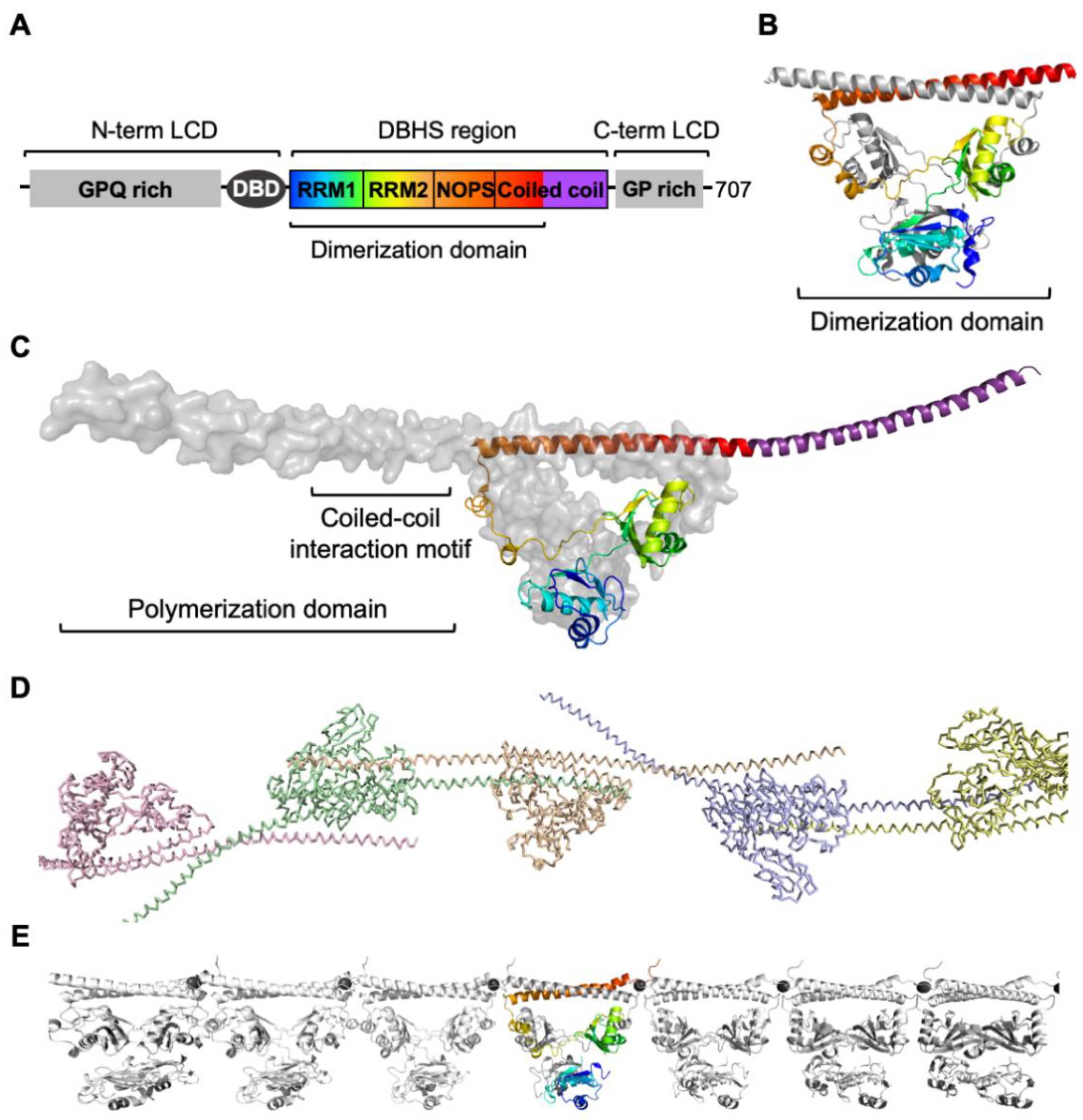
2.1. Structure of SFPQ
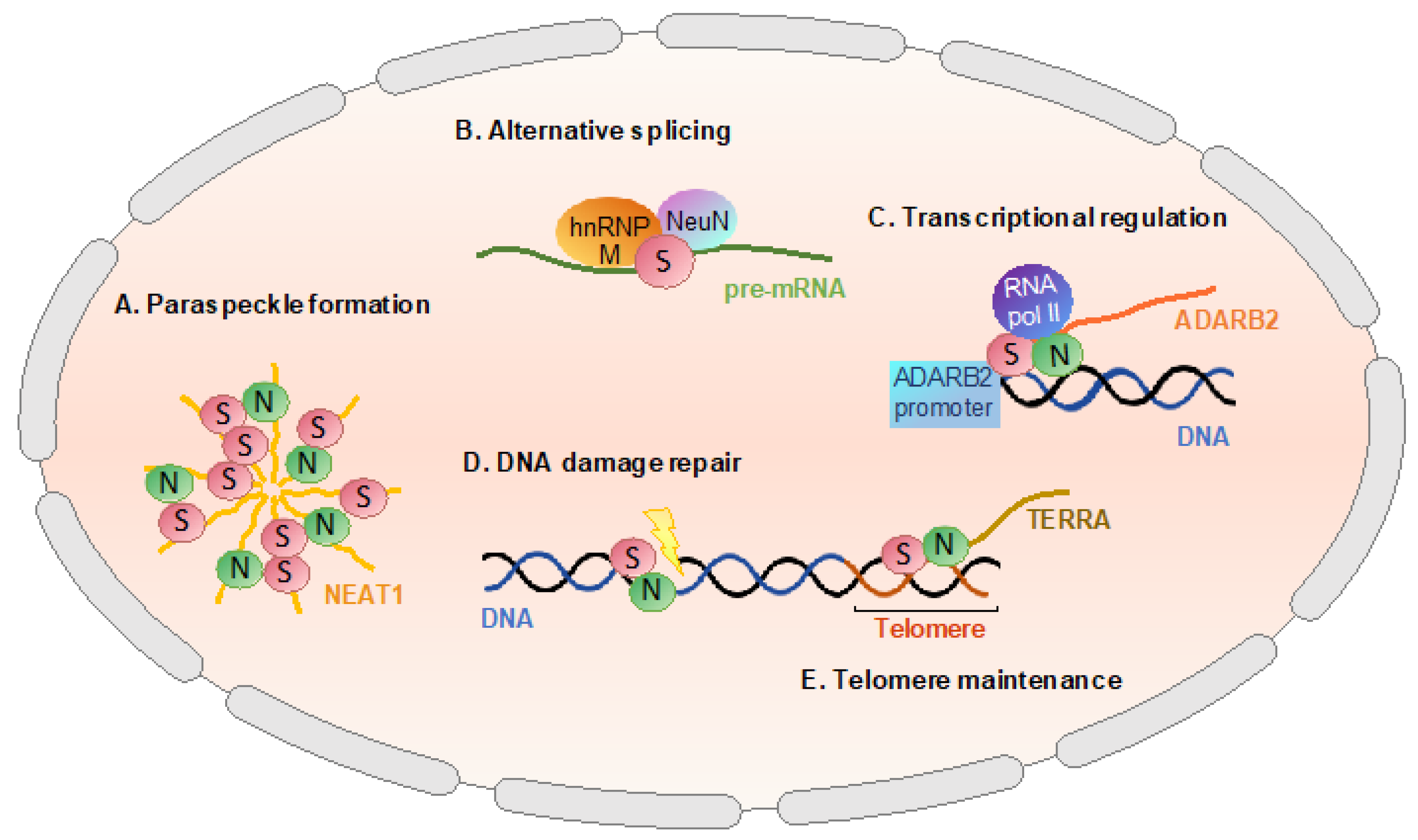
2.2. Reported Nuclear Function of SFPQ
2.2.1. Paraspeckle Formation
2.2.2. Alternative Splicing
2.2.3. Transcriptional Regulation
2.2.4. DNA Damage Repair and Maintenance of Genome Stability
3. SFPQ in Neuronal Development and Maintenance of Neuronal Function
3.1. Transcriptional and Post-Transcriptional Regulation to Develop and Maintain Neuronal Function
3.2. The Role of the Cytoplasmic Pool of SFPQ in Axon Growth and Survival
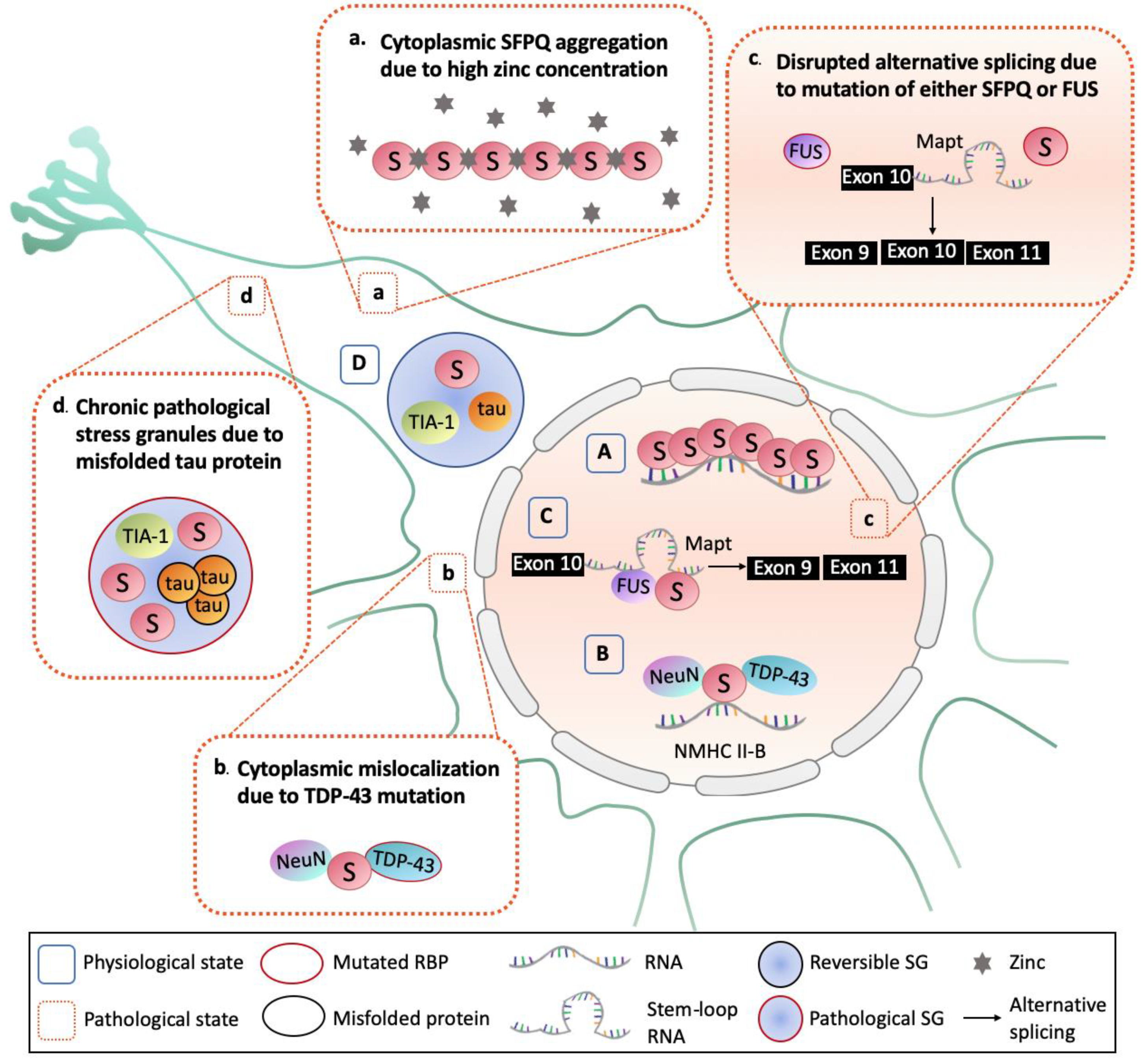
4. Implications of SFPQ in Neurodegenerative Diseases
4.1. Nuclear Depletion and Cytoplasmic Aggregation of SFPQ
4.2. Abnormal RNA Splicing
4.3. Persistent Pathological Stress Granules
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADARB2 | adenosine deaminase B2 |
| ALS | amyotrophic lateral sclerosis |
| AR | androgen receptor |
| ATM | ataxia telangiectasis mutated |
| CDK9 | cyclin-dependent kinase 9 |
| DBD | DNA-binding domain |
| DBHS | Drosophila behavior human splicing |
| ESS1 | exonic splicing silencer 1 |
| FTLD | frontotemporal lobar degeneration |
| FUS | fused in sarcoma |
| HDAC | histone deacetylase |
| hnRNP | heterogeneous nuclear ribonucleoprotein |
| HR | homologous recombination |
| iPSC | induced-pluripotent stem cell |
| LCD | low-complexity domain |
| MATR3 | Matrin 3 |
| NEAT1 | nuclear paraspeckle assembly transcript 1 |
| NeuN | neuronal nuclei |
| NHEJ | nonhomologous end-joining |
| NHR | nuclear hormone receptor |
| NMHC | nonmuscle myosin heavy chain |
| NONO | Non-POU domain-containing octamer-binding protein |
| NOPS | NonA/paraspeckles |
| PPAR | peroxisome proliferator-activated receptor |
| PR | progesterone receptor |
| PSPC1 | paraspeckle protein component 1 |
| PSF | PTB-associated splicing factor |
| PTB | polypyrimidine tract-binding protein |
| RBP | RNA-binding protein |
| RXR | retinoid X receptor |
| RRM | RNA-recognition motif |
| SFPQ | splicing factor proline- and glutamine-rich |
| STAT6 | signal transducer and activator of transcription 6 |
| TDP-43 | TAR DNA-binding protein 43 |
| TR | thyroid hormone receptor |
| TERRA | telomere repeat-containing long noncoding RNA |
