New Insights for BPIFB4 in Cardiovascular Therapy



Abstract
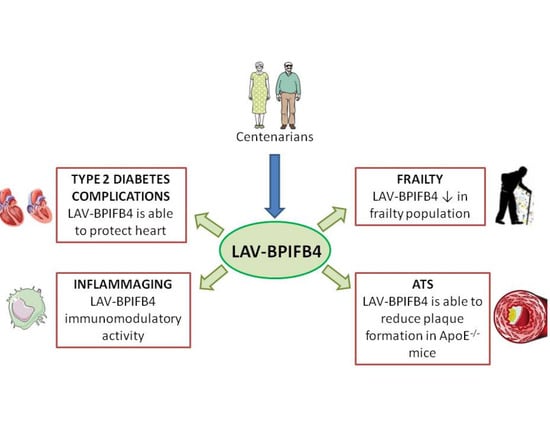
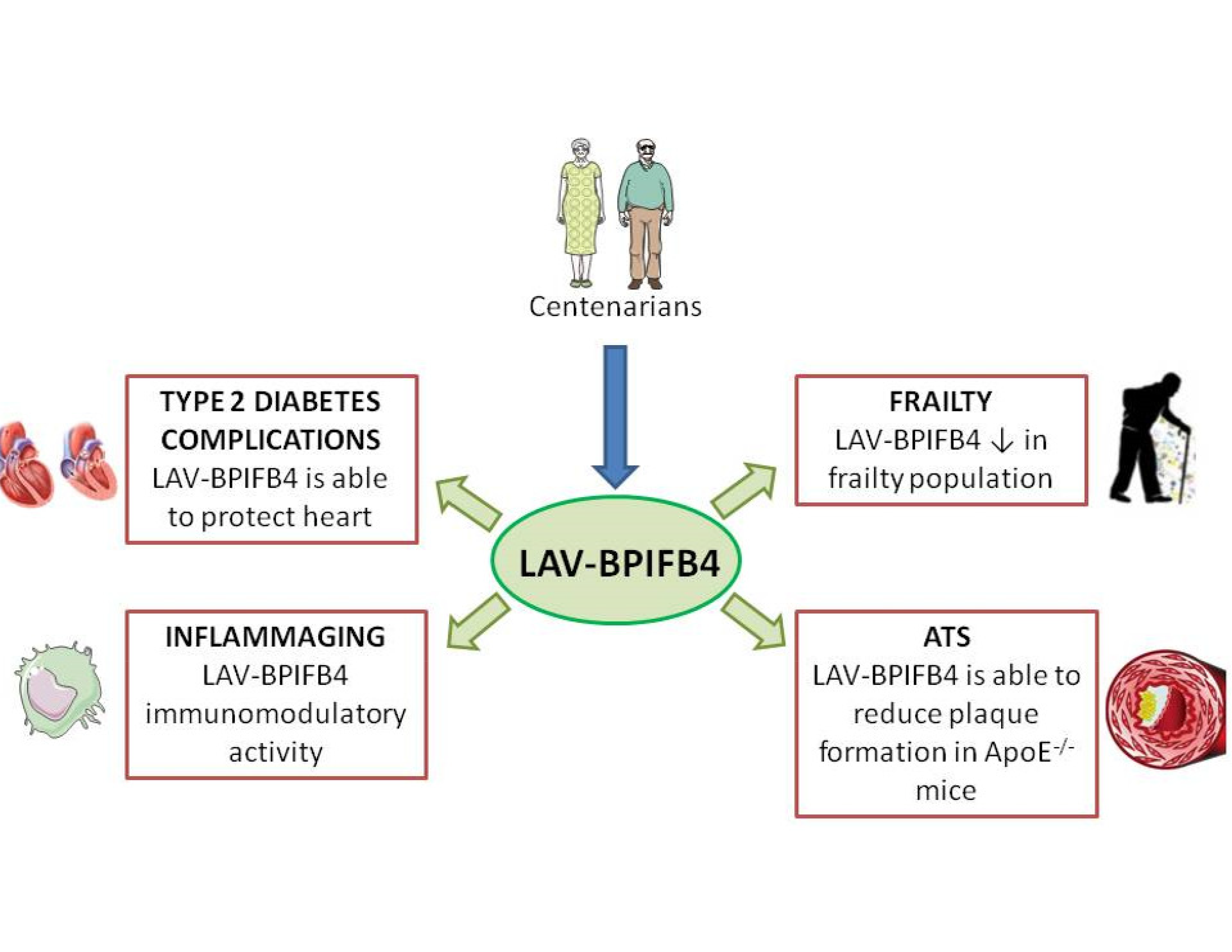
:
{kind=link}
{kind=link}
1. Introduction
2. Centenarians as a Model to Escape Aging
3. Characterization of Longevity Associated Variant of Bactericidal/Permeability-Increasing Fold-Containing Family B Member 4
4. BPIFB4 as a New Genetic Marker of Frailty
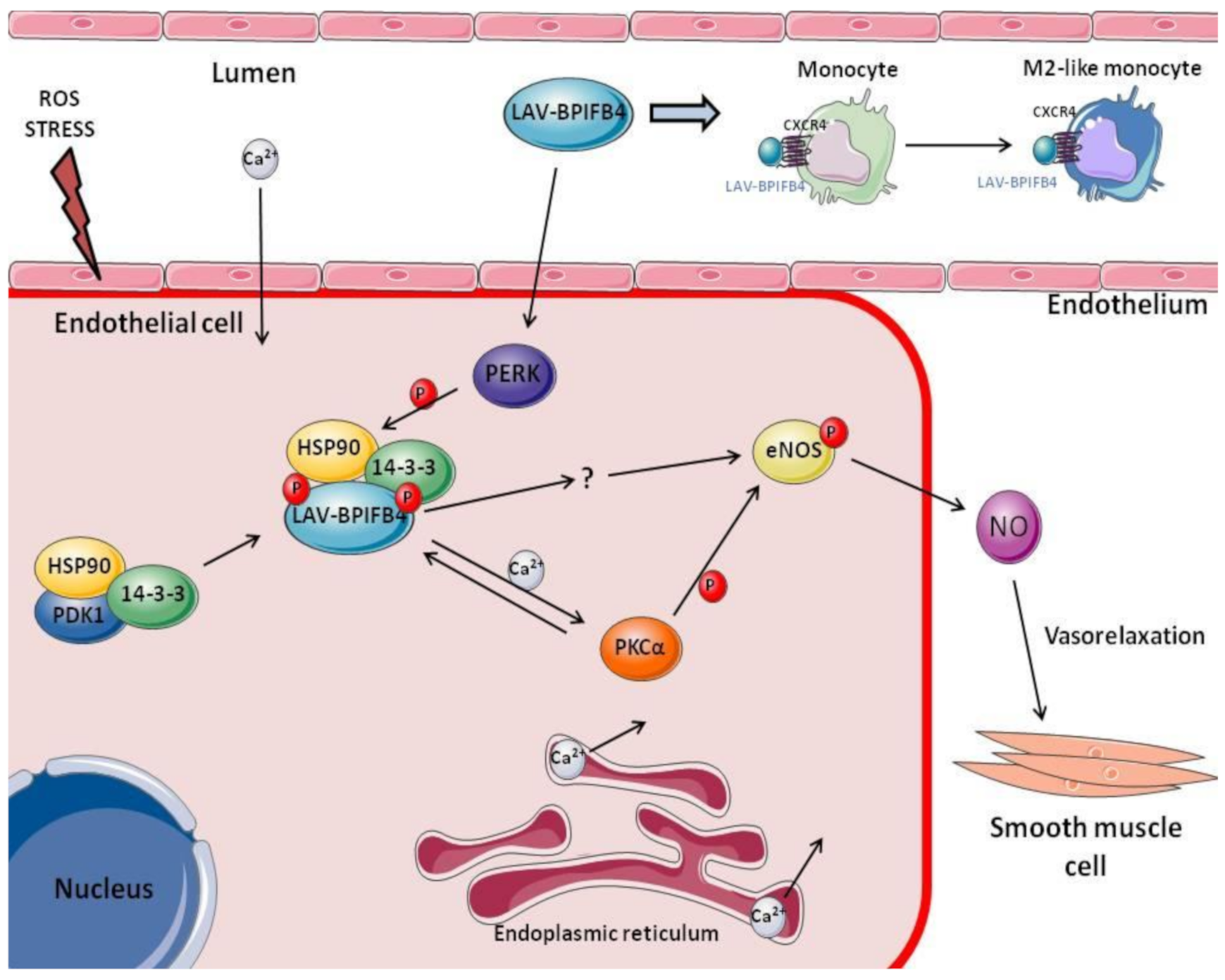
5. LAV-BPIFB4: A New Approach for Atherosclerosis Treatment
6. LAV-BPIFB4 as a Novel Treatment for Type 2 Diabetes Complications
7. The Role of LAV-BPIFB4 as an Immunoregulatory Driver in Age-Related CVD
8. Conclusions
9. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Antigen-presenting cells |
| ARDs | Age-related diseases |
| ATS | Atherosclerosis |
| BPIFB4 | Bactericidal/permeability-increasing fold-containing family B member 4 |
| CCL-2 | Chemokines chemokine CC-motif ligand 2 |
| CVDs | Cardiovascular diseases |
| CXCL-xx | Chemokine (C-X-C motif) ligand xx |
| EC | Endothelial cells |
| EPIC | Elderly prospective cohort study |
| GWAS | Genome wide association study |
| Hsp90 | Heat Shock Protein 90 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| ILx | Interleukin x |
| LAV | Longevity-associated variant |
| LLIs | Long-living individuals |
| LV | Left ventricular |
| MCP1 | Monocyte chemoattractant protein 1 |
| PERK | Protein kinase R (PKR)-like endoplasmic reticulum kinase |
| PKCα | Protein kinase C alpha |
| RANTES | Regulated upon activation, normal T cell expressed and secreted |
| ROS | Reactive oxygen species |
| RV | Rare variant |
| SICS | Southern Italian centenarian study |
| SOD3 | Superoxide dismutase 3 |
| T2D | Type 2 diabetes |
| TLR-4 | Toll-receptor like 4 |
| TNFα | Tumor necrosis factor α |
| VCAM1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
| VSMC | Vascular smooth muscle cells |
| WT | Wild type |
References
- Fleg, J.L.; Aronow, W.S.; Frishman, W.H. Cardiovascular drug therapy in the elderly: Benefits and challenges. Nat. Rev. Cardiol. 2010, 8, 13–28. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagiotakos, D.; Georgousopoulou, E.N.; Pitsavos, C.; Chrysohoou, C.; Metaxa, V.; Georgiopoulos, G.; Kalogeropoulou, K.; Tousoulis, D.; Stefanadis, C. Ten-year (2002–2012) cardiovascular disease incidence and all-cause mortality, in urban Greek population: The ATTICA Study. Int. J. Cardiol. 2015, 180, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Andrew, T.; Gardner, J.; Kimura, M.; Oelsner, E.; Cherkas, L.; Aviv, A.; Spector, T.D. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Strait, J.B.; Lakatta, E.G. Aging-Associated Cardiovascular Changes and Their Relationship to Heart Failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IDF releases report of global survey on access to medicines and supplies for people with diabetes. Diabetes Res. Clin. Pract. 2017, 129, 129–224.
- Bettaga, N.; Jäger, R.; Dünnes, S.; Groneberg, D.; Friebe, A. Cell-specific impact of nitric oxide-dependent guanylyl cyclase on arteriogenesis and angiogenesis in mice. Angiogenesis 2015, 18, 245–254. [Google Scholar] [CrossRef]
