Peroxisome Proliferator-Activated Receptor γ Coactivator 1α Activates Vascular Endothelial Growth Factor That Protects Against Neuronal Cell Death Following Status Epilepticus through PI3K/AKT and MEK/ERK Signaling

 , , ,
, , , 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Temporal Changes of PGC-1α Expression in the Hippocampal CA3 following Status Epilepticus
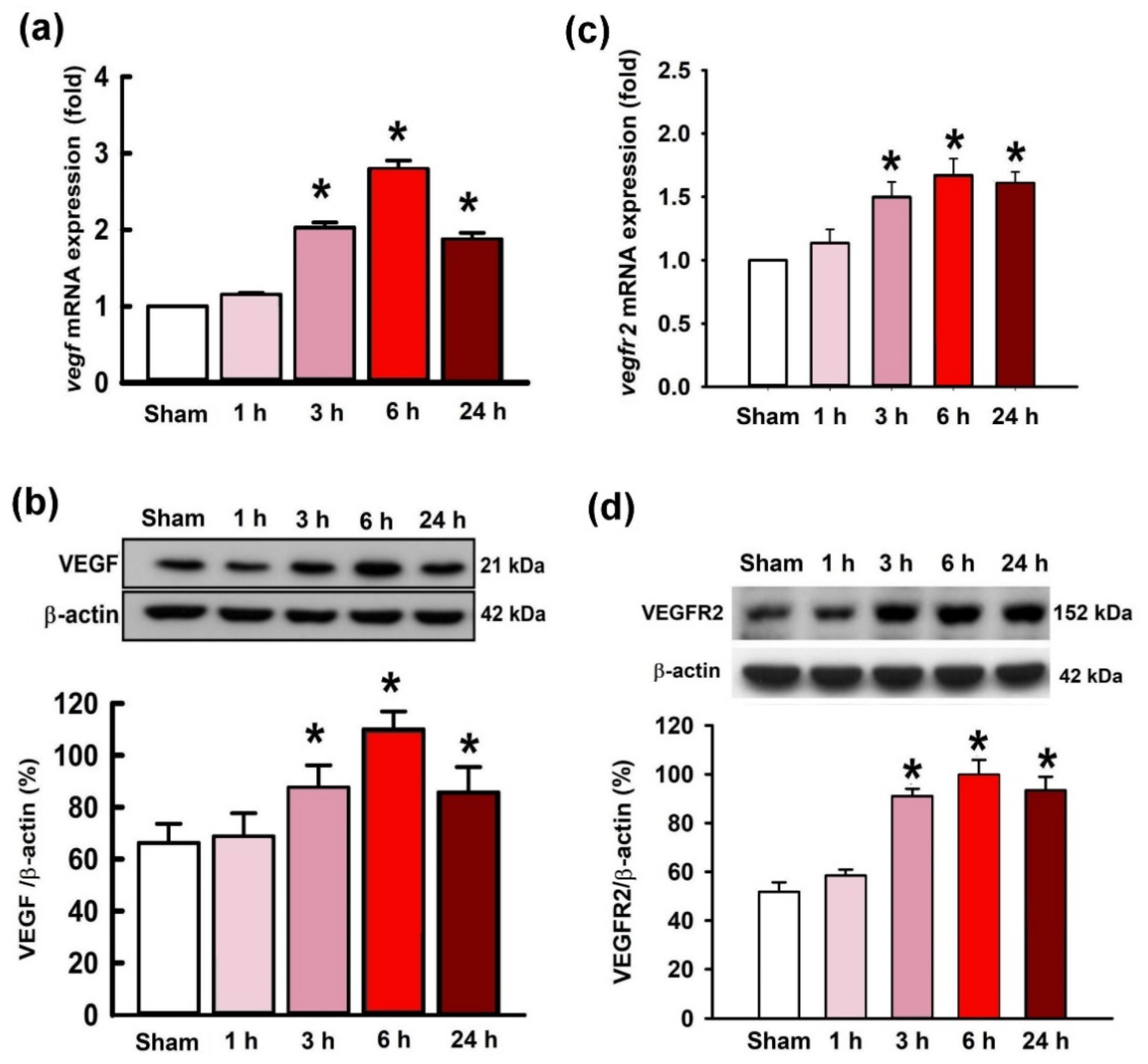
2.2. Temporal Changes of VEGF and VEGFR2 Expression in the Hippocampal CA3 Region following Status Epilepticus
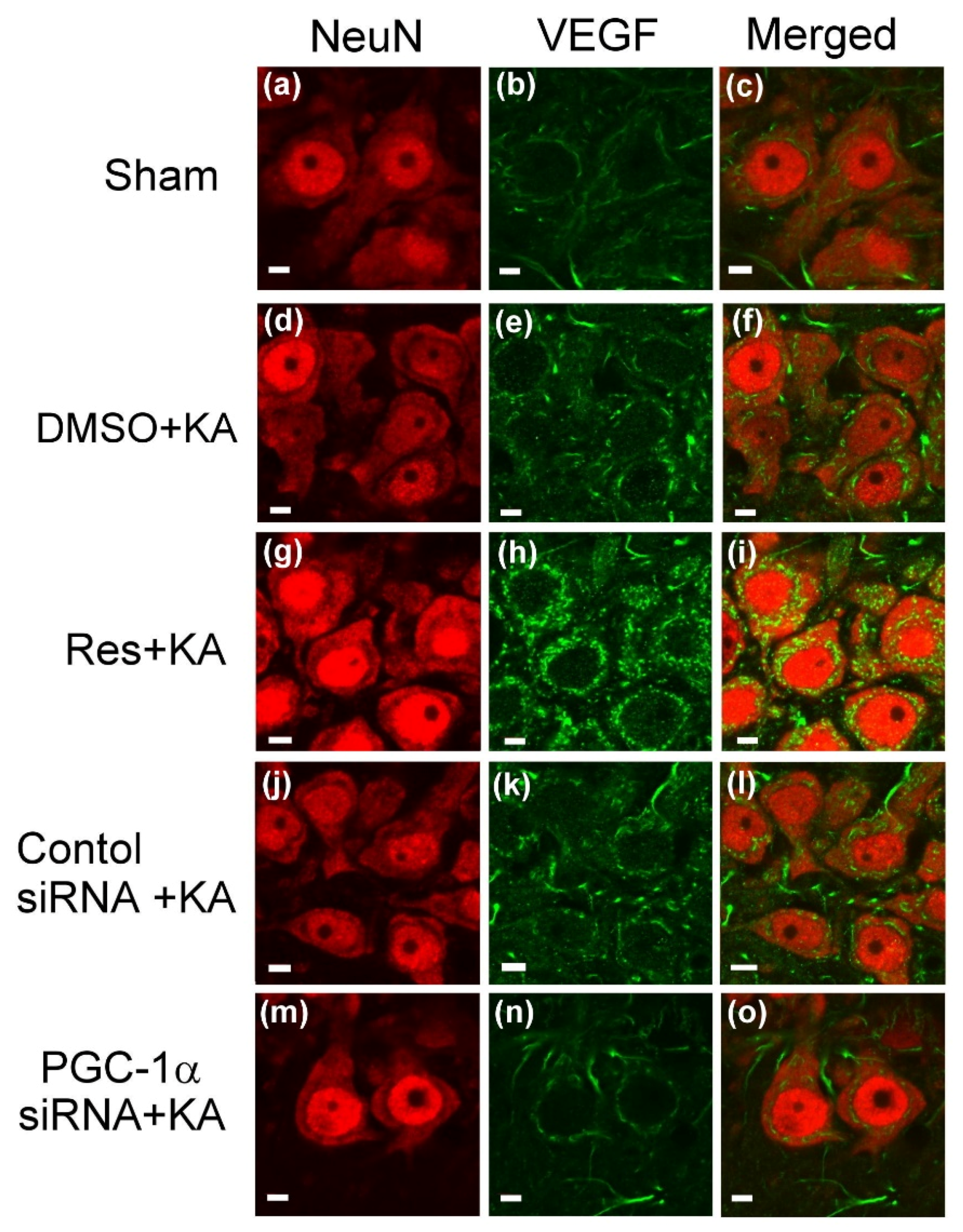
2.3. Effect of Resveratrol and Gene Knock-Down by Small Interfering RNA (siRNA) Against pgc-1α on VEGF Expression in the Hippocampus Following Experimental Status Epilepticus
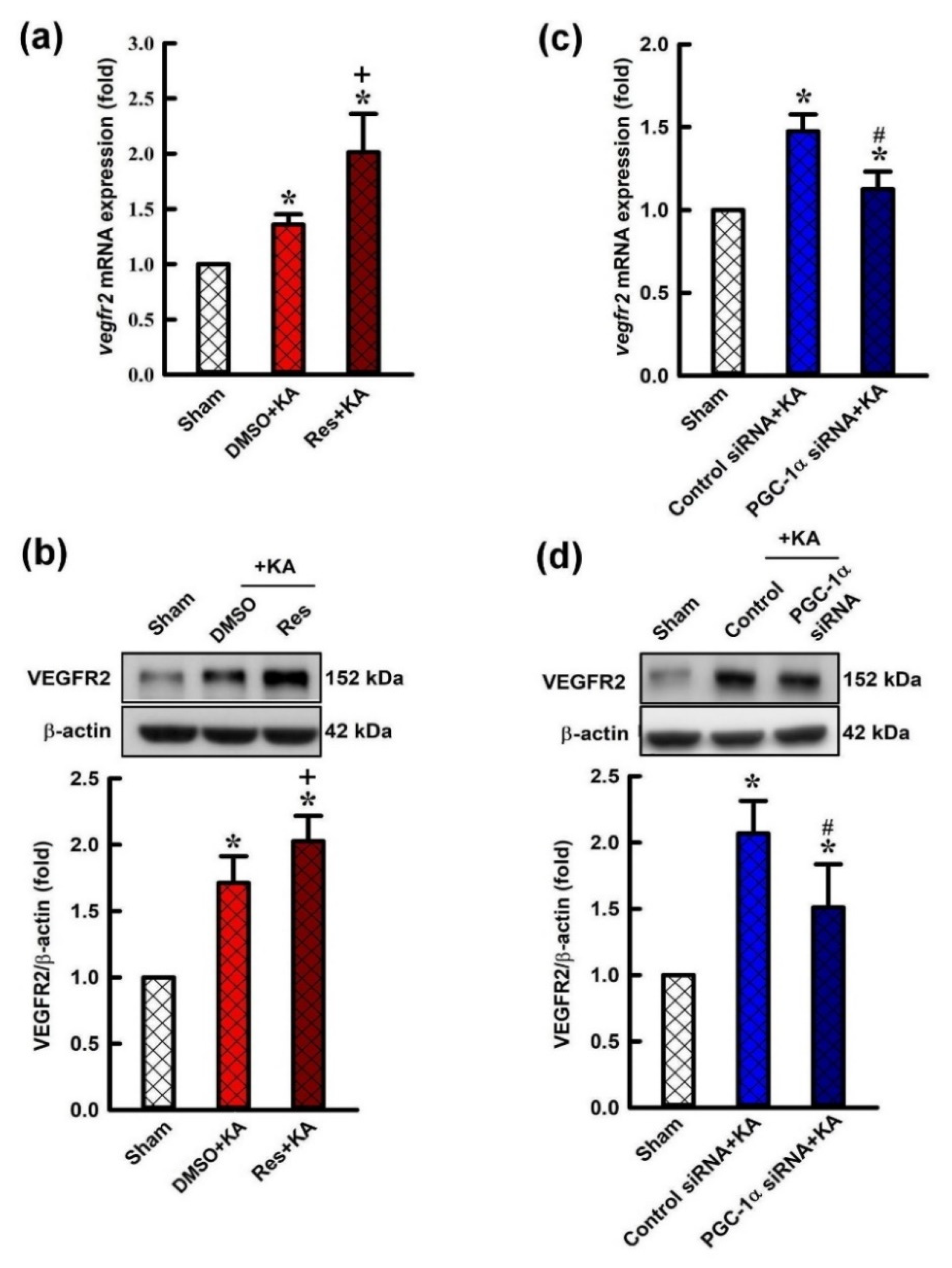
2.4. Effect of Resveratrol and Gene Knock-Down by Small Interfering RNA (siRNA) Against pgc-1α on Expression of VEGF Receptor 2 (VEGFR2) in the Hippocampus Following Experimental Status Epilepticus
2.5. Activation of VEGF/VEGFR2 Regulates the PI3K/Akt Survival Signaling Pathway in the Hippocampal Neurons Following Experimental Status Epilepticus
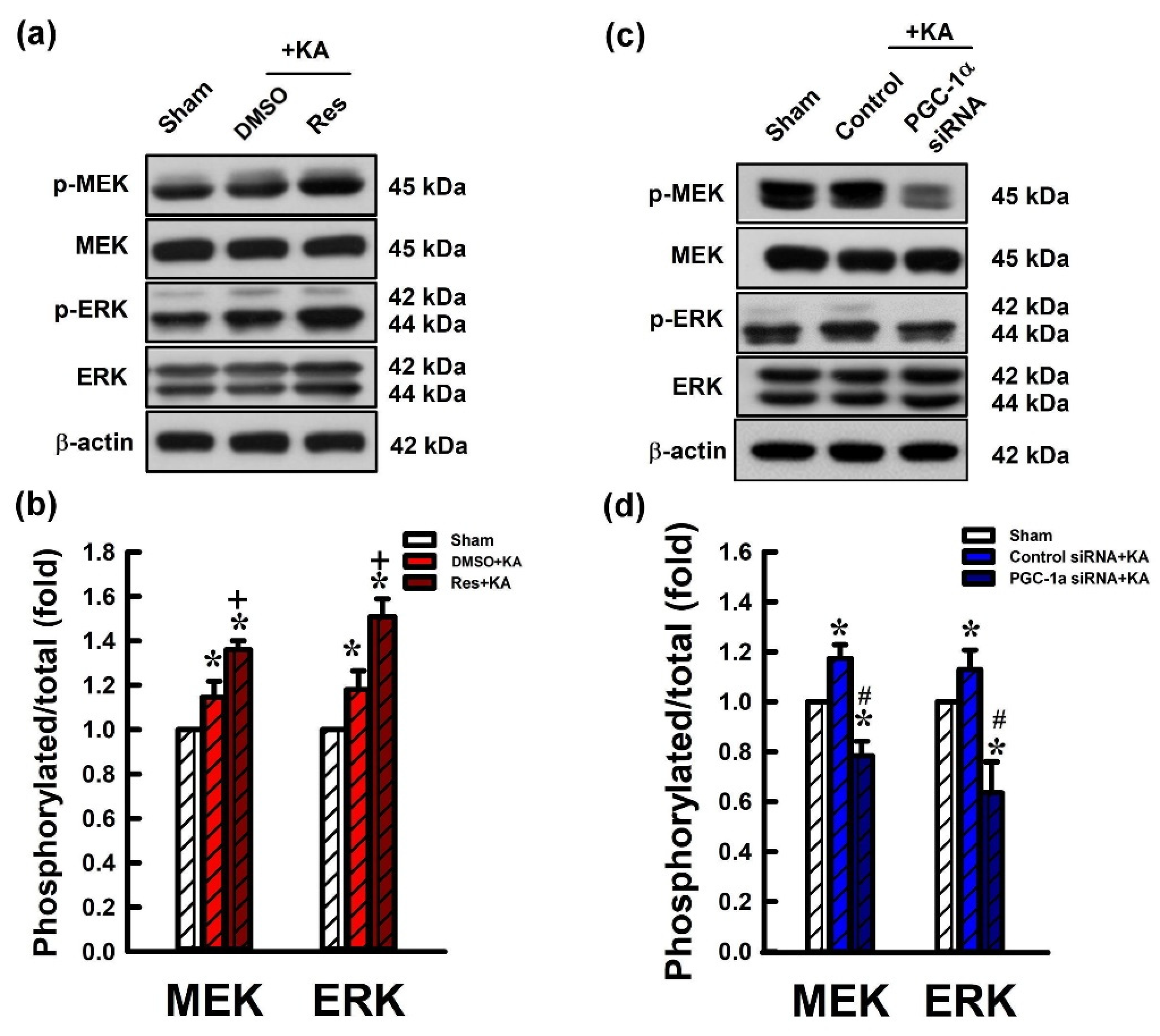
2.6. Activation VEGF/VEGFR2 Regulates the MEK/ERK Signaling Pathway in the Hippocampal Neurons Following Experimental Status Epilepticus
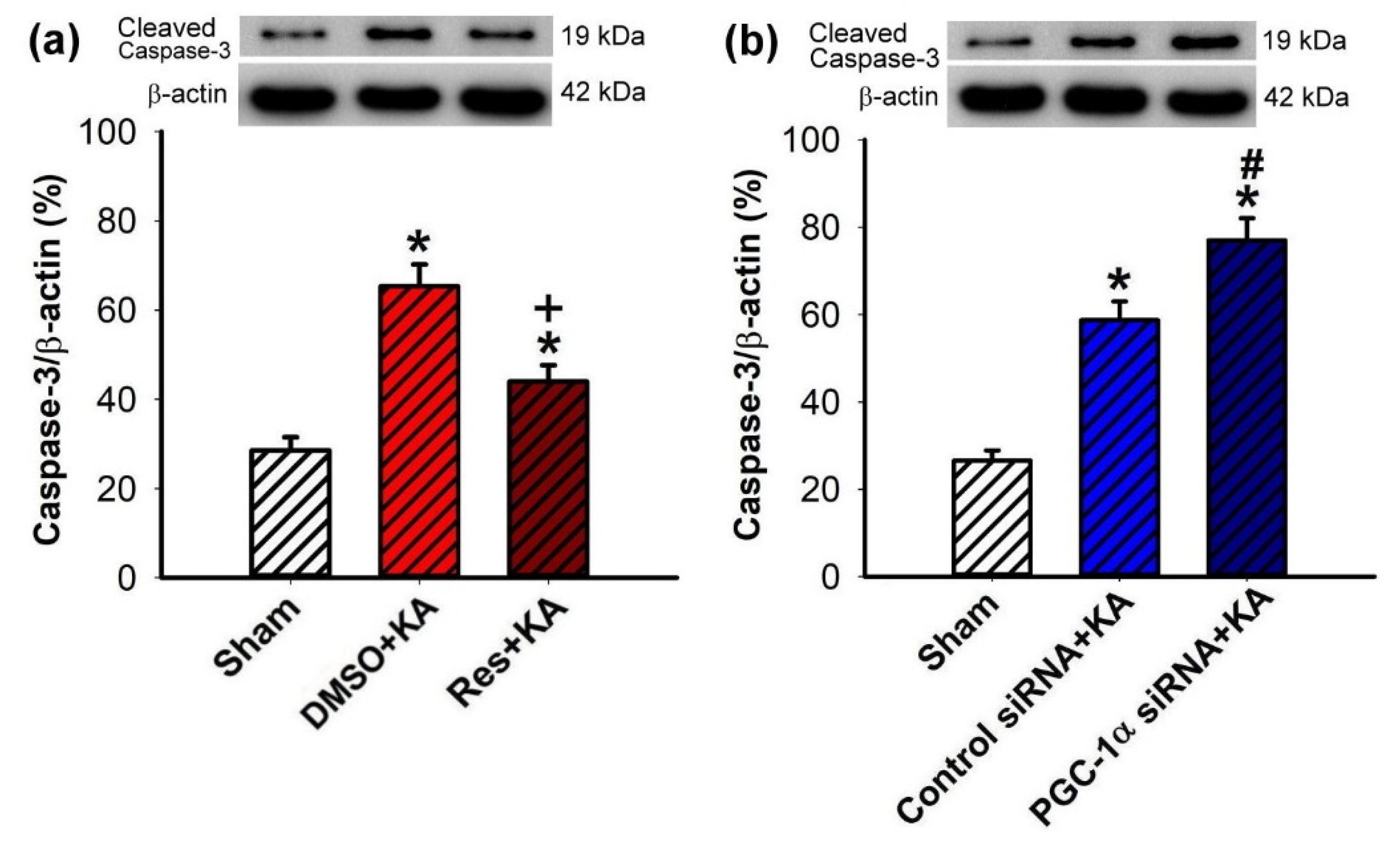
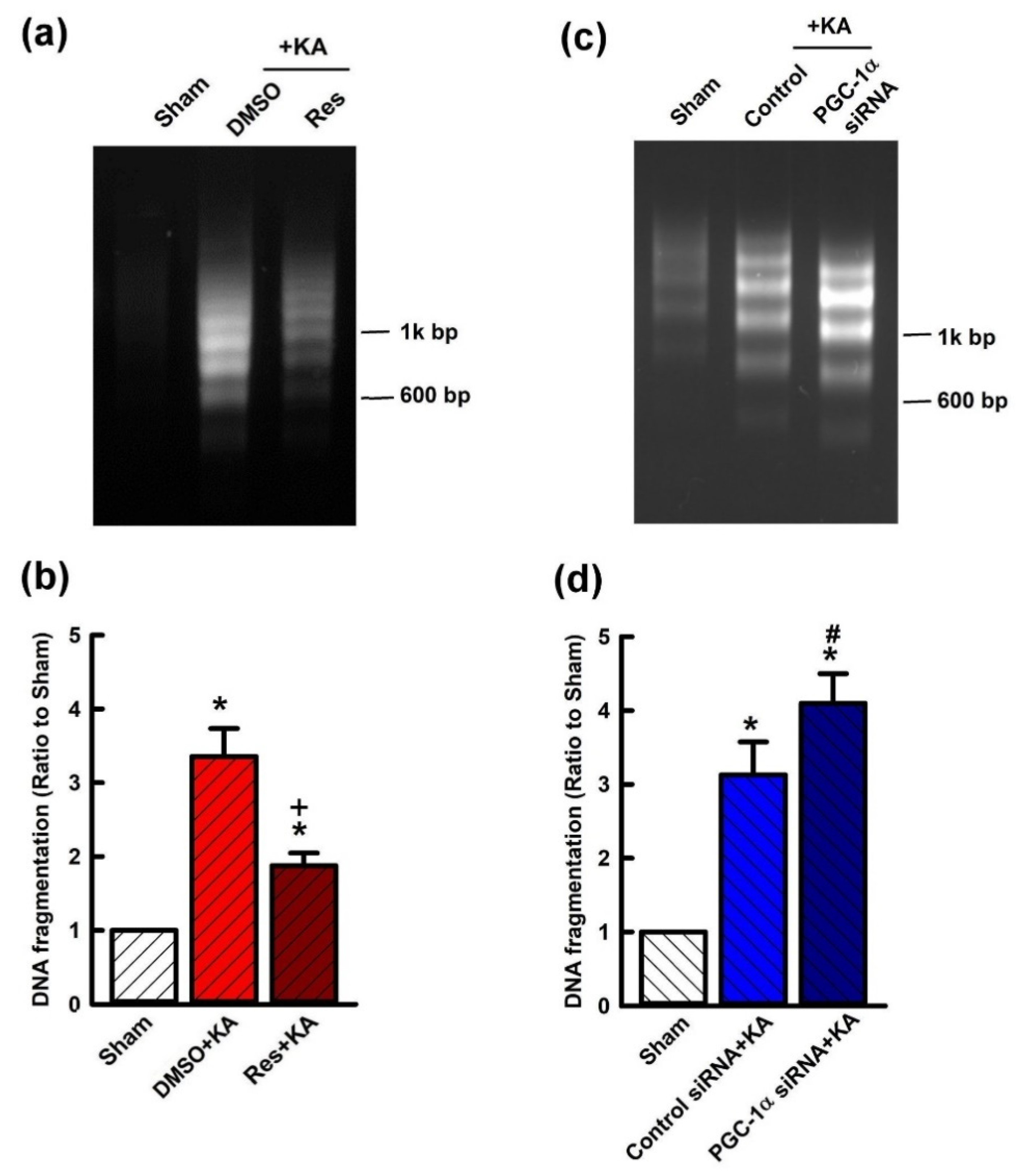
2.7. Effect of VEGF/VEGFR2 on Apoptosis and Neuronal Survival in the Hippocampal CA3 Subfield Following Experimental Status Epilepticus
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Status Epilepticus
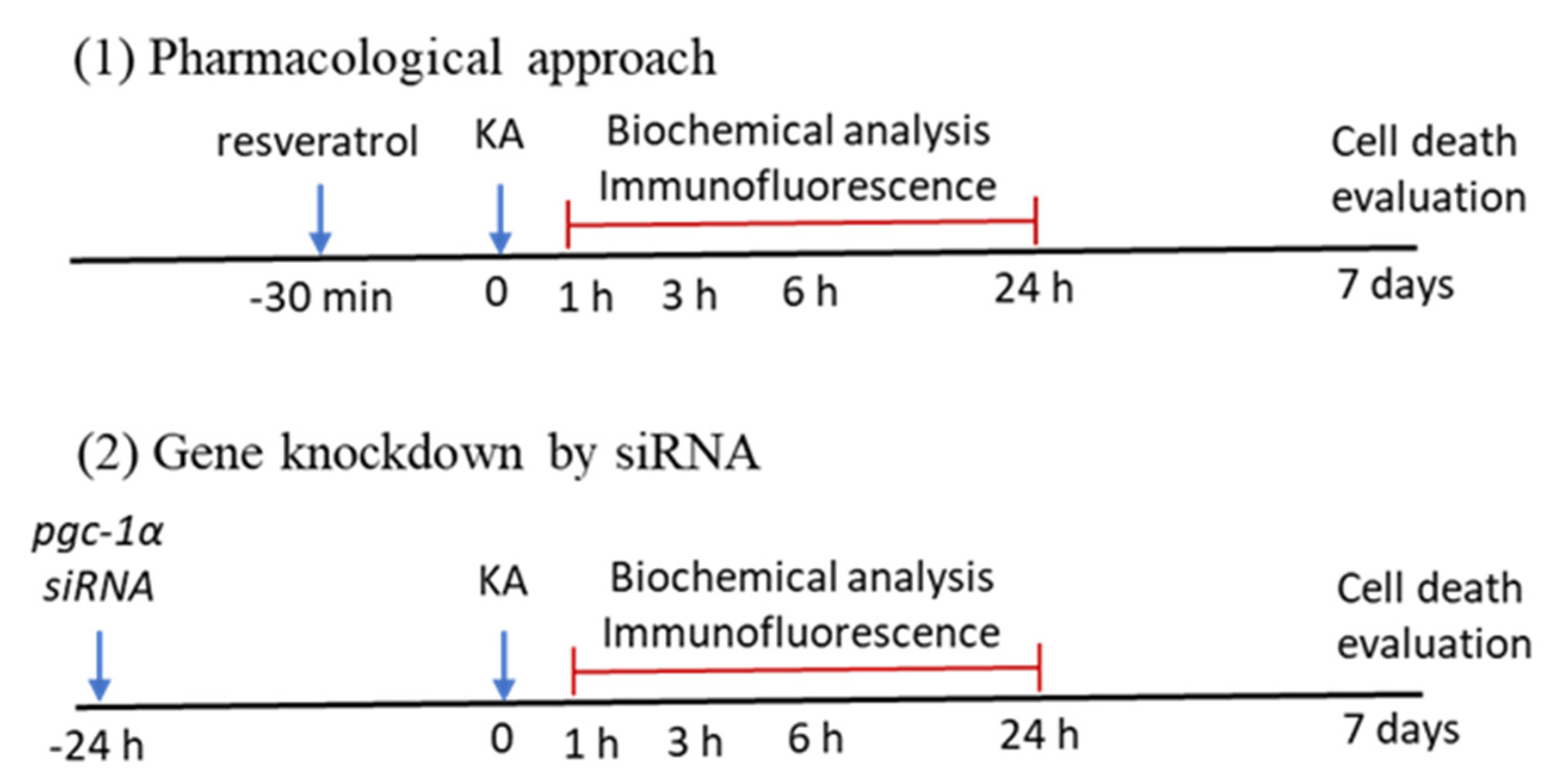
4.3. Pharmacological Pretreatments
4.4. Gene Knockdown by Microinjection of Small Interfering RNA (siRNA) Against pgc-1α into the Hippocampus
| pgc-1α | Sequence |
| Sequence 1 | 5′-CGGUGGAUGAAGACGGAUU-3′ |
| Sequence 2 | 5′-CAAUGAAUGCAGCGGUCUU-3′ |
| Sequence 3 | 5′-GAACAAGACUAUUGAGCGA -3′ |
| Sequence 4 | 5′-AUUCAAACUCAGACGAUUU-3′ |
4.5. Collection of Tissue Samples from the Hippocampus
4.6. RNA Isolation and Reverse Transcription Real-Time Polymerase Chain Reaction
| Gene | Forward Primer | Reverse Primer |
| pgc-1α | 5′-GTTTCATTACCTACCGTTACAC-3′ | 5′-ATCGTCTGAGTTTGAATCTAGG-3′ |
| vegf | 5′-GCAGATGTGAATGCAGACCA-3′ | 5′-TTTCCCTTTCCTCGAACTGA-3′ |
| vegfr2 | 5′-AAGCAAATGCTCAGCAGGAT-3′ | 5′-GAGGTAGGCAGGGAGAGTCC-3′ |
| Gapdh | 5′-AACGGCACAGTCAAGGCTGA-3′ | 5′- ACGCCAGTAGACTCCACGACAT -3′ |
4.7. Western Blotting Analysis
4.8. Double Immunofluorescence Staining and Laser Confocal Microscopy
4.9. Qualitative and Quantitative Analysis of DNA Fragmentation
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PGC1-α | Peroxisome proliferator-activated receptor γ coactivator 1α |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor |
| VEGFR2 | Vascular endothelial growth factor 2 |
| Flk1 | Fetal liver kinase receptor 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| AKT | Protein kinase B |
| MEK | Mitogen activated protein kinase |
| ERK | Extracellular signal-regulated kinase |
| CNS | Central nervous system |
| KA | Kainic acid |
| siRNA | Small Interfering RNA |
| hEEG | Hippocampal electroencephalogram |
| NeuN | Neuron-specific nuclear protein |
| UCP2 | Uncoupling protein 2 |
References
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Trinka, E.; Brigo, F.; Shorvon, S. Recent advances in status epilepticus. Curr. Opin. Neurol. 2016, 29, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Betjemann, J.P.; Lowenstein, D.H. Status epilepticus in adults. Lancet Neurol. 2015, 14, 615–624. [Google Scholar] [CrossRef]
- Chen, S.D.; Chang, A.Y.; Chuang, Y.C. The potential role of mitochondrial dysfunction in seizure-associated cell death in the hippocampus and epileptogenesis. J. Bioenerg. Biomembr. 2010, 42, 461–465. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Chen, S.D.; Jou, S.B.; Lin, T.K.; Chen, S.F.; Chen, N.C.; Hsu, C.Y. Sirtuin 1 regulates mitochondrial biogenesis and provides an endogenous neuroprotective mechanism against seizure-induced neuronal cell death in the hippocampus following status epilepticus. Int. J. Mol. Sci. 2019, 20, 3588. [Google Scholar] [CrossRef] [Green Version]
- Haut, S.R.; Veliskova, J.; Moshe, S.L. Susceptibility of immature and adult brains to seizure effects. Lancet Neurol. 2004, 3, 608–617. [Google Scholar] [CrossRef]
- Henshall, D.C.; Simon, R.P. Epilepsy and apoptosis pathways. J. Cereb. Blood Flow Metab. 2005, 25, 1557–1572. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Rosenstein, J.M.; Krum, J.M.; Ruhrberg, C. VEGF in the nervous system. Organogenesis 2010, 6, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Eichmann, A.; Simons, M. VEGF signaling inside vascular endothelial cells and beyond. Curr. Opin. Cell Biol. 2012, 24, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.W.; Madsen, J.R. VEGF Signaling in Neurological Disorders. Int. J. Mol. Sci. 2018, 19, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, N.M.; Lee, B.; Banasr, M.; Elsayed, M.; Duman, R.S. Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK-and PI3K/Akt-dependent signaling. Neuropharmacology 2012, 63, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitidou, L.; Kanter-Schlifke, I.; Dhondt, J.; Carmeliet, P.; Lambrechts, D.; Kokaia, M. VEGF receptor-2 (Flk-1) overexpression in mice counteracts focal epileptic seizures. PLoS ONE 2012, 7, e40535. [Google Scholar] [CrossRef] [Green Version]
- Zachary, I. Neuroprotective role of vascular endothelial growth factor: Signalling mechanisms, biological function and therapeutic potential. Neurosignals 2005, 14, 207–221. [Google Scholar] [CrossRef]