References
- Albert, F.W.; Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015, 16, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef] [PubMed]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Bond, C.S.; Fox, A.H. The DBHS proteins SFPQ, NONO and PSPC1: A multipurpose molecular scaffold. Nucleic Acids Res. 2016, 44, 3989–4004. [Google Scholar] [CrossRef]
- Ha, K.; Takeda, Y.; Dynan, W.S. Sequences in PSF/SFPQ mediate radioresistance and recruitment of PSF/SFPQ-containing complexes to DNA damage sites in human cells. DNA Repair (Amst) 2011, 10, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Sadowska, A.; Bekere, I.; Ho, D.; Gully, B.S.; Lu, Y.; Iyer, K.S.; Trewhella, J.; Fox, A.H.; Bond, C.S. The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation. Nucleic Acids Res. 2015, 43, 3826–3840. [Google Scholar] [CrossRef]
- Patton, J.G.; Porro, E.B.; Galceran, J.; Tempst, P.; Nadal-Ginard, B. Cloning and characterization of PSF a novel pre mRNA splicing factor. Genes Dev. 1993, 7, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Hewage, T.W.; Caria, S.; Lee, M. A new crystal structure and small-angle X-ray scattering analysis of the homodimer of human SFPQ. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2019, 75, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Ringuet, M.; Whitten, A.E.; Caria, S.; Lim, Y.W.; Badhan, R.; Anggono, V.; Lee, M. Structural basis of the zinc-induced cytoplasmic aggregation of the RNA-binding protein SFPQ. Nucleic Acids Res. 2020, 48, 3356–3365. [Google Scholar] [CrossRef] [Green Version]
- Thomas-Jinu, S.; Gordon, P.M.; Fielding, T.; Taylor, R.; Smith, B.N.; Snowden, V.; Blanc, E.; Vance, C.; Topp, S.; Wong, C.H.; et al. Non-nuclear Pool of Splicing Factor SFPQ Regulates Axonal Transcripts Required for Normal Motor Development. Neuron 2017, 94, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Passon, D.M.; Lee, M.; Rackham, O.; Stanley, W.A.; Sadowska, A.; Filipovska, A.; Fox, A.H.; Bond, C.S. Structure of the heterodimer of human NONO and paraspeckle protein component 1 and analysis of its role in subnuclear body formation. Proc. Natl. Acad. Sci. USA 2012, 109, 4846–4850. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Casas Garcia, G.P.; Perugini, M.A.; Fox, A.H.; Bond, C.S.; Lee, M. Crystal structure of a SFPQ/PSPC1 heterodimer provides insights into preferential heterodimerization of human DBHS family proteins. J. Biol. Chem. 2018, 293, 6593–6602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Passon, D.M.; Hennig, S.; Fox, A.H.; Bond, C.S. Construct optimization for studying protein complexes: Obtaining diffraction-quality crystals of the pseudosymmetric PSPC1-NONO heterodimer. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, S.; Ikei, A.; Taguchi, Y.; Tabuchi, Y.; Fujimoto, N.; Obinata, M.; Uesugi, S.; Kurihara, Y. PSPC1, NONO, and SFPQ are expressed in mouse Sertoli cells and may function as coregulators of androgen receptor-mediated transcription. Biol. Reprod. 2006, 75, 352–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shav-Tal, Y.; Zipori, D. PSF and p54(nrb)/NonO--multi-functional nuclear proteins. FEBS Lett. 2002, 531, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Yarosh, C.A.; Iacona, J.R.; Lutz, C.S.; Lynch, K.W. PSF: Nuclear busy-body or nuclear facilitator? Wiley Interdiscip. Rev. RNA 2015, 6, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.H.; Lam, Y.W.; Leung, A.K.; Lyon, C.E.; Andersen, J.; Mann, M.; Lamond, A.I. Paraspeckles: A novel nuclear domain. Curr. Biol. 2002, 12, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.T.; Ideue, T.; Sano, M.; Mituyama, T.; Hirose, T. MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl. Acad. Sci. USA 2009, 106, 2525–2530. [Google Scholar] [CrossRef] [Green Version]
- Naganuma, T.; Nakagawa, S.; Tanigawa, A.; Sasaki, Y.F.; Goshima, N.; Hirose, T. Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 2012, 31, 4020–4034. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Nakagawa, S.; Hirose, T. Architectural RNAs for Membraneless Nuclear Body Formation. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.H.; Nakagawa, S.; Hirose, T.; Bond, C.S. Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem. Sci. 2018, 43, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marko, M.; Leichter, M.; Patrinou-Georgoula, M.; Guialis, A. hnRNP M interacts with PSF and p54(nrb) and co-localizes within defined nuclear structures. Exp. Cell Res. 2010, 316, 390–400. [Google Scholar] [CrossRef]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.K.; Kim, Y.C.; Adelstein, R.S.; Kawamoto, S. Fox-3 and PSF interact to activate neural cell-specific alternative splicing. Nucleic Acids Res. 2011, 39, 3064–3078. [Google Scholar] [CrossRef] [Green Version]
