The Role of α1-Microglobulin (A1M) in Erythropoiesis and Erythrocyte Homeostasis—Therapeutic Opportunities in Hemolytic Conditions

 ,
,  ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
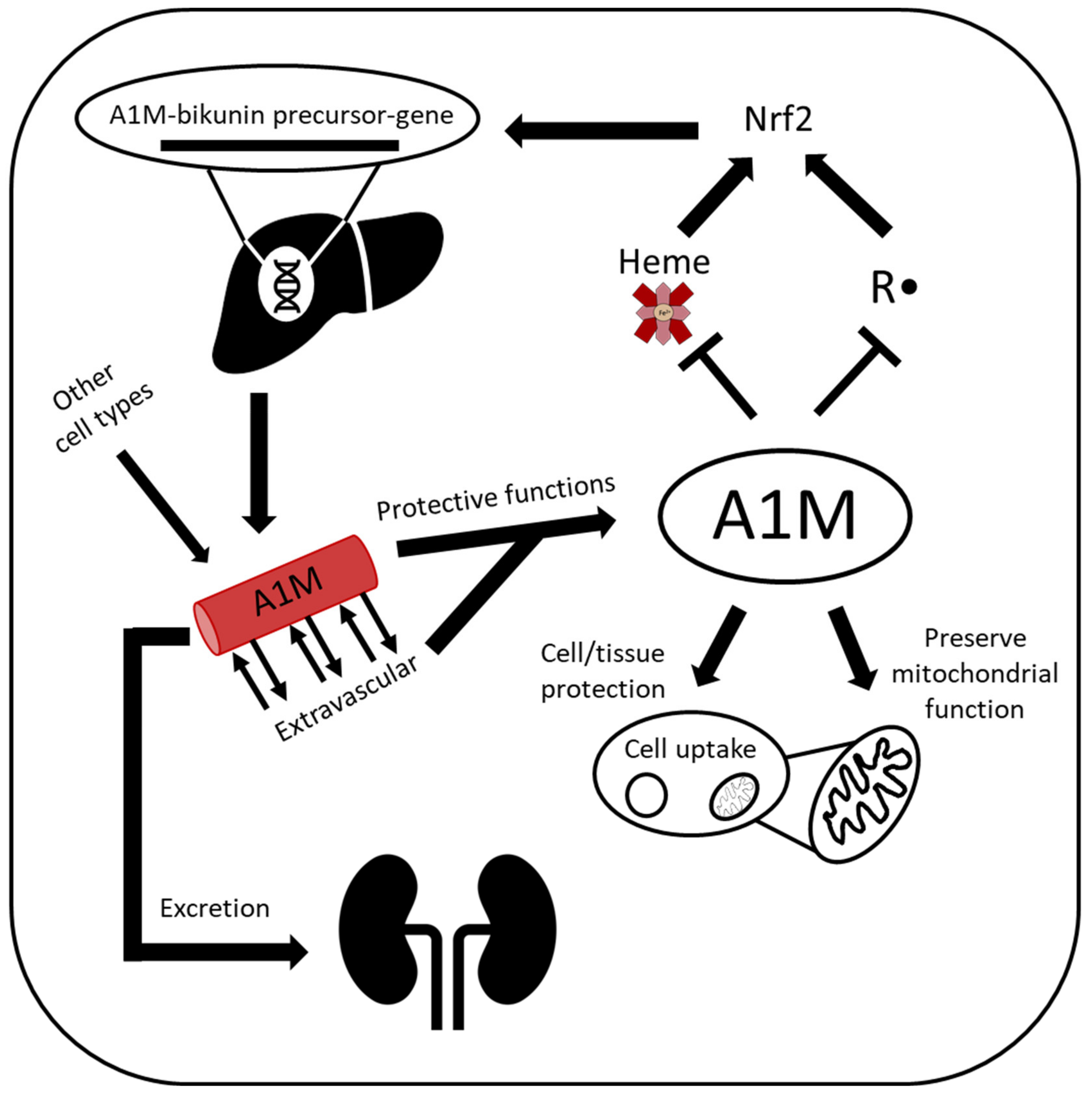
2. A1M
2.1. A1M Protein
2.2. A1M Protective Properties
2.3. In Vitro
2.4. In Vivo
2.5. RBC Protection/Homeostasis
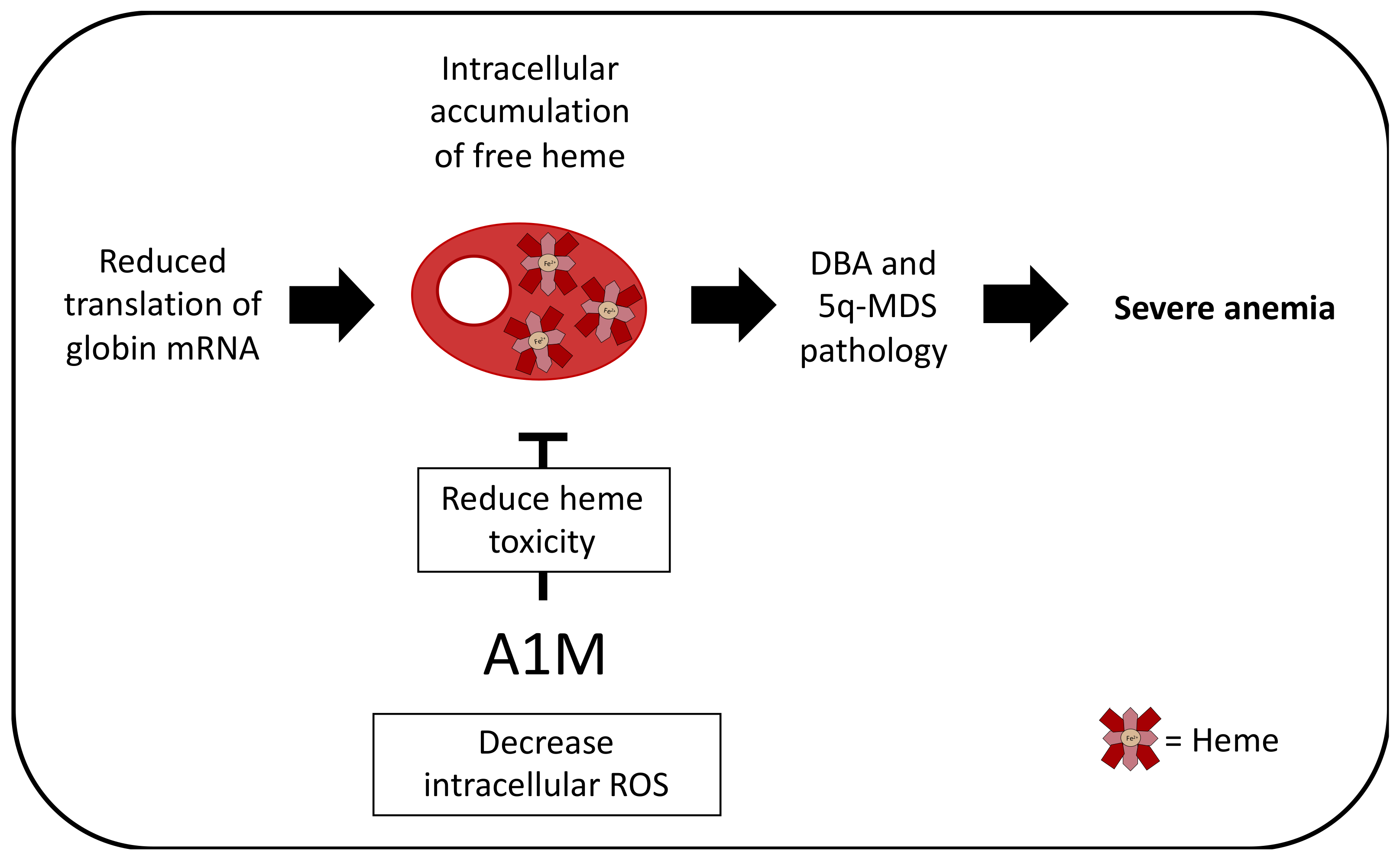
3. Erythropoietic Conditions
4. Hemolytic Conditions
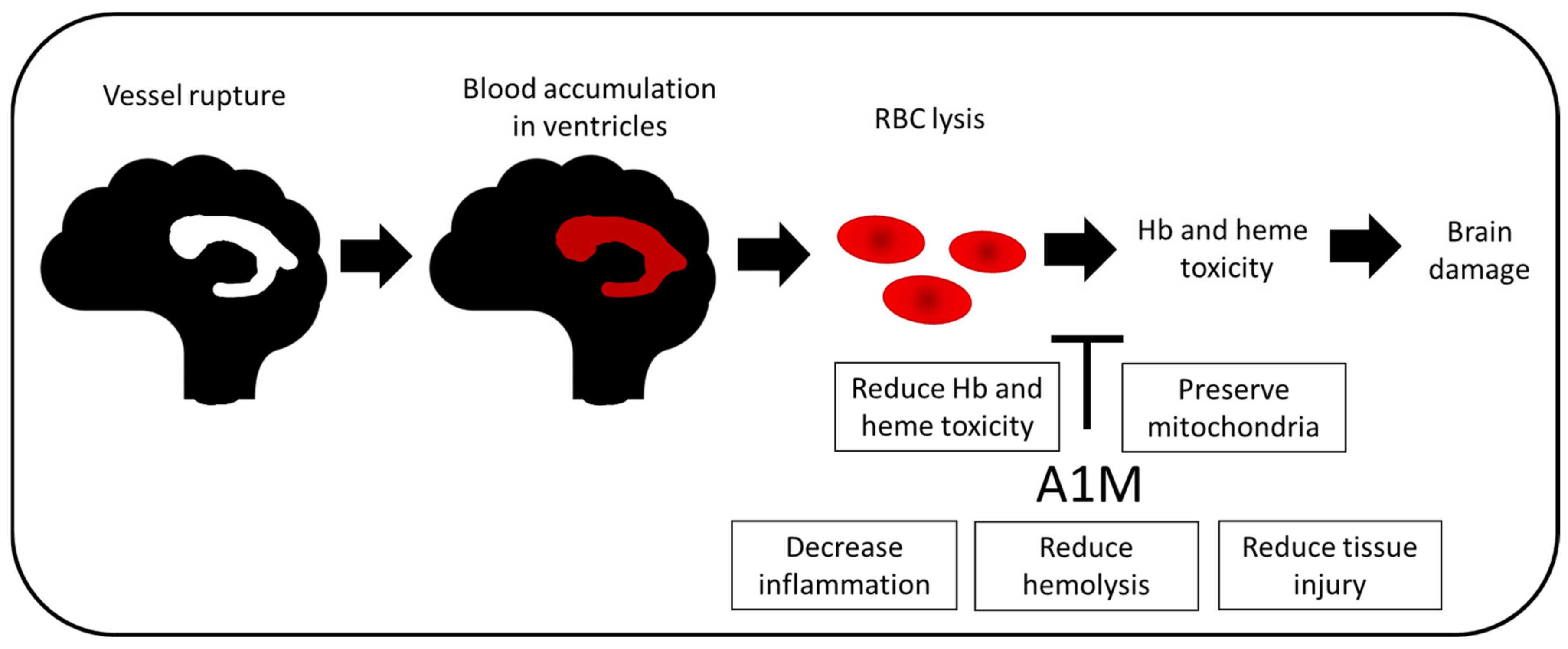
4.1. Intraventricular Hemorrhage in Preterm Infants
4.2. Blood Transfusion
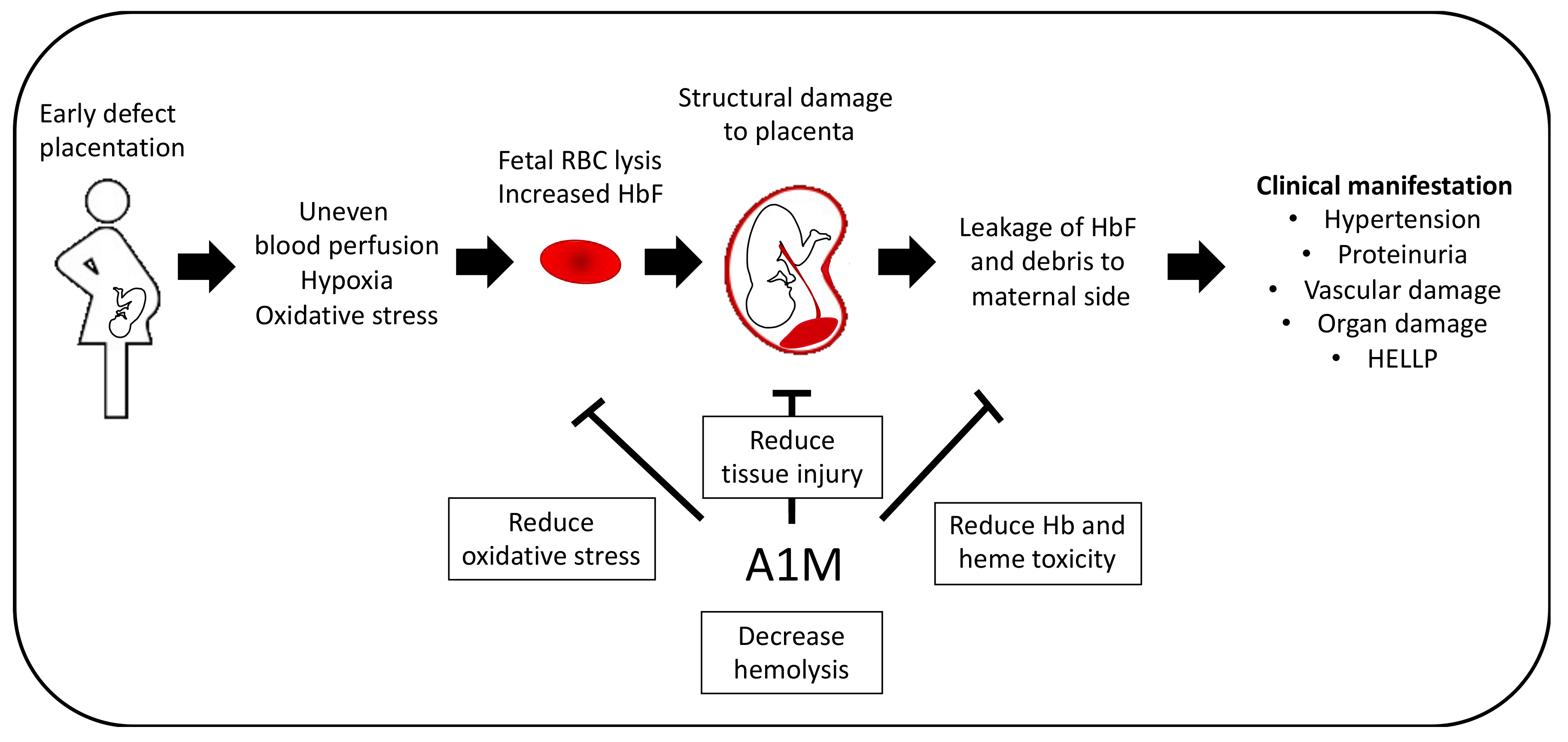
4.3. Preeclampsia
4.4. Atherosclerosis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Çimen, M.Y.B. Free radical metabolism in human erythrocytes. Clin. Chim. Acta 2008, 390, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.G.; Allhorn, M.; Bülow, L.; Hansson, S.R.; Ley, D.; Olsson, M.L.; Schmidtchen, A.; Akerström, B. Pathological conditions involving extracellular hemoglobin: Molecular mechanisms, clinical significance, and novel therapeutic opportunities for α(1)-microglobulin. Antioxid. Redox Signal. 2012, 17, 813–846. [Google Scholar] [CrossRef] [Green Version]
- Allhorn, M.; Berggård, T.; Nordberg, J.; Olsson, M.L.; Åkerström, B. Processing of the lipocalin α1-microglobulin by hemoglobin induces heme-binding and heme-degradation properties. Blood 2002, 99, 1894–1901. [Google Scholar] [CrossRef]
- Åkerström, B.; Maghzal, G.J.; Winterbourn, C.C.; Kettle, A.J. The lipocalin α1-microglobulin has radical scavenging activity. J. Biol. Chem. 2007, 282, 31493–31503. [Google Scholar] [CrossRef] [Green Version]
- Akerstrom, B.; Gram, M. A1M, an extravascular tissue cleaning and housekeeping protein. Free Radic. Biol. Med. 2014, 74, 274–282. [Google Scholar] [CrossRef]
- Kristiansson, A.; Bergwik, J.; Alattar, A.G.; Flygare, J.; Gram, M.; Hansson, S.R.; Olsson, M.L.; Storry, J.R.; Allhorn, M.; Åkerström, B. Human radical scavenger α(1)-microglobulin protects against hemolysis in vitro and α(1)-microglobulin knockout mice exhibit a macrocytic anemia phenotype. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Ekström, B.; Peterson, P.A.; Berggárd, I. A urinary and plasma α1-glycoprotein of low molecular weight: Isolation and some properties. Biochem. Biophys. Res. Commun. 1975, 65, 1427–1433. [Google Scholar] [CrossRef]
- Hanley, S.; Powell, R. Sequence of a cDNA clone encoding the Atlantic salmon alpha 1-microglobulin/bikunin protein. Gene 1994, 147, 297–298. [Google Scholar] [CrossRef]
- Ide, H.; Itoh, H.; Nawa, Y. Sequencing of cDNAs encoding alpha 1-microglobulin/bikunin of Mongolian gerbil and Syrian golden hamster in comparison with man and other species. Biochim. Biophys. Acta 1994, 1209, 286–292. [Google Scholar] [CrossRef]
- Kawahara, A.; Hikosaka, A.; Sasado, T.; Hirota, K. Thyroid hormone-dependent repression of α1-microglobulin/bikunin precursor (AMBP) gene expression during amphibian metamorphosis. Dev. Genes Evol. 1997, 206, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Åkerström, B. Bovine α1-microglobulin/bikunin. Isolation and characterization of liver cDNA and urinary α1-microglobulin. Biochim. Biophys. Acta (BBA)—Gene Struct. Expr. 1996, 1306, 98–106. [Google Scholar] [CrossRef]
- Ekstrom, B.; Berggard, I. Human α1 microglobulin: Purification procedure, chemical and physicochemical properties. J. Biol. Chem. 1977, 252, 8048–8057. [Google Scholar] [PubMed]
- Grzyb, J.; Latowski, D.; Strzałka, K. Lipocalins—A family portrait. J. Plant Physiol. 2006, 163, 895–915. [Google Scholar] [CrossRef]
