Neural Stem Cells and Cannabinoids in the Spotlight as Potential Therapy for Epilepsy

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Epilepsy and Epileptogenesis
3. Adult Neurogenesis and Neural Stem/Progenitor Cells
3.1. Neural Stem Cells in Epilepsy
3.2. Neural Stem Cell Therapies for Epilepsy
4. Cannabinoids and the Endocannabinoid System
4.1. The Endocannabinoid System in Physiological Conditions
4.2. Cannabinoid Pharmacology and Actions in Physiological Conditions
4.3. The Endocannabinoid System in Epilepsy
4.4. Cannabinoids and Neural Stem Cells
4.5. Current Cannabinoid Therapies for Epilepsy
5. Cannabinoids, Neural Stem Cells and Epilepsy: Perspectives and Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Magiorkinis, E.; Sidiropoulou, K.; Diamantis, A. Hallmarks in the history of epilepsy: Epilepsy in antiquity. Epilepsy Behav. 2010, 17, 103–108. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO|Atlas: Epilepsy Care in the World; Global Campaign against Epilepsy, International Bureau of Epilepsy, International League against Epilepsy, Eds.; WHO: Geneva, Switzerland, 2005; ISBN 978-92-4-156303-1. [Google Scholar]
- Perucca, P.; Gilliam, F.G. Adverse effects of antiepileptic drugs. Lancet Neurol. 2012, 11, 792–802. [Google Scholar] [CrossRef]
- Manford, M. Recent advances in epilepsy. J. Neurol. 2017, 264, 1811–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.M.; Kron, M.M. Neurogenesis and Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies; National Center for Biotechnology Information (US): Cary, NC, USA; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Zhong, Q.; Ren, B.-X.; Tang, F.-R. Neurogenesis in the Hippocampus of Patients with Temporal Lobe Epilepsy. Curr. Neurol. Neurosci. Rep. 2016, 16, 20. [Google Scholar] [CrossRef]
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2019, 24, 67–87. [Google Scholar] [CrossRef]
- Rosenberg, E.C.; Tsien, R.W.; Whalley, B.J.; Devinsky, O. Cannabinoids and Epilepsy. Neurotherapeutics 2015, 12, 747–768. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Cilio, M.R.; Cross, H.; Fernandez-Ruiz, J.; French, J.; Hill, C.; Katz, R.; Di Marzo, V.; Jutras-Aswad, D.; Notcutt, W.G.; et al. Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014, 55, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Gloss, D.; Vickrey, B. Cannabinoids for epilepsy. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Prenderville, J.A.; Kelly, Á.M.; Downer, E.J. The role of cannabinoids in adult neurogenesis: Cannabinoids and neurogenesis. Br. J. Pharmacol. 2015, 172, 3950–3963. [Google Scholar] [CrossRef] [Green Version]
- WHO. Epilepsy: A Public Health Imperative; WHO: Geneva, Switzerland, 2019; ISBN 978-92-4-151593-1. [Google Scholar]
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.-S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jetté, N. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology 2017, 88, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Abajobir, A.A.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abera, S.F.; Abyu, G.Y.; Ahmed, M.B.; Aichour, A.N.; Aichour, I.; et al. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J. Epileptic Seizures and Epilepsy: Definitions Proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.S.; Cross, J.H.; D’Souza, C.; French, J.A.; Haut, S.R.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia 2017, 58, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Pitkänen, A.; Lukasiuk, K.; Dudek, F.E.; Staley, K.J. Epileptogenesis. Cold Spring Harb. Perspect. Med. 2015, 5, a022822. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, E.M.; Coulter, D.A. Mechanisms of epileptogenesis: A convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Pitkänen, A.; Lukasiuk, K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011, 10, 173–186. [Google Scholar] [CrossRef]