- Damianov, A.; Ying, Y.; Lin, C.H.; Lee, J.A.; Tran, D.; Vashisht, A.A.; Bahrami-Samani, E.; Xing, Y.; Martin, K.C.; Wohlschlegel, J.A.; et al. Rbfox Proteins Regulate Splicing as Part of a Large Multiprotein Complex LASR. Cell 2016, 165, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Rothrock, C.; Cannon, B.; Hahm, B.; Lynch, K.W. A conserved signal-responsive sequence mediates activation-induced alternative splicing of CD45. Mol. Cell 2003, 12, 1317–1324. [Google Scholar] [CrossRef]
- Melton, A.A.; Jackson, J.; Wang, J.; Lynch, K.W. Combinatorial control of signal-induced exon repression by hnRNP L and PSF. Mol. Cell. Biol. 2007, 27, 6972–6984. [Google Scholar] [CrossRef] [Green Version]
- Heyd, F.; Lynch, K.W. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol. Cell 2010, 40, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Emili, A.; Shales, M.; McCracken, S.; Xie, W.; Tucker, P.W.; Kobayashi, R.; Blencowe, B.J.; Ingles, C.J. Splicing and transcription-associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. RNA (N. Y. NY) 2002, 8, 1102–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameoka, S.; Duque, P.; Konarska, M.M. p54(nrb) associates with the 5′ splice site within large transcription/splicing complexes. EMBO J. 2004, 23, 1782–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosonina, E.; Ip, J.Y.; Calarco, J.A.; Bakowski, M.A.; Emili, A.; McCracken, S.; Tucker, P.; Ingles, C.J.; Blencowe, B.J. Role for PSF in mediating transcriptional activator-dependent stimulation of pre-mRNA processing in vivo. Mol. Cell. Biol. 2005, 25, 6734–6746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, T.; Virnicchi, G.; Tanigawa, A.; Naganuma, T.; Li, R.; Kimura, H.; Yokoi, T.; Nakagawa, S.; Benard, M.; Fox, A.H.; et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell 2014, 25, 169–183. [Google Scholar] [CrossRef]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev. 1999, 20, 321–344. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Shylnova, O.; Challis, J.R.; Lye, S.J. Identification and characterization of the protein-associated splicing factor as a negative co-regulator of the progesterone receptor. J. Biol. Chem. 2005, 280, 13329–13340. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Sweet, J.; Challis, J.R.; Brown, T.; Lye, S.J. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol. Cell. Biol. 2007, 27, 4863–4875. [Google Scholar] [CrossRef] [Green Version]
- Mathur, M.; Tucker, P.W.; Samuels, H.H. PSF is a novel corepressor that mediates its effect through Sin3A and the DNA binding domain of nuclear hormone receptors. Mol. Cell. Biol. 2001, 21, 2298–2311. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Zhang, X.; Fu, X.; Zhang, X.; Gao, X.; Zhu, M.; Wang, X.; Yang, Z.; Jensen, O.N.; Saarikettu, J.; et al. PTB-associated splicing factor (PSF) functions as a repressor of STAT6-mediated Ig epsilon gene transcription by recruitment of HDAC1. J. Biol. Chem. 2011, 286, 3451–3459. [Google Scholar] [CrossRef] [Green Version]
- Bladen, C.L.; Udayakumar, D.; Takeda, Y.; Dynan, W.S. Identification of the polypyrimidine tract binding protein-associated splicing factor.p54(nrb) complex as a candidate DNA double-strand break rejoining factor. J. Biol. Chem. 2005, 280, 5205–5210. [Google Scholar] [CrossRef] [Green Version]
- de Silva, H.C.; Lin, M.Z.; Phillips, L.; Martin, J.L.; Baxter, R.C. IGFBP-3 interacts with NONO and SFPQ in PARP-dependent DNA damage repair in triple-negative breast cancer. Cell. Mol. Life Sci. 2019, 76, 2015–2030. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Lerenthal, Y.; Wang, S.Y.; Chen, D.J.; Shiloh, Y. Involvement of Matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle 2010, 9, 1568–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Sun, J.; Kinomura, A.; Fukuto, A.; Horikoshi, Y.; Tashiro, S. Matrin3 promotes homologous recombinational repair by regulation of RAD51. J. Biochem. 2019, 166, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, L.; Li, Z.; Li, S.; Dynan, W.S. SFPQ*NONO and XLF function separately and together to promote DNA double-strand break repair via canonical nonhomologous end joining. Nucleic Acids Res. 2017, 45, 1848–1859. [Google Scholar] [CrossRef]