- Rutardottir, S.; Nilsson, E.J.C.; Pallon, J.; Gram, M.; Åkerström, B. The cysteine 34 residue of A1M/α1-microglobulin is essential for protection of irradiated cell cultures and reduction of carbonyl groups. Free Radic. Res. 2013, 47, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Meining, W.; Skerra, A. The crystal structure of human α1-microglobulin reveals a potential haem-binding site. Biochem. J. 2012, 445, 175–182. [Google Scholar] [CrossRef]
- Kaumeyer, J.F.; Polazzi, J.O.; Kotick, M.P. The mRNA for a proteinase inhibitor related to the HI-30 domain of inter-alpha-trypsin inhibitor also encodes alpha-1-microglobulin (protein HC). Nucleic Acids Res. 1986, 14, 7839–7850. [Google Scholar] [CrossRef]
- Bratt, T.; Olsson, H.; Sjöberg, E.M.; Jergil, B.; Åkerström, B. Cleavage of the α1-microglobulin-bikunin precursor is localized to the Golgi apparatus of rat liver cells. Biochim. Biophys. Acta (BBA)—Gen. Subj. 1993, 1157, 147–154. [Google Scholar] [CrossRef]
- Bergwik, J.; Kristiansson, A.; Welinder, C.; Göransson, O.; Hansson, S.R.; Gram, M.; Erlandsson, L.; Åkerström, B. Knockout of the radical scavenger α1-microglobulin in mice results in defective bikunin synthesis, endoplasmic reticulum stress and increased body weight. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Sekikawa, S.; Onda, T.; Miura, N.; Nomura, T.; Takano, N.; Shibahara, T.; Honda, K. Underexpression of α-1-microglobulin/bikunin precursor predicts a poor prognosis in oral squamous cell carcinoma. Int. J. Oncol. 2018, 53, 2605–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayraktar, E.; Igci, M.; Erturhan, S.; Igci, Y.Z.; Karakok, M.; Gogebakan, B.; Ulasli, M.; Cakmak, E.A.; Arslan, A. Reduced gene expression of bikunin as a prognostic marker for renal cell carcinoma. Exp. Oncol. 2014, 36, 107–111. [Google Scholar]
- Eatemadi, A.; Aiyelabegan, H.T.; Negahdari, B.; Mazlomi, M.A.; Daraee, H.; Daraee, N.; Eatemadi, R.; Sadroddiny, E. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed. Pharmacother. 2017, 86, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Suzuki, M.; Hirashima, Y.; Terao, T. The protease inhibitor bikunin, a novel anti-metastatic agent. Biol. Chem. 2003, 384, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.G.; Allhorn, M.; Olofsson, T.; Åkerström, B. Up-regulation of α1-microglobulin by hemoglobin and reactive oxygen species in hepatoma and blood cell lines. Free Radic. Biol. Med. 2007, 42, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.G.; Allhorn, M.; Larsson, J.; Cederlund, M.; Lundqvist, K.; Schmidtchen, A.; Sørensen, O.E.; Mörgelin, M.; Åkerström, B. Up-Regulation of A1M/α1-Microglobulin in Skin by Heme and Reactive Oxygen Species Gives Protection from Oxidative Damage. PLoS ONE 2011, 6, e27505. [Google Scholar] [CrossRef]
- Kristiansson, A.; Davidsson, S.; Johansson, M.E.; Piel, S.; Elmér, E.; Hansson, M.J.; Åkerström, B.; Gram, M. α1-Microglobulin (A1M) Protects Human Proximal Tubule Epithelial Cells from Heme-Induced Damage In Vitro. Int. J. Mol. Sci. 2020, 21, 5825. [Google Scholar] [CrossRef]