- Izzo, C.; Carrizzo, A.; Alfano, A.; Virtuoso, N.; Capunzo, M.; Calabrese, M.; De Simone, E.; Sciarretta, S.; Frati, G.; Oliveti, M.; et al. The Impact of Aging on Cardio and Cerebrovascular Diseases. Int. J. Mol. Sci. 2018, 19, 481. [Google Scholar] [CrossRef] [Green Version]
- Villa, F.; Carrizzo, A.; Spinelli, C.C.; Ferrario, A.; Malovini, A.; Maciąg, A.; Damato, A.; Auricchio, A.; Spinetti, G.; Sangalli, E.; et al. Genetic Analysis Reveals a Longevity-Associated Protein Modulating Endothelial Function and Angiogenesis. Circ. Res. 2015, 117, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Trichopoulou, A.; Orfanos, P.; Norat, T.; Bueno-De-Mesquita, B.; Ocke, M.C.; Peeters, P.H.; Van Der Schouw, Y.T.; Boeing, H.; Hoffmann, K.; Boffetta, P.; et al. Modified Mediterranean diet and survival: EPIC-elderly prospective cohort study. BMJ 2005, 330, 991. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, A.; Villa, F.; Malovini, A.; Araniti, F.; Puca, A.A. The application of genetics approaches to the study of exceptional longevity in humans: Potential and limitations. Immun. Ageing 2012, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, P.; Solovieff, N.; Dewan, A.; Walsh, K.M.; Puca, A.; Hartley, S.W.; Melista, E.; Andersen, S.L.; Dworkis, D.A.; Wilk, J.B.; et al. Genetic Signatures of Exceptional Longevity in Humans. PLoS ONE 2012, 7, e29848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, F.; Carrizzo, A.; Ferrario, A.; Maciag, A.; Cattaneo, M.; Spinelli, C.C.; Montella, F.; Damato, A.; Ciaglia, E.; Puca, A.A. A Model of Evolutionary Selection: The Cardiovascular Protective Function of the Longevity Associated Variant of BPIFB4. Int. J. Mol. Sci. 2018, 19, 3229. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, P.; Andersen, S.L.; Puca, A.; Atzmon, G.; Barzilai, N.; Sebastiani, P. APOE Alleles and Extreme Human Longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Garatachea, N.; Marín, P.J.; Santos-Lozano, A.; Sanchis-Gomar, F.; Emanuele, E.; Lucia, A. TheApoEGene Is Related with Exceptional Longevity: A Systematic Review and Meta-Analysis. Rejuvenation Res. 2015, 18, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Kato, T.; Atsumi, A.; Yamamoto, T.; Inoue, N.; Ishikawa, M.; Okada, S.; Ishigaki, N.; et al. Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nat. Med. 2007, 13, 1193–1202. [Google Scholar] [CrossRef]
- Rizza, S.; Cardaci, S.; Montagna, C.; Di Giacomo, G.; De Zio, D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A.; et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397. [Google Scholar] [CrossRef] [Green Version]
- Puca, A.A.; Andrew, P.; Novelli, V.; Anselmi, C.V.; Somalvico, F.; Cirillo, N.A.; Chatgilialoglu, C.; Ferreri, C. Fatty Acid Profile of Erythrocyte Membranes As Possible Biomarker of Longevity. Rejuvenation Res. 2008, 11, 63–72. [Google Scholar] [CrossRef]