- Morozumi, Y.; Takizawa, Y.; Takaku, M.; Kurumizaka, H. Human PSF binds to RAD51 and modulates its homologous-pairing and strand-exchange activities. Nucleic Acids Res. 2009, 37, 4296–4307. [Google Scholar] [CrossRef] [Green Version]
- Petti, E.; Buemi, V.; Zappone, A.; Schillaci, O.; Broccia, P.V.; Dinami, R.; Matteoni, S.; Benetti, R.; Schoeftner, S. SFPQ and NONO suppress RNA:DNA-hybrid-related telomere instability. Nat. Commun. 2019, 10, 1001. [Google Scholar] [CrossRef] [Green Version]
- Brookes, E.; Riccio, A. Location, location, location: Nuclear structure regulates gene expression in neurons. Curr. Opin. Neurobiol. 2019, 59, 16–25. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, A.; Iida, K.; Tsubota, T.; Hosokawa, M.; Denawa, M.; Brown, J.B.; Ninomiya, K.; Ito, M.; Kimura, H.; Abe, T.; et al. Loss of Sfpq Causes Long-Gene Transcriptopathy in the Brain. Cell Rep. 2018, 23, 1326–1341. [Google Scholar] [CrossRef]
- Iida, K.; Hagiwara, M.; Takeuchi, A. Multilateral Bioinformatics Analyses Reveal the Function-Oriented Target Specificities and Recognition of the RNA-Binding Protein SFPQ. iScience 2020, 23, 101325. [Google Scholar] [CrossRef]
- Wang, G.; Yang, H.; Yan, S.; Wang, C.E.; Liu, X.; Zhao, B.; Ouyang, Z.; Yin, P.; Liu, Z.; Zhao, Y.; et al. Cytoplasmic mislocalization of RNA splicing factors and aberrant neuronal gene splicing in TDP-43 transgenic pig brain. Mol. Neurodegener. 2015, 10, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigaki, S.; Fujioka, Y.; Okada, Y.; Riku, Y.; Udagawa, T.; Honda, D.; Yokoi, S.; Endo, K.; Ikenaka, K.; Takagi, S.; et al. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Rep. 2017, 18, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Rubin, J.; Sive, H. Whitesnake/sfpq is required for cell survival and neuronal development in the zebrafish. Dev. Dyn. 2007, 236, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.T.; Sakamoto, H.; Inoue, K. Interaction and colocalization of HERMES/RBPMS with NonO, PSF, and G3BP1 in neuronal cytoplasmic RNP granules in mouse retinal line cells. Genes Cells 2015, 20, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Dohmae, N.; Hirokawa, N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron 2004, 43, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Cosker, K.E.; Fenstermacher, S.J.; Pazyra-Murphy, M.F.; Elliott, H.L.; Segal, R.A. The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 2016, 19, 690–696. [Google Scholar] [CrossRef]