- Campbell, M.R.; Karaca, M.; Adamski, K.N.; Chorley, B.N.; Wang, X.; Bell, D.A. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxidative Med. Cell. Longev. 2013, 2013, 120305. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, J.; Wingardh, K.; Berggard, T.; Davies, J.R.; Logdberg, L.; Strand, S.E.; Akerstrom, B. Distribution of iodine 125-labeled alpha1-microglobulin in rats after intravenous injection. J. Lab. Clin. Med. 2001, 137, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Berggard, T.; Enghild, J.J.; Badve, S.; Salafia, C.M.; Logdberg, L.; Akerstrom, B. Histologic distribution and biochemical properties of alpha 1-microglobulin in human placenta. Am. J. Reprod. Immunol. 1999, 41, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Wester, L.; Fast, J.; Labuda, T.; Cedervall, T.; Wingårdh, K.; Olofsson, T.; Åkerström, B. Carbohydrate groups of α1-microglobulin are important for secretion and tissue localization but not for immunological properties. Glycobiology 2000, 10, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berggard, T.; Thelin, N.; Falkenberg, C.; Enghild, J.J.; Akerstrom, B. Prothrombin, albumin and immunoglobulin A form covalent complexes with alpha1-microglobulin in human plasma. Eur. J. Biochem. 1997, 245, 676–683. [Google Scholar] [CrossRef]
- Yu, H.; Yanagisawa, Y.; Forbes, M.A.; Cooper, E.H.; Crockson, R.A.; MacLennan, I.C. Alpha-1-microglobulin: An indicator protein for renal tubular function. J. Clin. Pathol. 1983, 36, 253–259. [Google Scholar] [CrossRef]
- Nordberg, J.; Allhorn, M.; Winqvist, I.; Åkerström, B.; Olsson, M.L. Quantitative and qualitative evaluation of plasma and urine α1-microglobulin in healthy donors and patients with different haemolytic disorders and haemochromatosis. Clin. Chim. Acta 2007, 386, 31–37. [Google Scholar] [CrossRef]
- Terzi, I.; Papaioannou, V.; Papanas, N.; Dragoumanis, C.; Petala, A.; Theodorou, V.; Gioka, T.; Vargemezis, V.; Maltezos, E.; Pneumatikos, I. Alpha1-microglobulin as an early biomarker of sepsis-associated acute kidney injury: A prospective cohort study. Hippokratia 2014, 18, 262–268. [Google Scholar]
- Fernando, B.; Alli-Shaik, A.; Hemage, R.K.D.; Badurdeen, Z.; Hettiarachchi, T.W.; Abeysundara, H.T.K.; Abeysekara, T.D.J.; Wazil, A.; Rathnayake, S.; Gunaratne, J.; et al. Pilot Study of Renal Urinary Biomarkers for Diagnosis of CKD of Uncertain Etiology. Kidney Int. Rep. 2019, 4, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Hansson, M.; Gustafsson, R.; Jacquet, C.; Chebaane, N.; Satchell, S.; Thunberg, T.; Ahlm, C.; Fors Connolly, A.M. Cystatin C and α-1-Microglobulin Predict Severe Acute Kidney Injury in Patients with Hemorrhagic Fever with Renal Syndrome. Pathogens 2020, 9. [Google Scholar] [CrossRef]
- Olsson, M.G.; Rosenlof, L.W.; Kotarsky, H.; Olofsson, T.; Leanderson, T.; Morgelin, M.; Fellman, V.; Akerstrom, B. The radical-binding lipocalin A1M binds to a Complex I subunit and protects mitochondrial structure and function. Antioxid. Redox Signal. 2013, 18, 2017–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allhorn, M.; Klapyta, A.; Åkerström, B. Redox properties of the lipocalin α1-microglobulin: Reduction of cytochrome c, hemoglobin, and free iron. Free Radic. Biol. Med. 2005, 38, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, J.; Allhorn, M.; Kerstrom, B. The lipocalin alpha(1)-microglobulin binds heme in different species. Arch. Biochem. Biophys. 2004, 432, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Siebel, J.F.; Kosinsky, R.L.; Akerstrom, B.; Knipp, M. Insertion of heme b into the structure of the Cys34-carbamidomethylated human lipocalin alpha(1)-microglobulin: Formation of a [(heme)(2) (alpha(1)-Microglobulin)](3) complex. Chembiochem 2012, 13, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.G.; Olofsson, T.; Tapper, H.; Akerstrom, B. The lipocalin alpha1-microglobulin protects erythroid K562 cells against oxidative damage induced by heme and reactive oxygen species. Free Radic. Res. 2008, 42, 725–736. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawal, H.A.; Asghar, K.; Bureik, M.; Jalal, N. Bystander signaling via oxidative metabolism. OncoTargets Ther. 2017, 10, 3925–3940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, M.G.; Nilsson, E.J.C.; Rutardóttir, S.; Paczesny, J.; Pallon, J.; Åkerström, B. Bystander Cell Death and Stress Response is Inhibited by the Radical Scavenger α1-Microglobulin in Irradiated Cell Cultures. Radiat. Res. 2010, 174, 590–600. [Google Scholar] [CrossRef]
- Bergwik, J.; Åkerström, B. α1-Microglobulin Binds Illuminated Flavins and Has a Protective Effect Against Sublethal Riboflavin-Induced Damage in Retinal Epithelial Cells. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Cederlund, M.; Ghosh, F.; Arnér, K.; Andréasson, S.; Åkerström, B. Vitreous levels of oxidative stress biomarkers and the radical-scavenger α1-microglobulin/A1M in human rhegmatogenous retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Akerstrom, B.; Rosenlof, L.; Hagerwall, A.; Rutardottir, S.; Ahlstedt, J.; Johansson, M.E.; Erlandsson, L.; Allhorn, M.; Gram, M. rA1M-035, a Physicochemically Improved Human Recombinant alpha1-Microglobulin, Has Therapeutic Effects in Rhabdomyolysis-Induced Acute Kidney Injury. Antioxid. Redox Signal. 2019, 30, 489–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunnarsson, R.; Åkerström, B.; Hansson, S.R.; Gram, M. Recombinant alpha-1-microglobulin: A potential treatment for preeclampsia. Drug Discov. Today 2017, 22, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Erlandsson, L.; Ducat, A.; Castille, J.; Zia, I.; Kalapotharakos, G.; Hedström, E.; Vilotte, J.-L.; Vaiman, D.; Hansson, S.R. Alpha-1 microglobulin as a potential therapeutic candidate for treatment of hypertension and oxidative stress in the STOX1 preeclampsia mouse model. Sci. Rep. 2019, 9, 8561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wester-Rosenlof, L.; Casslen, V.; Axelsson, J.; Edstrom-Hagerwall, A.; Gram, M.; Holmqvist, M.; Johansson, M.E.; Larsson, I.; Ley, D.; Marsal, K.; et al. A1M/alpha1-microglobulin protects from heme-induced placental and renal damage in a pregnant sheep model of preeclampsia. PLoS ONE 2014, 9, e86353. [Google Scholar] [CrossRef]
- Naav, A.; Erlandsson, L.; Axelsson, J.; Larsson, I.; Johansson, M.; Wester-Rosenlof, L.; Morgelin, M.; Casslen, V.; Gram, M.; Akerstrom, B.; et al. A1M Ameliorates Preeclampsia-Like Symptoms in Placenta and Kidney Induced by Cell-Free Fetal Hemoglobin in Rabbit. PLoS ONE 2015, 10, e0125499. [Google Scholar] [CrossRef] [Green Version]
- Sverrisson, K.; Axelsson, J.; Rippe, A.; Gram, M.; Akerstrom, B.; Hansson, S.R.; Rippe, B. Extracellular fetal hemoglobin induces increases in glomerular permeability: Inhibition with alpha1-microglobulin and tempol. Am. J. Physiol. Ren. Physiol. 2014, 306, F442–F448. [Google Scholar] [CrossRef] [Green Version]
- Romantsik, O.; Agyemang, A.A.; Sveinsdóttir, S.; Rutardóttir, S.; Holmqvist, B.; Cinthio, M.; Mörgelin, M.; Gumus, G.; Karlsson, H.; Hansson, S.R.; et al. The heme and radical scavenger α1-microglobulin (A1M) confers early protection of the immature brain following preterm intraventricular hemorrhage. J. Neuroinflamm. 2019, 16, 122. [Google Scholar] [CrossRef] [Green Version]