- Appiah, D.; Baumgartner, R.N. The Influence of Education and Apolipoprotein epsilon4 on Mortality in Community-Dwelling Elderly Men and Women. J. Aging Res. 2018, 2018, 6037058. [Google Scholar] [CrossRef]
- Poirier, J.; Miron, J.; Picard, C.; Gormley, P.; Théroux, L.; Breitner, J.; Dea, D. Apolipoprotein E and lipid homeostasis in the etiology and treatment of sporadic Alzheimer’s disease. Neurobiol. Aging 2014, 35, S3–S10. [Google Scholar] [CrossRef] [Green Version]
- Bingle, C.D.; Craven, C.J. PLUNC: A novel family of candidate host defence proteins expressed in the upper airways and nasopharynx. Hum. Mol. Genet. 2002, 11, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, C.; Villa, F.; Carrizzo, A.; Spinelli, C.C.; Damato, A.; Ambrosio, M.; Ferrario, A.; Madonna, M.; Uccellatore, A.; Lupini, S.; et al. A rare genetic variant of BPIFB4 predisposes to high blood pressure via impairment of nitric oxide signaling. Sci. Rep. 2017, 7, 9706. [Google Scholar] [CrossRef] [PubMed]
- Bingle, C.D.; Seal, R.L.; Craven, C.J. Systematic nomenclature for the PLUNC/PSP/BSP30/SMGB proteins as a subfamily of the BPI fold-containing superfamily. Biochem. Soc. Trans. 2011, 39, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, C.C.; Carrizzo, A.; Ferrario, A.; Villa, F.; Damato, A.; Ambrosio, M.; Madonna, M.; Frati, G.; Fucile, S.; Sciaccaluga, M.; et al. LAV-BPIFB4 isoform modulates eNOS signalling through Ca2+/PKC-alpha-dependent mechanism. Cardiovasc. Res. 2017, 113, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Fried, L.P.; Tangen, C.M.; Walston, J.; Newman, A.B.; Hirsch, C.; Gottdiener, J.; Seeman, T.; Tracy, R.; Kop, W.J.; Burke, G.; et al. Frailty in older adults: Evidence for a phenotype. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2001, 56, M146–M157. [Google Scholar] [CrossRef]
- Rockwood, K.; Song, X.; Macknight, C.; Bergman, H.; Hogan, D.; McDowell, I.; Mitnitski, A. A global clinical measure of fitness and frailty in elderly people. Can. Med Assoc. J. CMAJ 2005, 173, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Rockwood, K.; Mitnitski, A. How Might Deficit Accumulation Give Rise to Frailty? J. Frailty Aging 2012, 1, 8–12. [Google Scholar] [PubMed]
- Cesari, M.; Gambassi, G.; Van Kan, G.A.; Vellas, B. The frailty phenotype and the frailty index: Different instruments for different purposes. Age Ageing 2014, 43, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Campo, G.; Maietti, E.; Tonet, E.; Biscaglia, S.; Ariza-Solè, A.; Pavasini, R.; Tebaldi, M.; Cimaglia, P.; Bugani, G.; Serenelli, M.; et al. The Assessment of Scales of Frailty and Physical Performance Improves Prediction of Major Adverse Cardiac Events in Older Adults with Acute Coronary Syndrome. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 75, 1113–1119. [Google Scholar] [CrossRef]
- Tonet, E.; Pavasini, R.; Biscaglia, S.; Campo, G. Frailty in patients admitted to hospital for acute coronary syndrome: When, how and why? J. Geriatr. Cardiol. 2019, 16, 129–137. [Google Scholar]