- Fukuda, Y.; Pazyra-Murphy, M.F.; Tasdemir-Yilmaz, O.E.; Li, Y.; Rose, L.; Yeoh, Z.C.; Vangos, N.E.; Geffken, E.A.; Seo, H.S.; Adelmant, G.; et al. Fast Transport of RNA Granules by Direct Interactions with KIF5A/KLC1 Motors Prevents Axon Degeneration. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Shu, R.; Zhu, Y. Dysregulation and Dislocation of SFPQ Disturbed DNA Organization in Alzheimer’s Disease and Frontotemporal Dementia. J. Alzheimer’s Dis. 2018, 61, 1311–1321. [Google Scholar] [CrossRef]
- Ke, Y.D.; Dramiga, J.; Schutz, U.; Kril, J.J.; Ittner, L.M.; Schroder, H.; Gotz, J. Tau-mediated nuclear depletion and cytoplasmic accumulation of SFPQ in Alzheimer’s and Pick’s disease. PLoS ONE 2012, 7, e35678. [Google Scholar] [CrossRef]
- Luisier, R.; Tyzack, G.E.; Hall, C.E.; Mitchell, J.S.; Devine, H.; Taha, D.M.; Malik, B.; Meyer, I.; Greensmith, L.; Newcombe, J.; et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat. Commun. 2018, 9, 2010. [Google Scholar] [CrossRef] [PubMed]
- Younas, N.; Zafar, S.; Shafiq, M.; Noor, A.; Siegert, A.; Arora, A.S.; Galkin, A.; Zafar, A.; Schmitz, M.; Stadelmann, C.; et al. SFPQ and Tau: Critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol. 2020, 140, 317–339. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Riku, Y.; Fujioka, Y.; Endo, K.; Iwade, N.; Kawai, K.; Ishibashi, M.; Yokoi, S.; Katsuno, M.; Watanabe, H.; et al. Aberrant interaction between FUS and SFPQ in neurons in a wide range of FTLD spectrum diseases. Brain 2020, 143, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Heemels, M.T. Neurodegenerative diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Elbaum-Garfinkle, S. Matter over mind: Liquid phase separation and neurodegeneration. J. Biol. Chem. 2019, 294, 7160–7168. [Google Scholar] [CrossRef] [Green Version]