- Geiseler, S.J.; Morland, C. The Janus face of VEGF in stroke. Int. J. Mol. Sci. 2018, 19, 1362. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, B.; Zhu, Y.; Li, Y.; Liu, P.; Gao, B.; Tian, S.; Du, L.; Bai, Y. Post-stroke Constraint-induced movement therapy increases functional recovery, angiogenesis, and neurogenesis with enhanced expression of HIF-1alpha and VEGF. Curr. Neurovasc. Res. 2017, 14, 368–377. [Google Scholar] [CrossRef]
- Ma, Y.; Qu, Y.; Fei, Z. Vascular endothelial growth factor in cerebral ischemia. J. Neurosci. Res. 2011, 89, 969–978. [Google Scholar] [CrossRef]
- Nicoletti, J.N.; Lenzer, J.; Salerni, E.A.; Shah, S.K.; Elkady, A.; Khalid, S.; Quinteros, D.; Rotella, F.; Betancourth, D.; Croll, S.D. Vascular endothelial growth factor attenuates status epilepticus-induced behavioral impairments in rats. Epilepsy Behav. 2010, 19, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Kulshreshtha, D.; Vijayalakshmi, K.; Alladi, P.A.; Sathyaprabha, T.N.; Nalini, A.; Raju, T.R. Vascular endothelial growth factor attenuates neurodegenerative changes in the NSC-34 motor neuron cell line induced by cerebrospinal fluid of sporadic amyotrophic lateral sclerosis patients. Neurodegener. Dis. 2011, 8, 322–330. [Google Scholar] [CrossRef]
- Harris, R.; Miners, J.S.; Allen, S.; Love, S. VEGFR1 and VEGFR2 in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 741–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Chen, Z.; Wei, X.; Chen, Z.; Fu, Y.; Yang, X.; Chen, D.; Wang, R.; Jenner, P.; Lu, J.H.; et al. Cystatin C as a potential therapeutic mediator against Parkinson’s disease via VEGF-induced angiogenesis and enhanced neuronal autophagy in neurovascular units. Cell Death Dis. 2017, 8, e2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, J.N.; Shah, S.K.; McCloskey, D.P.; Goodman, J.H.; Elkady, A.; Atassi, H.; Hylton, D.; Rudge, J.S.; Scharfman, H.E.; Croll, S.D. Vascular endothelial growth factor is up-regulated after status epilepticus and protects against seizure-induced neuronal loss in hippocampus. Neuroscience 2008, 151, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Rigau, V.; Morin, M.; Rousset, M.C.; de Bock, F.; Lebrun, A.; Coubes, P.; Picot, M.C.; Baldy-Moulinier, M.; Bockaert, J.; Crespel, A.; et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain 2007, 130, 1942–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, S.S.; Collier, E.F.; Hunsberger, J.; Adams, D.; Terwilliger, R.; Selvanayagam, E.; Duman, R.S. Gene profile of electroconvulsive seizures: Induction of neurotrophic and angiogenic factors. J. Neurosci. 2003, 23, 10841–10851. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Chen, S.D.; Hsu, C.Y.; Chen, S.F.; Chen, N.C.; Jou, S.B. Resveratrol promotes mitochondrial biogenesis and protects against seizure-induced neuronal cell damage in the hippocampus following status epilepticus by activation of the PGC-1alpha signaling pathway. Int. J. Mol. Sci. 2019, 20, 998. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; St-Pierre, J. PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1alpha and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Lu, J.; Mori, T.; Smith-Powell, L.; Synold, T.W.; Chen, S.; Wen, W. Baicalin increases VEGF expression and angiogenesis by activating the ERR{alpha}/PGC-1{alpha} pathway. Cardiovasc. Res. 2011, 89, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Thom, R.; Rowe, G.C.; Jang, C.; Safdar, A.; Arany, Z. Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor gamma coactivator (PGC)-1alpha. J. Biol. Chem. 2014, 289, 8810–8817. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Quintans, N.; Prieto, I.; Sanchez-Ramos, C.; Luque, A.; Arza, E.; Olmos, Y.; Monsalve, M. Regulation of endothelial dynamics by PGC-1alpha relies on ROS control of VEGF-A signaling. Free Radic. Biol. Med. 2016, 93, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, U.; Kilic, E.; Jarve, A.; Guo, Z.; Spudich, A.; Bieber, K.; Barzena, U.; Bassetti, C.L.; Marti, H.H.; Hermann, D.M. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating ERK-1/2 and Akt pathways. J. Neurosci. 2006, 26, 12439–12446. [Google Scholar] [CrossRef] [PubMed]
- Ogaki, A.; Ikegaya, Y.; Koyama, R. Vascular abnormalities and the role of vascular endothelial growth factor in the epileptic brain. Front. Pharmacol. 2020, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Abid, M.R.; Guo, S.; Minami, T.; Spokes, K.C.; Ueki, K.; Skurk, C.; Walsh, K.; Aird, W.C. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, F.; Hu, B.; Cheng, J.W.; Sun, Y.F.; Zhou, K.Q.; Wang, P.X.; Guo, W.; Zhou, J.; Fan, J.; Chen, Z.; et al. Anlotinib suppresses tumor progression via blocking the VEGFR2/PI3K/AKT cascade in intrahepatic cholangiocarcinoma. Cell Death Dis. 2020, 11, 573. [Google Scholar] [CrossRef]
- Song, M.; Finley, S.D. Mechanistic insight into activation of MAPK signaling by pro-angiogenic factors. BMC Syst. Biol. 2018, 12, 145. [Google Scholar] [CrossRef] [Green Version]
- Irusta, G.; Abramovich, D.; Parborell, F.; Tesone, M. Direct survival role of vascular endothelial growth factor (VEGF) on rat ovarian follicular cells. Mol. Cell. Endocrinol. 2010, 325, 93–100. [Google Scholar] [CrossRef]
- Walker, M.C.; White, H.S.; Sander, J.W. Disease modification in partial epilepsy. Brain 2002, 125, 1937–1950. [Google Scholar] [CrossRef]
- Walker, M. Neuroprotection in epilepsy. Epilepsia 2007, 48 (Suppl. S8), 66–68. [Google Scholar] [CrossRef]