- Leng, S.; Chaves, P.; Koenig, K.; Walston, J. Serum interleukin-6 and hemoglobin as physiological correlates in the geriatric syndrome of frailty: A pilot study. J. Am. Geriatr. Soc. 2002, 50, 1268–1271. [Google Scholar] [CrossRef]
- Leng, S.X.; Cappola, A.R.; Andersen, R.E.; Blackman, M.R.; Koenig, K.; Blair, M.; Walston, J.D. Serum levels of insulin-like growth factor-I (IGF-I) and dehydroepiandrosterone sulfate (DHEA-S), and their relationships with serum interleukin-6, in the geriatric syndrome of frailty. Aging Clin. Exp. Res. 2004, 16, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Walston, J. Frailty—The Search For Underlying Causes. Sci. Aging Knowl. Environ. 2004, 2004, 4. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J. Frailty—and Its Dangerous Effects—Might Be Preventable. Ann. Intern. Med. 2004, 141, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Lipsitz, L.A. Physiological Complexity, Aging, and the Path to Frailty. Sci. Aging Knowl. Environ. 2004, 2004, pe16. [Google Scholar] [CrossRef]
- Montesanto, A.; Lagani, V.; Martino, C.; Dato, S.; De Rango, F.; Berardelli, M.; Corsonello, A.; Mazzei, B.; Mari, V.; Lattanzio, F.; et al. A novel, population-specific approach to define frailty. Age 2010, 32, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Malavolta, M.; Dato, S.; Villa, F.; De Rango, F.; Iannone, F.; Ferrario, A.; Maciag, A.; Ciaglia, E.; D’Amato, A.; Carrizzo, A.; et al. LAV-BPIFB4 associates with reduced frailty in humans and its transfer prevents frailty progression in old mice. Aging 2019, 11, 6555–6568. [Google Scholar] [CrossRef]
- Farhat, N.; Thorin-Trescases, N.; Voghel, G.; Villeneuve, L.; Mamarbachi, M.; Perrault, L.P.; Carrier, M.; Thorin, E. Stress-induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Can. J. Physiol. Pharmacol. 2008, 86, 761–769. [Google Scholar] [CrossRef]
- Niemann, B.; Chen, Y.; Teschner, M.; Li, L.; Silber, R.-E.; Rohrbach, S. Obesity induces signs of premature cardiac aging in younger patients: The role of mitochondria. J. Am. Coll. Cardiol. 2011, 57, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Bennett, M.R. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Puca, A.A.; Carrizzo, A.; Spinelli, C.C.; Damato, A.; Ambrosio, M.; Villa, F.; Ferrario, A.; Maciag, A.; Fornai, F.; Lenzi, P.; et al. Single systemic transfer of a human gene associated with exceptional longevity halts the progression of atherosclerosis and inflammation in ApoE knockout mice through a CXCR4-mediated mechanism. Eur. Heart J. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witztum, J.L.; Lichtman, A.H. The Influence of Innate and Adaptive Immune Responses on Atherosclerosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 73–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Shen, H.; Schenten, M.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2011, 32, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.R.; Evan, G.I.; Schwartz, S.M. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J. Clin. Investig. 1995, 95, 2266–2274. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Weile, C.; Sondergaard, C.S.; Hindkjaer, J.; Kassem, M.; Falk, E. Smooth Muscle Cells in Atherosclerosis Originate From the Local Vessel Wall and Not Circulating Progenitor Cells in ApoE Knockout Mice. Arter. Thromb. Vasc. Biol. 2006, 26, 2696–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M.R. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Lutgens, E.; De Muinck, E.D.; Kitslaar, P.J.; Tordoir, J.H.; Wellens, H.J.; Daemen, M.J. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc. Res. 1999, 41, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Guntani, A.; Matsumoto, T.; Kyuragi, R.; Iwasa, K.; Onohara, T.; Itoh, H.; Katušić, Z.S.; Maehara, Y. Reduced Proliferation of Aged Human Vascular Smooth Muscle Cells—Role of Oxygen-Derived Free Radicals and BubR1 Expression. J. Surg. Res. 2011, 170, 143–149. [Google Scholar] [CrossRef]
- Moon, S.-K.; Thompson, L.J.; Madamanchi, N.R.; Ballinger, S.; Papaconstantinou, J.; Horaist, C.; Runge, M.S.; Patterson, C. Aging, oxidative responses, and proliferative capacity in cultured mouse aortic smooth muscle cells. Am. J. Physiol. Circ. Physiol. 2001, 280, H2779–H2788. [Google Scholar] [CrossRef]
- Martinet, W.; Knaapen, M.W.; De Meyer, G.; Herman, A.G.; Kockx, M. Elevated Levels of Oxidative DNA Damage and DNA Repair Enzymes in Human Atherosclerotic Plaques. Circulation 2002, 106, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Tatarková, Z.; Kuka, S.; Račay, P.; Lehotský, J.; Dobrota, D.; Mištuna, D.; Kaplán, P. Effects of Aging on Activities of Mitochondrial Electron Transport Chain Complexes and Oxidative Damage in Rat Heart. Physiol. Res. 2011, 60, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Breitschopf, K.; Zeiher, A.M.; Dimmeler, S. Pro-atherogenic factors induce telomerase inactivation in endothelial cells through an Akt-dependent mechanism. FEBS Lett. 2001, 493, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Kavurma, M.M.; Figg, N.; Bennett, M.R.; Mercer, J.; Khachigian, L.M.; Littlewood, T.D. Oxidative stress regulates IGF1R expression in vascular smooth-muscle cells via p53 and HDAC recruitment. Biochem. J. 2007, 407, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrman, B.; Volkova, N.; Aviram, M. Oxidative stress increases the expression of the CD36 scavenger receptor and the cellular uptake of oxidized low-density lipoprotein in macrophages from atherosclerotic mice: Protective role of antioxidants and of paraoxonase. Atherosclerosis 2002, 161, 307–316. [Google Scholar] [CrossRef]
- Asai, K.; Kudej, R.K.; Shen, Y.-T.; Yang, G.-P.; Takagi, G.; Kudej, A.B.; Geng, Y.-J.; Sato, N.; Nazareno, J.B.; Vatner, R.E.; et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arter. Thromb. Vasc. Biol. 2000, 20, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Khaidakov, M.; Wang, X.; Mehta, J.L. Potential Involvement of LOX-1 in Functional Consequences of Endothelial Senescence. PLoS ONE 2011, 6, e20964. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Aida, K.; VandeBerg, J.L.; Wang, X.L. Passage-Dependent Changes in Baboon Endothelial Cells—Relevance to In Vitro Aging. DNA Cell Biol. 2004, 23, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Hampel, B.; Bernhard, D.; Hala, M.; Zwerschke, W.; Jansen-Dürr, P. Replicative senescence of human endothelial cells in vitro involves G1 arrest, polyploidization and senescence-associated apoptosis. Exp. Gerontol. 2001, 36, 1327–1347. [Google Scholar] [CrossRef]