- Maziuk, B.; Ballance, H.I.; Wolozin, B. Dysregulation of RNA Binding Protein Aggregation in Neurodegenerative Disorders. Front. Mol. Neurosci. 2017, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, S.S.; Botelho, H.M.; Gomes, C.M. Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 2012, 256, 2253–2270. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef]
- Caragounis, A.; Price, K.A.; Soon, C.P.; Filiz, G.; Masters, C.L.; Li, Q.X.; Crouch, P.J.; White, A.R. Zinc induces depletion and aggregation of endogenous TDP-43. Free Radic. Biol. Med. 2010, 48, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Garnier, C.; Devred, F.; Byrne, D.; Puppo, R.; Roman, A.Y.; Malesinski, S.; Golovin, A.V.; Lebrun, R.; Ninkina, N.N.; Tsvetkov, P.O. Zinc binding to RNA recognition motif of TDP-43 induces the formation of amyloid-like aggregates. Sci. Rep. 2017, 7, 6812. [Google Scholar] [CrossRef] [Green Version]
- Tyzack, G.E.; Luisier, R.; Taha, D.M.; Neeves, J.; Modic, M.; Mitchell, J.S.; Meyer, I.; Greensmith, L.; Newcombe, J.; Ule, J.; et al. Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain 2019, 142, 2572–2580. [Google Scholar] [CrossRef] [Green Version]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [Green Version]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Pluckthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Y.W.; James, D.; Huang, J.; Lee, M. The Emerging Role of the RNA-Binding Protein SFPQ in Neuronal Function and Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 7151. https://doi.org/10.3390/ijms21197151
Lim YW, James D, Huang J, Lee M. The Emerging Role of the RNA-Binding Protein SFPQ in Neuronal Function and Neurodegeneration. International Journal of Molecular Sciences. 2020; 21(19):7151. https://doi.org/10.3390/ijms21197151
Chicago/Turabian StyleLim, Yee Wa, Dylan James, Jie Huang, and Mihwa Lee. 2020. "The Emerging Role of the RNA-Binding Protein SFPQ in Neuronal Function and Neurodegeneration" International Journal of Molecular Sciences 21, no. 19: 7151. https://doi.org/10.3390/ijms21197151
APA StyleLim, Y. W., James, D., Huang, J., & Lee, M. (2020). The Emerging Role of the RNA-Binding Protein SFPQ in Neuronal Function and Neurodegeneration. International Journal of Molecular Sciences, 21(19), 7151. https://doi.org/10.3390/ijms21197151