- Klein, P.; Dingledine, R.; Aronica, E.; Bernard, C.; Blümcke, I.; Boison, D.; Brodie, M.J.; Brooks-Kayal, A.R.; Engel, J.; Forcelli, P.A.; et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 2018, 59, 37–66. [Google Scholar] [CrossRef]
- Rao, M.S.; Hattiangady, B.; Reddy, D.S.; Shetty, A.K. Hippocampal neurodegeneration, spontaneous seizures, and mossy fiber sprouting in the F344 rat model of temporal lobe epilepsy. J. Neurosci. Res. 2006, 83, 1088–1105. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Buckmaster, P.S. Reduced Inhibition of Dentate Granule Cells in a Model of Temporal Lobe Epilepsy. J. Neurosci. 2003, 23, 2440–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muro-García, T.; Martín-Suárez, S.; Espinosa, N.; Valcárcel-Martín, R.; Marinas, A.; Zaldumbide, L.; Galbarriatu, L.; Sierra, A.; Fuentealba, P.; Encinas, J.M. Reactive Disruption of the Hippocampal Neurogenic Niche After Induction of Seizures by Injection of Kainic Acid in the Amygdala. Front. Cell Dev. Biol. 2019, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; da Costa Araujo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J.A. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks-Kayal, A.R.; Shumate, M.D.; Jin, H.; Rikhter, T.Y.; Coulter, D.A. Selective changes in single cell GABA A receptor subunit expression and function in temporal lobe epilepsy. Nat. Med. 1998, 4, 1166–1172. [Google Scholar] [CrossRef]
- Montroull, L.E.; Danelon, V.; Cragnolini, A.B.; Mascó, D.H. Loss of TrkB Signaling Due to Status Epilepticus Induces a proBDNF-Dependent Cell Death. Front. Cell. Neurosci. 2019, 13, 4. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, M.; Avoli, M. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 2013, 37, 2887–2899. [Google Scholar] [CrossRef] [Green Version]
- Scharfman, H.E.; Myers, C.E. Hilar mossy cells of the dentate gyrus: A historical perspective. Front. Neural Circuits 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Reddy, D.S.; Kuruba, R. Experimental models of status epilepticus and neuronal injury for evaluation of therapeutic interventions. Int. J. Mol. Sci. 2013, 14, 18284–18318. [Google Scholar] [CrossRef]
- Swissa, E.; Serlin, Y.; Vazana, U.; Prager, O.; Friedman, A. Blood–brain barrier dysfunction in status epileptics: Mechanisms and role in epileptogenesis. Epilepsy Behav. 2019, 101, 106285. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef] [PubMed]
- Clossen, B.L.; Reddy, D.S. Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1519–1538. [Google Scholar] [CrossRef] [PubMed]
- Billakota, S.; Devinsky, O.; Kim, K.-W. Why we urgently need improved epilepsy therapies for adult patients. Neuropharmacology 2019, 18, 107855. [Google Scholar] [CrossRef] [PubMed]
- Kandratavicius, L.; Alves Balista, P.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, H.; Garcia-Cairasco, N.; Soares Bueno-Junior, L.; Pereira Leite, J. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef] [Green Version]
- Raol, Y.H.; Brooks-Kayal, A.R. Experimental Models of Seizures and Epilepsies. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2012; Volume 105, pp. 57–82. ISBN 978-0-12-394596-9. [Google Scholar]
- Löshcer, W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002, 50, 105–123. [Google Scholar]
- Sarkisian, M.R. Overview of the Current Animal Models for Human Seizure and Epileptic Disorders. Epilepsy Behav. 2001, 2, 201–216. [Google Scholar] [CrossRef]
- Avoli, M.; Jefferys, J.G.R. Models of drug-induced epileptiform synchronization in vitro. J. Neurosci. Methods 2016, 260, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef]