- Ahlstedt, J.; Tran, T.A.; Strand, F.; Holmqvist, B.; Strand, S.-E.; Gram, M.; Åkerström, B. Biodistribution and pharmacokinetics of recombinant α1-microglobulin and its potential use in radioprotection of kidneys. Am. J. Nucl. Med. Mol. Imaging 2015, 5, 333–347. [Google Scholar]
- Kristiansson, A.; Ahlstedt, J.; Holmqvist, B.; Brinte, A.; Tran, T.A.; Forssell-Aronsson, E.; Strand, S.E.; Gram, M.; Akerstrom, B. Protection of Kidney Function with Human Antioxidation Protein alpha1-Microglobulin in a Mouse (177)Lu-DOTATATE Radiation Therapy Model. Antioxid. Redox Signal. 2019, 30, 1746–1759. [Google Scholar] [CrossRef] [Green Version]
- Andersson, C.K.; Shubbar, E.; Schüler, E.; Åkerström, B.; Gram, M.; Forssell-Aronsson, E.B. Recombinant α1-Microglobulin Is a Potential Kidney Protector in 177Lu-Octreotate Treatment of Neuroendocrine Tumors. J. Nucl. Med. 2019, 60, 1600–1604. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar] [PubMed]
- Ofori-Acquah, S.F.; Hazra, R.; Orikogbo, O.O.; Crosby, D.; Flage, B.; Ackah, E.B.; Lenhart, D.; Tan, R.J.; Vitturi, D.A.; Paintsil, V.; et al. Hemopexin deficiency promotes acute kidney injury in sickle cell disease. Blood 2020, 135, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.L. Heme A1M’ed at the kidney in sickle cell disease. Blood 2020, 135, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Mosialou, I.; Shikhel, S.; Liu, J.M.; Maurizi, A.; Luo, N.; He, Z.; Huang, Y.; Zong, H.; Friedman, R.A.; Barasch, J.; et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 2017, 543, 385–390. [Google Scholar] [CrossRef]
- Mera, P.; Ferron, M.; Mosialou, I. Regulation of Energy Metabolism by Bone-Derived Hormones. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Flygare, J.; Karlsson, S. Diamond-Blackfan anemia: Erythropoiesis lost in translation. Blood 2007, 109, 3152–3154. [Google Scholar] [CrossRef] [Green Version]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [Green Version]
- Doty, R.T.; Phelps, S.R.; Shadle, C.; Sanchez-Bonilla, M.; Keel, S.B.; Abkowitz, J.L. Coordinate expression of heme and globin is essential for effective erythropoiesis. J. Clin. Investig. 2015, 125, 4681–4691. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Keel, S.B.; Shimamura, A.; Liu, L.; Gerds, A.T.; Li, H.Y.; Wood, B.L.; Scott, B.L.; Abkowitz, J.L. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med. 2016, 8, 338ra367. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, S.; Aspesi, A.; Silengo, L.; Altruda, F.; Dianzani, I.; Chiabrando, D. Alteration of heme metabolism in a cellular model of Diamond-Blackfan anemia. Eur. J. Haematol. 2016, 96, 367–374. [Google Scholar] [CrossRef]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rio, S.; Gastou, M.; Karboul, N.; Derman, R.; Suriyun, T.; Manceau, H.; Leblanc, T.; El Benna, J.; Schmitt, C.; Azouzi, S.; et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood 2019, 133, 1358–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doty, R.T.; Yan, X.; Lausted, C.; Munday, A.D.; Yang, Z.; Yi, D.; Jabbari, N.; Liu, L.; Keel, S.B.; Tian, Q.; et al. Single-cell analyses demonstrate that a heme-GATA1 feedback loop regulates red cell differentiation. Blood 2019, 133, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A. Alpha 1 Microglobulin: A Potentially Paradoxical Anti-Oxidant Agent. Adv. Tech. Biol. Med. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Shim, S.Y.; Sung, I.K. Intraventricular Hemorrhage and Post Hemorrhagic Hydrocephalus among Very-Low-Birth-Weight Infants in Korea. J. Korean Med. Sci. 2015, 30 (Suppl. 1), S52–S58. [Google Scholar] [CrossRef]