- Sutula, T.P.; Hagen, J.; Pitkanen, A. Do epileptic seizures damage the brain? Curr. Opin. Neurol. 2003, 16, 189–195. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Lin, T.K.; Huang, H.Y.; Chang, W.N.; Liou, C.W.; Chen, S.D.; Chang, A.Y.; Chan, S.H. Peroxisome proliferator-activated receptors gamma/mitochondrial uncoupling protein 2 signaling protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus. J. Neuroinflamm. 2012, 9, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, R.; Henshall, D.; Stoehr, S.; Meller, R. Endogenous mechanisms of neuroprotection. Epilepsia 2007, 48 (Suppl. S8), 72–73. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lui, C.C.; Lee, C.C.; Chen, S.D.; Chang, W.N.; Lu, C.H.; Chen, N.C.; Chang, A.Y.; Chan, S.H.; Chuang, Y.C. Clinical significance of serological biomarkers and neuropsychological performances in patients with temporal lobe epilepsy. BMC Neurol. 2012, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chen, S.D.; Lin, T.K.; Chang, W.N.; Liou, C.W.; Chang, A.Y.; Chan, S.H.; Chuang, Y.C. Heat shock protein 70 protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus via inhibition of nuclear factor-kappaB activation-induced nitric oxide synthase II expression. Neurobiol. Dis. 2014, 62, 241–249. [Google Scholar] [CrossRef]
- Yang, J.L.; Lin, Y.T.; Chuang, P.C.; Bohr, V.A.; Mattson, M.P. BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromol. Med. 2014, 16, 161–174. [Google Scholar] [CrossRef]
- Chen, S.D.; Zhen, Y.Y.; Lin, J.W.; Lin, T.K.; Huang, C.W.; Liou, C.W.; Chan, S.H.; Chuang, Y.C. Dynamin-related protein 1 promotes mitochondrial fission and contributes to the hippocampal neuronal cell death following experimental status epilepticus. CNS Neurosci. Ther. 2016, 22, 988–999. [Google Scholar] [CrossRef]
- Chen, S.D.; Lin, T.K.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Protective effects of peroxisome proliferator-activated receptors gamma coactivator-1alpha against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res. 2010, 88, 605–613. [Google Scholar] [CrossRef]
- Zhang, Q.; Lei, Y.H.; Zhou, J.P.; Hou, Y.Y.; Wan, Z.; Wang, H.L.; Meng, H. Role of PGC-1alpha in Mitochondrial Quality Control in Neurodegenerative Diseases. Neurochem. Res. 2019, 44, 2031–2043. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Sweeney, G.; Song, J. The association between PGC-1alpha and Alzheimer’s disease. Anat. Cell Biol. 2016, 49, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Tsunemi, T.; La Spada, A.R. PGC-1alpha at the intersection of bioenergetics regulation and neuron function: From Huntington’s disease to Parkinson’s disease and beyond. Progr. Neurobiol. 2012, 97, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ham, P.B., III; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Progr Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef] [PubMed]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef]