- Rodrigues, R.; Lourenço, D.; Paulo, S.; Mateus, J.; Ferreira, M.; Mouro, F.; Moreira, J.; Ribeiro, F.; Sebastião, A.; Xapelli, S. Cannabinoid Actions on Neural Stem Cells: Implications for Pathophysiology. Molecules 2019, 24, 1350. [Google Scholar] [CrossRef] [Green Version]
- Amaral, D.G.; Scharfman, H.E.; Lavenex, P. The dentate gyrus: Fundamental neuroanatomical organization (dentate gyrus for dummies). In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2007; Volume 163, pp. 3–790. ISBN 978-0-444-53015-8. [Google Scholar]
- Santos, V.R.; santana Melo, I.; Pacheco, A.L.D.; de Castro, O.W. Life and death in the hippocampus: What’s bad? Epilepsy Behav. 2019, 106595. [Google Scholar] [CrossRef]
- Song, J.; Zhong, C.; Bonaguidi, M.A.; Sun, G.J.; Hsu, D.; Gu, Y.; Meletis, K.; Huang, Z.J.; Ge, S.; Enikolopov, G.; et al. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 2012, 489, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Malva, J.O.; Bernardino, L.; Gray, W.; Overall, R.W. Different Mechanisms Must Be Considered to Explain the Increase in Hippocampal Neural Precursor Cell Proliferation by Physical Activity. Front. Neurosci. 2016, 1, 362. [Google Scholar] [CrossRef] [Green Version]
- Tozuka, Y.; Fukuda, S.; Namba, T.; Seki, T.; Hisatsune, T. GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron 2005, 47, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.; Goh, E.L.K.; Sailor, K.A.; Kitabatake, Y.; Ming, G.; Song, H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, H.G.; Biebl, M.; Wilhelm, D.; Li, M.; Friedlander, R.M.; Winkler, J. Increased generation of granule cells in adult Bcl-2-overexpressing mice: A role for cell death during continued hippocampal neurogenesis. Eur. J. Neurosci. 2005, 22, 1907–1915. [Google Scholar] [CrossRef]
- Biebl, M.; Cooper, C.M.; Winkler, J.; Kuhn, H.G. Analysis of neurogenesis and programmed cell death reveals a self- renewing capacity in the adult rat brain. Neurosci. Lett. 2000, 291, 17–20. [Google Scholar] [CrossRef]
- Wu, M.V.; Sahay, A.; Duman, R.S.; Hen, R. Functional Differentiation of Adult-Born Neurons along the Septotemporal Axis of the Dentate Gyrus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018978. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef]
- Bielefeld, P.; Durá, I.; Danielewicz, J.; Lucassen, P.J.; Baekelandt, V.; Abrous, D.N.; Encinas, J.M.; Fitzsimons, C.P. Insult-induced aberrant hippocampal neurogenesis: Functional consequences and possible therapeutic strategies. Behav. Brain Res. 2019, 372, 112032. [Google Scholar] [CrossRef]
- Jessberger, S.; Parent, J.M. Epilepsy and Adult Neurogenesis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020677. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, A.; Kann, G.; Flubacher, A.; Heinrich, C.; Freiman, T.M.; Zentner, J.; Frotscher, M.; Haas, C.A. Granule cell dispersion is not accompanied by enhanced neurogenesis in temporal lobe epilepsy patients. Exp. Neurol. 2007, 203, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Blümcke, I.; Schewe, J.-C.; Normann, S.; Brüstle, O.; Schramm, J.; Elger, C.E.; Wiestler, O.D. Increase of nestin-immunoreactive neural precursor cells in the dentate gyrus of pediatric patients with early-onset temporal lobe epilepsy: Increased Hippocampal Neurogenesis in Human TLE. Hippocampus 2001, 11, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bielefeld, P.; Schouten, M.; Meijer, G.M.; Breuk, M.J.; Geijtenbeek, K.; Karayel, S.; Tiaglik, A.; Vuuregge, A.H.; Willems, R.A.L.; Witkamp, D.; et al. Co-administration of Anti microRNA-124 and -137 Oligonucleotides Prevents Hippocampal Neural Stem Cell Loss Upon Non-convulsive Seizures. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra, A.; Martín-Suárez, S.; Valcárcel-Martín, R.; Pascual-Brazo, J.; Aelvoet, S.-A.; Abiega, O.; Deudero, J.J.; Brewster, A.L.; Bernales, I.; Anderson, A.E.; et al. Neuronal Hyperactivity Accelerates Depletion of Neural Stem Cells and Impairs Hippocampal Neurogenesis. Cell Stem Cell 2015, 16, 488–503. [Google Scholar] [CrossRef] [Green Version]