- Handley, S.C.; Passarella, M.; Lee, H.C.; Lorch, S.A. Incidence Trends and Risk Factor Variation in Severe Intraventricular Hemorrhage across a Population Based Cohort. J. Pediatr. 2018, 200, 24–29. [Google Scholar] [CrossRef]
- Schmidt, B.; Asztalos, E.V.; Roberts, R.S.; Robertson, C.M.T.; Sauve, R.S.; Whitfield, M.F.; for the Trial of Indomethacin Prophylaxis in Preterms (TIPP). Impact of Bronchopulmonary Dysplasia, Brain Injury, and Severe Retinopathy on the Outcome of Extremely Low-Birth-Weight Infants at 18 MonthsResults From the Trial of Indomethacin Prophylaxis in Preterms. JAMA 2003, 289, 1124–1129. [Google Scholar] [CrossRef] [Green Version]
- Holsti, A.; Serenius, F.; Farooqi, A. Impact of major neonatal morbidities on adolescents born at 23–25 weeks of gestation. Acta Paediatr. 2018, 107, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Reeder, J.D.; Kaude, J.V.; Setzer, E.S. Choroid plexus hemorrhage in premature neonates: Recognition by sonography. AJNR Am. J. Neuroradiol. 1982, 3, 619–622. [Google Scholar]
- Ballabh, P. Intraventricular hemorrhage in premature infants: Mechanism of disease. Pediatr. Res. 2010, 67, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ley, D.; Romantsik, O.; Vallius, S.; Sveinsdóttir, K.; Sveinsdóttir, S.; Agyemang, A.A.; Baumgarten, M.; Mörgelin, M.; Lutay, N.; Bruschettini, M.; et al. High Presence of Extracellular Hemoglobin in the Periventricular White Matter Following Preterm Intraventricular Hemorrhage. Front. Physiol. 2016, 7, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.D.T.G.; Eggert, A.; Boerman, O.C.; Oyen, W.J.G.; Verhofstad, A.; Abraham, N.G.; Adema, G.; van Kooyk, Y.; de Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001, 98, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Sveinsdottir, S.; Gram, M.; Cinthio, M.; Sveinsdottir, K.; Mörgelin, M.; Ley, D. Altered expression of aquaporin 1 and 5 in the choroid plexus following preterm intraventricular hemorrhage. Int. J. Dev. Neurosci. 2014, 36, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Gram, M.; Sveinsdottir, S.; Cinthio, M.; Sveinsdottir, K.; Hansson, S.R.; Mörgelin, M.; Åkerström, B.; Ley, D. Extracellular hemoglobin—Mediator of inflammation and cell death in the choroid plexus following preterm intraventricular hemorrhage. J. Neuroinflamm. 2014, 11, 200. [Google Scholar] [CrossRef] [Green Version]
- Gram, M.; Sveinsdottir, S.; Ruscher, K.; Hansson, S.R.; Cinthio, M.; Akerström, B.; Ley, D. Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J. Neuroinflamm. 2013, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Agyemang, A.A.; Sveinsdóttir, K.; Vallius, S.; Sveinsdóttir, S.; Bruschettini, M.; Romantsik, O.; Hellström, A.; Smith, L.E.H.; Ohlsson, L.; Holmqvist, B.; et al. Cerebellar Exposure to Cell-Free Hemoglobin Following Preterm Intraventricular Hemorrhage: Causal in Cerebellar Damage? Transl. Stroke Res. 2017, 8, 461–473. [Google Scholar] [CrossRef]
- Giblett, E.R. Blood group alloantibodies: An assessment of some laboratory practices. Transfusion 1977, 17, 299–308. [Google Scholar] [CrossRef]
- Tormey, C.A.; Fisk, J.; Stack, G. Red blood cell alloantibody frequency, specificity, and properties in a population of male military veterans. Transfusion 2008, 48, 2069–2076. [Google Scholar] [CrossRef]
- Poles, D.; on behalf of the Serious Hazards of Transfusion (SHOT) Steering Group. The 2019 Annual SHOT Report (2020). Narayan, S., Ed.; 2020. Available online: www.shot.org (accessed on 29 September 2020).