- Folbergrova, J.; Jesina, P.; Kubova, H.; Otahal, J. Effect of Resveratrol on oxidative stress and mitochondrial dysfunction in immature brain during epileptogenesis. Mol. Neurobiol. 2018, 55, 7512–7522. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xu, Q.; Zhang, L.; Kong, D.; Ma, R.; Wang, L. Protective effect of resveratrol against kainate-induced temporal lobe epilepsy in rats. Neurochem. Res. 2009, 34, 1393–1400. [Google Scholar] [CrossRef]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef]
- Mesquita-Britto, M.H.R.; Mendonca, M.C.P.; Soares, E.S.; Sakane, K.K.; da Cruz-Hofling, M.A. Inhibition of VEGF-Flk-1 binding induced profound biochemical alteration in the hippocampus of a rat model of BBB breakdown by spider venom. A preliminary assessment using FT-IR spectroscopy. Neurochem. Int. 2018, 120, 64–74. [Google Scholar] [CrossRef]
- Shimotake, J.; Derugin, N.; Wendland, M.; Vexler, Z.S.; Ferriero, D.M. Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke 2010, 41, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Sondell, M.; Lundborg, G.; Kanje, M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J. Neurosci. 1999, 19, 5731–5740. [Google Scholar] [CrossRef]
- Ruiz de Almodovar, C.; Lambrechts, D.; Mazzone, M.; Carmeliet, P. Role and therapeutic potential of VEGF in the nervous system. Physiol. Rev. 2009, 89, 607–648. [Google Scholar] [CrossRef]
- Ju, S.; Xu, C.; Wang, G.; Zhang, L. VEGF-C induces alternative activation of microglia to promote recovery from traumatic brain injury. J. Alzheimers Dis. 2019, 68, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chen, Y.C.; Huang, C.H.; Chen, C.C.; Hsu, Y.H.; Chen, H.M.; Chiu, F.L.; Kuo, H.C.; Chang, C.; Chern, Y. Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann. Neurol. 2015, 78, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.L.; Lin, K.J.; Wang, P.W.; Chuang, J.H.; Lin, H.Y.; Chen, S.D.; Chuang, Y.C.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy. Free Radic. Res. 2018, 52, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Claesson-Welsh, L. VEGF receptor signal transduction. Sci. STKE 2001, 2001, re21. [Google Scholar] [CrossRef]
- Hao, T.; Rockwell, P. Signaling through the vascular endothelial growth factor receptor VEGFR-2 protects hippocampal neurons from mitochondrial dysfunction and oxidative stress. Free Radic. Biol. Med. 2013, 63, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: Therapeutic perspectives. Pharmacol. Ther. 2014, 144, 202–225. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhang, Y.; Shi, Z.; Lu, D.; Li, T.; Ding, Y.; Ruan, Y.; Xu, A. The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci. Rep. 2016, 6, 26859. [Google Scholar] [CrossRef]