- Sato, I.; Morita, I.; Kaji, K.; Ikeda, M.; Nagao, M.; Murota, S. Reduction of Nitric Oxide Producing Activity Associated with in Vitro Aging in Cultured Human Umbilical Vein Endothelial Cell. Biochem. Biophys. Res. Commun. 1993, 195, 1070–1076. [Google Scholar] [CrossRef]
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, U.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol. Circ. Physiol. 2009, 297, H425–H432. [Google Scholar] [CrossRef] [Green Version]
- Busillo, J.M.; Armando, S.; Sengupta, R.; Meucci, O.; Bouvier, M.; Benovic, J.L. Site-specific Phosphorylation of CXCR4 Is Dynamically Regulated by Multiple Kinases and Results in Differential Modulation of CXCR4 Signaling. J. Biol. Chem. 2010, 285, 7805–7817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merckelbach, S.; Van Der Vorst, E.P.C.; Kallmayer, M.; Rischpler, C.; Burgkart, R.; Döring, Y.; De Borst, G.-J.; Schwaiger, M.; Eckstein, H.-H.; Weber, C.; et al. Expression and Cellular Localization of CXCR4 and CXCL12 in Human Carotid Atherosclerotic Plaques. Thromb. Haemost. 2018, 118, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Qin, J.J.; Zhang, Y.; Cheng, W.L.; Ji, Y.X.; Gong, F.H.; Zhu, X.Y.; She, Z.G.; Huang, Z.; Li, H.; et al. LILRB4 deficiency aggravates the development of atherosclerosis and plaque instability by increasing the macrophage inflammatory response via NF-kappaB signaling. Clin. Sci. 2017, 131, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Molica, F.; Meens, M.J.; Dubrot, J.; Ehrlich, A.; Roth, C.L.; Morel, S.; Pelli, G.; Vinet, L.; Braunersreuther, V.; Ratib, O.; et al. Pannexin1 links lymphatic function to lipid metabolism and atherosclerosis. Sci. Rep. 2017, 7, 13706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Report on Ddiabetes; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.K.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. BMJ 2015, 351, h3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Demarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2015, 12, 144–153. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Dang, Z.; Avolio, E.; Thomas, A.C.; Faulkner, A.; Beltrami, A.P.; Cervellin, C.; Carrizzo, A.; Maciag, A.; Gu, Y.; Ciaglia, E.; et al. Transfer of a human gene variant associated with exceptional longevity improves cardiac function in obese type 2 diabetic mice through induction of the SDF-1/CXCR4 signalling pathway. Eur. J. Heart Fail. 2020. [Google Scholar] [CrossRef]
- Rundell, V.L.M.; Geenen, D.L.; Buttrick, P.M.; De Tombe, P.P. Depressed cardiac tension cost in experimental diabetes is due to altered myosin heavy chain isoform expression. Am. J. Physiol. Circ. Physiol. 2004, 287, H408–H413. [Google Scholar] [CrossRef]