- Pirenne, F. The cause and pathogenesis of hemolytic transfusion reactions in sickle-cell disease. Curr. Opin. Hematol. 2019, 26, 488–494. [Google Scholar] [CrossRef]
- Balbuena-Merle, R.; Hendrickson, J.E. Red blood cell alloimmunization and delayed hemolytic transfusion reactions in patients with sickle cell disease. Transfus. Clin. Biol. 2019, 26, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Adkins, B.D.; Sharma, D.; Eichbaum, Q. Can we better predict delayed hemolytic transfusion reactions and hyperhemolysis in sickle cell disease? Transfus. Apher. Sci. 2020, 59, 102681. [Google Scholar] [CrossRef] [PubMed]
- Dean, C.L.; Maier, C.L.; Chonat, S.; Chang, A.; Carden, M.A.; El Rassi, F.; McLemore, M.L.; Stowell, S.R.; Fasano, R.M. Challenges in the treatment and prevention of delayed hemolytic transfusion reactions with hyperhemolysis in sickle cell disease patients. Transfusion 2019, 59, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Seheult, J.N.; Bahr, M.P.; Spinella, P.C.; Triulzi, D.J.; Yazer, M.H. The Dead Sea needs salt water… massively bleeding patients need whole blood: The evolution of blood product resuscitation. Transfus. Clin. Biol. 2019, 26, 174–179. [Google Scholar] [CrossRef]
- Avery, P.; Morton, S.; Tucker, H.; Green, L.; Weaver, A.; Davenport, R. Whole blood transfusion versus component therapy in adult trauma patients with acute major haemorrhage. Emerg. Med. J. 2020, 37, 370–378. [Google Scholar] [CrossRef]
- Doughty, H.; Maguire, A.; Fitchett, G.; Parker, P. Group O low titre only emergency donor panels for small combat teams. J. R. Army Med. Corps 2017, 163, 401–404. [Google Scholar] [CrossRef]
- Yoshida, T.; Prudent, M.; D’Alessandro, A. Red blood cell storage lesion: Causes and potential clinical consequences. Blood Transfus. 2019, 17, 27–52. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Seghatchian, J. Hitchhiker’s guide to the red cell storage galaxy: Omics technologies and the quality issue. Transfus. Apher. Sci. 2017, 56, 248–253. [Google Scholar] [CrossRef]
- Mays, J.A.; Hess, J.R. Modelling the effects of blood component storage lesions on the quality of haemostatic resuscitation in massive transfusion for trauma. Blood Transfus. 2017, 15, 153–157. [Google Scholar] [CrossRef]
- Dumont, L.J.; AuBuchon, J.P. Evaluation of proposed FDA criteria for the evaluation of radiolabeled red cell recovery trials. Transfusion 2008, 48, 1053–1060. [Google Scholar] [CrossRef]
- Mangalmurti, N.S.; Chatterjee, S.; Cheng, G.; Andersen, E.; Mohammed, A.; Siegel, D.L.; Schmidt, A.M.; Albelda, S.M.; Lee, J.S. Advanced glycation end products on stored red blood cells increase endothelial reactive oxygen species generation through interaction with receptor for advanced glycation end products. Transfusion 2010, 50, 2353–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Available online: http://www.who.int/reproductivehealth/publications/monitoring/maternal-mortality-2015/en/ (accessed on 1 September 2020).
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brosens, J.J.; Pijnenborg, R.; Brosens, I.A. The myometrial junctional zone spiral arteries in normal and abnormal pregnancies: A review of the literature. Am. J. Obstet. Gynecol. 2002, 187, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Jauniaux, E. Placental oxidative stress: From miscarriage to preeclampsia. J. Soc. Gynecol. Investig. 2004, 11, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Hubel, C.A. Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet 1999, 354, 788–789. [Google Scholar] [CrossRef]
- Centlow, M.; Wingren, C.; Borrebaeck, C.; Brownstein, M.J.; Hansson, S.R. Differential gene expression analysis of placentas with increased vascular resistance and pre-eclampsia using whole-genome microarrays. J. Pregnancy 2011, 2011, 472354. [Google Scholar] [CrossRef] [PubMed]
- Hansson, S.R.; Naav, A.; Erlandsson, L. Oxidative stress in preeclampsia and the role of free fetal hemoglobin. Front. Physiol. 2014, 5, 516. [Google Scholar] [CrossRef] [Green Version]
- Centlow, M.; Carninci, P.; Nemeth, K.; Mezey, E.; Brownstein, M.; Hansson, S.R. Placental expression profiling in preeclampsia: Local overproduction of hemoglobin may drive pathological changes. Fertil. Steril. 2008, 90, 1834–1843. [Google Scholar] [CrossRef] [Green Version]
- Masoumi, Z.; Familari, M.; Källén, K.; Ranstam, J.; Olofsson, P.; Hansson, S.R. Fetal hemoglobin in umbilical cord blood in preeclamptic and normotensive pregnancies: A cross-sectional comparative study. PLoS ONE 2017, 12, e0176697. [Google Scholar] [CrossRef]
- Kalapotharakos, G.; Murtoniemi, K.; Åkerström, B.; Hämäläinen, E.; Kajantie, E.; Räikkönen, K.; Villa, P.; Laivuori, H.; Hansson, S.R. Plasma Heme Scavengers Alpha-1-Microglobulin and Hemopexin as Biomarkers in High-Risk Pregnancies. Front. Physiol. 2019, 10, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, U.D.; Jälmby, M.; Faas, M.M.; Hansson, S.R. The hemoglobin degradation pathway in patients with preeclampsia—Fetal hemoglobin, heme, heme oxygenase-1 and hemopexin—Potential diagnostic biomarkers? Pregnancy Hypertens 2018, 14, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Brook, A.; Hoaksey, A.; Gurung, R.; Yoong, E.E.C.; Sneyd, R.; Baynes, G.C.; Bischof, H.; Jones, S.; Higgins, L.E.; Jones, C.; et al. Cell free hemoglobin in the fetoplacental circulation: A novel cause of fetal growth restriction? FASEB J. 2018, 32, 5436–5446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C. Oxidative reactions of hemoglobin. Methods Enzymol. 1990, 186, 265–272. [Google Scholar]
- Gram, M.; Anderson, U.D.; Johansson, M.E.; Edstrom-Hagerwall, A.; Larsson, I.; Jalmby, M.; Hansson, S.R.; Akerstrom, B. The Human Endogenous Protection System against Cell-Free Hemoglobin and Heme Is Overwhelmed in Preeclampsia and Provides Potential Biomarkers and Clinical Indicators. PLoS ONE 2015, 10, e0138111. [Google Scholar] [CrossRef] [Green Version]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Anderson, U.D.; Olsson, M.G.; Rutardottir, S.; Centlow, M.; Kristensen, K.H.; Isberg, P.E.; Thilaganathan, B.; Akerstrom, B.; Hansson, S.R. Fetal hemoglobin and alpha1-microglobulin as first- and early second-trimester predictive biomarkers for preeclampsia. Am. J. Obstet. Gynecol. 2011, 204, 520–e521. [Google Scholar] [CrossRef]
- Olsson, M.G.; Centlow, M.; Rutardottir, S.; Stenfors, I.; Larsson, J.; Hosseini-Maaf, B.; Olsson, M.L.; Hansson, S.R.; Akerstrom, B. Increased levels of cell-free hemoglobin, oxidation markers, and the antioxidative heme scavenger alpha(1)-microglobulin in preeclampsia. Free Radic. Biol. Med. 2010, 48, 284–291. [Google Scholar] [CrossRef] [Green Version]