- Yang, J.L.; Chen, W.Y.; Chen, S.D. The emerging role of GLP-1 receptors in DNA repair: Implications in neurological disorders. Int. J. Mol. Sci. 2017, 18, 1861. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef]
- Sun, J.; Ren, D.D.; Wan, J.Y.; Chen, C.; Chen, D.; Yang, H.; Feng, C.L.; Gao, J. Desensitizing mitochondrial permeability transition by ERK-cyclophilin D axis contributes to the neuroprotective effect of gallic acid against cerebral ischemia/reperfusion injury. Front. Pharmacol. 2017, 8, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caglayan, B.; Caglayan, A.B.; Beker, M.C.; Yalcin, E.; Beker, M.; Kelestemur, T.; Sertel, E.; Ozturk, G.; Kilic, U.; Sahin, F.; et al. Evidence that activation of P2×7R does not exacerbate neuronal death after optic nerve transection and focal cerebral ischemia in mice. Exp. Neurol. 2017, 296, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alegria, K.; Flores-Leon, M.; Avila-Munoz, E.; Rodriguez-Corona, N.; Arias, C. pi3k signaling in neurons: A central node for the control of multiple functions. Int. J. Mol. Sci. 2018, 19, 3725. [Google Scholar] [CrossRef] [Green Version]
- Jha, S.K.; Jha, N.K.; Kar, R.; Ambasta, R.K.; Kumar, P. p38 MAPK and PI3K/AKT signalling cascades in Parkinson’s disease. Int. J. Mol. Cell. Med. 2015, 4, 67–86. [Google Scholar] [PubMed]
- Matsuda, S.; Ichimura, M.; Ogino, M.; Nakano, N.; Minami, A.; Murai, T.; Kitagishi, Y. Effective PI3K modulators for improved therapy against malignant tumors and for neuroprotection of brain damage after tumor therapy (Review). Int. J. Oncol. 2016, 49, 1785–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Wang, H.G. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 2001, 20, 7779–7786. [Google Scholar] [CrossRef] [Green Version]
- Korhonen, L.; Belluardo, N.; Mudo, G.; Lindholm, D. Increase in Bcl-2 phosphorylation and reduced levels of BH3-only Bcl-2 family proteins in kainic acid-mediated neuronal death in the rat brain. Eur. J. Neurosci. 2003, 18, 1121–1134. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.D.; Cruz, F. The ERK 1 and 2 pathway in the nervous system: From basic aspects to possible clinical applications in pain and visceral dysfunction. Curr. Neuropharmacol. 2007, 5, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target (Review). Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef] [Green Version]
- Chico, L.K.; Van Eldik, L.J.; Watterson, D.M. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discov. 2009, 8, 892–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Yuan, X.; Hu, Z.; Liu, S.; Li, H.; Wu, M.; Yuan, J.; Zhao, Z.; Su, J.; Wang, X.; et al. Valproic acid protects primary dopamine neurons from MPP(+)-induced neurotoxicity: Involvement of GSK3beta phosphorylation by Akt and ERK through the mitochondrial intrinsic Apoptotic pathway. Biomed. Res. Int. 2017, 2017, 8124501. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Chang, A.Y.; Lin, J.W.; Hsu, S.P.; Chan, S.H. Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia 2004, 45, 1202–1209. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Chen, S.D.; Lin, T.K.; Liou, C.W.; Chang, W.N.; Chan, S.H.; Chang, A.Y. Upregulation of nitric oxide synthase II contributes to apoptotic cell death in the hippocampal CA3 subfield via a cytochrome c/caspase-3 signaling cascade following induction of experimental temporal lobe status epilepticus in the rat. Neuropharmacology 2007, 52, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C.; Chen, S.D.; Liou, C.W.; Lin, T.K.; Chang, W.N.; Chan, S.H.; Chang, A.Y. Contribution of nitric oxide, superoxide anion, and peroxynitrite to activation of mitochondrial apoptotic signaling in hippocampal CA3 subfield following experimental temporal lobe status epilepticus. Epilepsia 2009, 50, 731–746. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.-B.; Hsu, S.-P.; Pan, H.-Y.; Chen, S.-D.; Chen, S.-F.; Lin, T.-K.; Liu, X.-P.; Li, J.-H.; Chen, N.-C.; Liou, C.-W.; et al. Peroxisome Proliferator-Activated Receptor γ Coactivator 1α Activates Vascular Endothelial Growth Factor That Protects Against Neuronal Cell Death Following Status Epilepticus through PI3K/AKT and MEK/ERK Signaling. Int. J. Mol. Sci. 2020, 21, 7247. https://doi.org/10.3390/ijms21197247
Huang J-B, Hsu S-P, Pan H-Y, Chen S-D, Chen S-F, Lin T-K, Liu X-P, Li J-H, Chen N-C, Liou C-W, et al. Peroxisome Proliferator-Activated Receptor γ Coactivator 1α Activates Vascular Endothelial Growth Factor That Protects Against Neuronal Cell Death Following Status Epilepticus through PI3K/AKT and MEK/ERK Signaling. International Journal of Molecular Sciences. 2020; 21(19):7247. https://doi.org/10.3390/ijms21197247
Chicago/Turabian StyleHuang, Jyun-Bin, Shih-Pin Hsu, Hsiu-Yung Pan, Shang-Der Chen, Shu-Fang Chen, Tsu-Kung Lin, Xuan-Ping Liu, Jie-Hau Li, Nai-Ching Chen, Chia-Wei Liou, and et al. 2020. "Peroxisome Proliferator-Activated Receptor γ Coactivator 1α Activates Vascular Endothelial Growth Factor That Protects Against Neuronal Cell Death Following Status Epilepticus through PI3K/AKT and MEK/ERK Signaling" International Journal of Molecular Sciences 21, no. 19: 7247. https://doi.org/10.3390/ijms21197247
APA StyleHuang, J. -B., Hsu, S. -P., Pan, H. -Y., Chen, S. -D., Chen, S. -F., Lin, T. -K., Liu, X. -P., Li, J. -H., Chen, N. -C., Liou, C. -W., Hsu, C. -Y., Chuang, H. -Y., & Chuang, Y. -C. (2020). Peroxisome Proliferator-Activated Receptor γ Coactivator 1α Activates Vascular Endothelial Growth Factor That Protects Against Neuronal Cell Death Following Status Epilepticus through PI3K/AKT and MEK/ERK Signaling. International Journal of Molecular Sciences, 21(19), 7247. https://doi.org/10.3390/ijms21197247