- Korte, F.S.; Herron, T.J.; Rovetto, M.J.; McDonald, K.S. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am. J. Physiol. Circ. Physiol. 2005, 289, H801–H812. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-L.; Zhu, L.-Y.; Han, R.; Sun, L.-L.; Li, J.-X.; Dou, J.-T. Pathophysiology of peripheral arterial disease in diabetes mellitus. J. Diabetes 2016, 9, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Vanhoutte, P.M. Cellular signaling and NO production. Pflügers Archiv. Eur. J. Physiol. 2010, 459, 807–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivieri, F.; Prattichizzo, F.; Grillari, J.; Balistreri, C.R. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediat. Inflamm. 2018, 2018, 1–6. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.I.; Kim, H.-S. Emerging role of sirtuins on tumorigenesis: Possible link between aging and cancer. BMB Rep. 2013, 46, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [Green Version]
- Moskalev, A.; Shaposhnikov, M.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Yanai, H.; Fraifeld, V.E.; Zhavoronkov, A. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 2013, 12, 661–684. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Raza, S.T.; Mahdi, F. Telomere length variations in aging and age-related diseases. Curr. Aging Sci. 2014, 7, 161–167. [Google Scholar] [CrossRef]
- Wu, L.E.; Gomes, A.P.; Sinclair, D.A. Geroncogenesis: Metabolic changes during aging as a driver of tumorigenesis. Cancer Cell 2014, 25, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar]
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fülöp, T. Innate immunosenescence: Effect of aging on cells and receptors of the innate immune system in humans. Semin. Immunol. 2012, 24, 331–341. [Google Scholar] [CrossRef]
- Carta, S.; Penco, F.; Lavieri, R.; Martini, A.; Dinarello, C.A.; Gattorno, M.; Rubartelli, A. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc. Natl. Acad. Sci. USA 2015, 112, 2835–2840. [Google Scholar] [CrossRef] [Green Version]
- Dall’Olio, F.; Vanhooren, V.; Chen, C.C.; Slagboom, E.P.; Wuhrer, M.; Franceschi, C. N-glycomic biomarkers of biological aging and longevity: A link with inflammaging. Ageing Res. Rev. 2013, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Deepa, S.S.; Unnikrishnan, A.; Matyi, S.; Hadad, N.; Richardson, A. Necroptosis increases with age and is reduced by dietary restriction. Aging Cell 2018, 17, e12770. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.M.; Liu, Y.-J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Jakubzick, C.V.; Randolph, G.J.; Henson, P.M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 2017, 17, 349–362. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, W.; Hao, D.; He, B.; Kong, L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int. Immunopharmacol. 2019, 70, 459–466. [Google Scholar] [CrossRef]