- May, K.; Rosenlof, L.; Olsson, M.G.; Centlow, M.; Morgelin, M.; Larsson, I.; Cederlund, M.; Rutardottir, S.; Siegmund, W.; Schneider, H.; et al. Perfusion of human placenta with hemoglobin introduces preeclampsia-like injuries that are prevented by alpha1-microglobulin. Placenta 2011, 32, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Youssef, L.; Erlandsson, L.; Åkerström, B.; Miranda, J.; Paules, C.; Crovetto, F.; Crispi, F.; Gratacos, E.; Hansson, S.R. Hemopexin and α1-microglobulin heme scavengers with differential involvement in preeclampsia and fetal growth restriction. PLoS ONE 2020, 15, e0239030. [Google Scholar] [CrossRef]
- Shahal, Y.; Bauminger, E.R.; Zmora, E.; Katz, M.; Mazor, D.; Horn, S.; Meyerstein, N. Oxidative Stress in Newborn Erythrocytes. Pediatr. Res. 1991, 29, 119–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracci, R.; Buonocore, G. The antioxidant status of erythrocytes in preterm and term infants. Semin. Neonatol. 1998, 3, 191–197. [Google Scholar] [CrossRef]
- Norwitz, E.R.; Tsen, L.C.; Park, J.S.; Fitzpatrick, P.A.; Dorfman, D.M.; Saade, G.R.; Buhimschi, C.S.; Buhimschi, I.A. Discriminatory proteomic biomarker analysis identifies free hemoglobin in the cerebrospinal fluid of women with severe preeclampsia. Am. J. Obstet. Gynecol. 2005, 193, 957–964. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, C.B.; Duvekot, J.J.; Güzel, C.; Hansson, S.R.; de Leeuw, T.G.; Steegers, E.A.; Versendaal, J.; Luider, T.M.; Stoop, M.P. Elevated levels of protein AMBP in cerebrospinal fluid of women with preeclampsia compared to normotensive pregnant women. Proteom. Clin. Appl. 2017, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haram, K.; Svendsen, E.; Abildgaard, U. The HELLP syndrome: Clinical issues and management. A Review. BMC Pregnancy Childbirth 2009, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Insull, W., Jr. The Pathology of Atherosclerosis: Plaque Development and Plaque Responses to Medical Treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef]
- Davies, M.J. The pathophysiology of acute coronary syndromes. Heart 2000, 83, 361. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.M. Atherothrombosis and ischaemic stroke. BMJ 2007, 334, 379–380. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.P.; Moreno, P.R. Intraplaque hemorrhage. Curr. Mol. Med. 2006, 6, 479–488. [Google Scholar] [CrossRef]
- Moreno, P.R.; Purushothaman, M.; Purushothaman, K.R. Plaque neovascularization: Defense mechanisms, betrayal, or a war in progress. Ann. N. Y. Acad. Sci. 2012, 1254, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, G.; Balla, J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol. 2014, 5, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tziakas, D.N.; Chalikias, G.K.; Stakos, D.; Tentes, I.K.; Thomaidi, A.; Chatzikyriakou, S.; Mitrousi, K.; Kortsaris, A.X.; Kaski, J.C.; Boudoulas, H.; et al. Statin use is associated with a significant reduction in cholesterol content of erythrocyte membranes. A novel pleiotropic effect? Cardiovasc. Drugs Ther. 2009, 23, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Akyea, R.K.; Kai, J.; Qureshi, N.; Iyen, B.; Weng, S.F. Sub-optimal cholesterol response to initiation of statins and future risk of cardiovascular disease. Heart 2019, 105, 975–981. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, Y.; Kondo, K.; Momiyama, Y. The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases. Int. J. Mol. Sci. 2019, 20, 3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlak, T.W.; Snyder, S.H. Bilirubin Benefits: Cellular Protection by a Biliverdin Reductase Antioxidant Cycle. Pediatrics 2004, 113, 1776. [Google Scholar] [CrossRef] [PubMed]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef] [Green Version]
- Juan, S.-H.; Lee, T.-S.; Tseng, K.-W.; Liou, J.-Y.; Shyue, S.-K.; Wu Kenneth, K.; Chau, L.-Y. Adenovirus-Mediated Heme Oxygenase-1 Gene Transfer Inhibits the Development of Atherosclerosis in Apolipoprotein E–Deficient Mice. Circulation 2001, 104, 1519–1525. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Cederlund, M.; Deronic, A.; Pallon, J.; Sørensen, O.E.; Åkerström, B. A1M/α1-microglobulin is proteolytically activated by myeloperoxidase, binds its heme group and inhibits low density lipoprotein oxidation. Front. Physiol. 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Wójcik-Cichy, K.; Koślińska-Berkan, E.; Piekarska, A. The influence of NAFLD on the risk of atherosclerosis and cardiovascular diseases. Clin. Exp. Hepatol. 2018, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sandfort, V.; Lai, S.; Ahlman, M.A.; Mallek, M.; Liu, S.; Sibley, C.T.; Turkbey, E.B.; Lima, J.A.C.; Bluemke, D.A. Obesity Is Associated With Progression of Atherosclerosis During Statin Treatment. J. Am. Heart Assoc. 2016, 5, e003621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirier, P.; Giles Thomas, D.; Bray George, A.; Hong, Y.; Stern Judith, S.; Pi-Sunyer, F.X.; Eckel Robert, H. Obesity and Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 968–976. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kristiansson, A.; Gram, M.; Flygare, J.; Hansson, S.R.; Åkerström, B.; Storry, J.R. The Role of α1-Microglobulin (A1M) in Erythropoiesis and Erythrocyte Homeostasis—Therapeutic Opportunities in Hemolytic Conditions. Int. J. Mol. Sci. 2020, 21, 7234. https://doi.org/10.3390/ijms21197234
Kristiansson A, Gram M, Flygare J, Hansson SR, Åkerström B, Storry JR. The Role of α1-Microglobulin (A1M) in Erythropoiesis and Erythrocyte Homeostasis—Therapeutic Opportunities in Hemolytic Conditions. International Journal of Molecular Sciences. 2020; 21(19):7234. https://doi.org/10.3390/ijms21197234
Chicago/Turabian StyleKristiansson, Amanda, Magnus Gram, Johan Flygare, Stefan R. Hansson, Bo Åkerström, and Jill R. Storry. 2020. "The Role of α1-Microglobulin (A1M) in Erythropoiesis and Erythrocyte Homeostasis—Therapeutic Opportunities in Hemolytic Conditions" International Journal of Molecular Sciences 21, no. 19: 7234. https://doi.org/10.3390/ijms21197234
APA StyleKristiansson, A., Gram, M., Flygare, J., Hansson, S. R., Åkerström, B., & Storry, J. R. (2020). The Role of α1-Microglobulin (A1M) in Erythropoiesis and Erythrocyte Homeostasis—Therapeutic Opportunities in Hemolytic Conditions. International Journal of Molecular Sciences, 21(19), 7234. https://doi.org/10.3390/ijms21197234