- Costantini, A.; Viola, N.; Berretta, A.; Galeazzi, R.; Matacchione, G.; Sabbatinelli, J.; Storci, G.; De Matteis, S.; Butini, L.; Rippo, M.R.; et al. Age-related M1/M2 phenotype changes in circulating monocytes from healthy/unhealthy individuals. Aging 2018, 10, 1268–1280. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Zawada, A.M.; Rogacev, K.S.; Schirmer, S.H.; Sester, M.; Böhm, M.; Fliser, D.; Heine, G.H. Monocyte heterogeneity in human cardiovascular disease. Immunobiology 2012, 217, 1273–1284. [Google Scholar] [CrossRef]
- Spinetti, G.; Sangalli, E.; Specchia, C.; Villa, F.; Spinelli, C.C.; Pipolo, R.; Carrizzo, A.; Greco, S.; Voellenkle, C.; Vecchione, C.; et al. The expression of the BPIFB4 and CXCR4 associates with sustained health in long-living individuals from Cilento-Italy. Aging 2017, 9, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Galkina, E.V.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, K.; Vengrenyuk, Y.; Ramsey, S.A.; Vila, N.R.; Girgis, N.M.; Liu, J.; Gusarova, V.; Gromada, J.; Weinstock, A.; Moore, K.J.; et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Investig. 2017, 127, 2904–2915. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, L.; Estecha, A.; Samaniego, R.; Sánchez-Ramón, S.; Vega, M.A.; Sánchez-Mateos, P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood 2011, 117, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Ransohoff, R.M. The roles of chemokine CXCL12 in embryonic and brain tumor angiogenesis. Semin. Cancer Biol. 2009, 19, 111–115. [Google Scholar] [CrossRef]
- Karin, N. The multiple faces of CXCL12 (SDF-1α) in the regulation of immunity during health and disease. J. Leukoc. Biol. 2010, 88, 463–473. [Google Scholar] [CrossRef]
- Terry, D.F.; Sebastiani, P.; Andersen, S.L.; Perls, T.T. Disentangling the Roles of Disability and Morbidity in Survival to Exceptional Old Age. Arch. Intern. Med. 2008, 168, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Di Pardo, A.; Ciaglia, E.; Cattaneo, M.; Maciag, A.; Montella, F.; Lopardo, V.; Ferrario, A.; Villa, F.; Madonna, M.; Amico, E.; et al. The longevity-associated variant of BPIFB4 improves a CXCR4-mediated striatum–microglia crosstalk preventing disease progression in a mouse model of Huntington’s disease. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dossena, M.; Ferrario, A.; Lopardo, V.; Ciaglia, E.; Puca, A.A. New Insights for BPIFB4 in Cardiovascular Therapy. Int. J. Mol. Sci. 2020, 21, 7163. https://doi.org/10.3390/ijms21197163
Dossena M, Ferrario A, Lopardo V, Ciaglia E, Puca AA. New Insights for BPIFB4 in Cardiovascular Therapy. International Journal of Molecular Sciences. 2020; 21(19):7163. https://doi.org/10.3390/ijms21197163
Chicago/Turabian StyleDossena, Marta, Anna Ferrario, Valentina Lopardo, Elena Ciaglia, and Annibale Alessandro Puca. 2020. "New Insights for BPIFB4 in Cardiovascular Therapy" International Journal of Molecular Sciences 21, no. 19: 7163. https://doi.org/10.3390/ijms21197163
APA StyleDossena, M., Ferrario, A., Lopardo, V., Ciaglia, E., & Puca, A. A. (2020). New Insights for BPIFB4 in Cardiovascular Therapy. International Journal of Molecular Sciences, 21(19), 7163. https://doi.org/10.3390/ijms21197163