- Botterill, J.J.; Brymer, K.J.; Caruncho, H.J.; Kalynchuk, L.E. Aberrant hippocampal neurogenesis after limbic kindling: Relationship to BDNF and hippocampal-dependent memory. Epilepsy Behav. 2015, 47, 83–92. [Google Scholar] [CrossRef]
- Ribak, C.E.; Tran, P.H.; Spigelman, I.; Okazaki, M.M.; Victor Nadler, J. Status epilepticus-induced hilar basal dendrites on rodent granule cells contribute to recurrent excitatory circuitry. J. Comp. Neurol. 2000, 428, 240–253. [Google Scholar] [CrossRef]
- Shapiro, L.A.; Ribak, C.E. Newly born dentate granule neurons after pilocarpine-induced epilepsy have hilar basal dendrites with immature synapses. Epilepsy Res. 2006, 69, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Parent, J.M.; Yu, T.W.; Leibowitz, R.T.; Geschwind, D.H.; Sloviter, R.S.; Lowenstein, D.H. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. 1997, 17, 3727–3738. [Google Scholar] [CrossRef]
- Jessberger, S.; Römer, B.; Babu, H.; Kempermann, G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp. Neurol. 2005, 196, 342–351. [Google Scholar] [CrossRef]
- Gong, C.; Wang, T.-W.; Huang, H.S.; Parent, J.M. Reelin Regulates Neuronal Progenitor Migration in Intact and Epileptic Hippocampus. J. Neurosci. 2007, 27, 1803–1811. [Google Scholar] [CrossRef] [Green Version]
- Scharfman, H.E.; Sollas, A.E.; Berger, R.E.; Goodman, J.H.; Pierce, J.P. Perforant path activation of ectopic granule cells that are born after pilocarpine-induced seizures. Neuroscience 2003, 121, 1017–1029. [Google Scholar] [CrossRef]
- Scharfman, H.E.; Goodman, J.H.; Sollas, A.L. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: Functional implications of seizure-induced neurogenesis. J. Neurosci. 2000, 20, 6144–6158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessberger, S.; Nakashima, K.; Clemenson, G.D.; Mejia, E.; Mathews, E.; Ure, K.; Ogawa, S.; Sinton, C.M.; Gage, F.H.; Hsieh, J. Epigenetic Modulation of Seizure-Induced Neurogenesis and Cognitive Decline. J. Neurosci. 2007, 27, 5967–5975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, B.; Belbezier, A.; Haesebaert, J.; Rheims, S.; Ducray, F.; Picard, G.; Rogemond, V.; Psimaras, D.; Berzero, G.; Desestret, V.; et al. Long-term outcomes in temporal lobe epilepsy with glutamate decarboxylase antibodies. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Helmstaedter, C. Effects of chronic epilepsy on declarative memory systems. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2002; Volume 135, pp. 439–453. ISBN 978-0-444-50814-0. [Google Scholar]
- Jakubs, K.; Nanobashvili, A.; Bonde, S.; Ekdahl, C.T.; Kokaia, Z.; Kokaia, M.; Lindvall, O. Environment Matters: Synaptic Properties of Neurons Born in the Epileptic Adult Brain Develop to Reduce Excitability. Neuron 2006, 52, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Raedt, R.; Boon, P.; Persson, A.; Alborn, A.-M.; Boterberg, T.; Van Dycke, A.; Linder, B.; De Smedt, T.; Wadman, W.J.; Ben-Menachem, E.; et al. Radiation of the Rat Brain Suppresses Seizure-Induced Neurogenesis and Transiently Enhances Excitability during Kindling Acquisition. Epilepsia 2007, 48, 1952–1963. [Google Scholar] [CrossRef]
- Pekcec, A.; Lüpke, M.; Baumann, R.; Seifert, H.; Potschka, H. Modulation of neurogenesis by targeted hippocampal irradiation fails to affect kindling progression. Hippocampus 2011, 21, 866–876. [Google Scholar] [CrossRef]
- Jung, K.-H.; Chu, K.; Kim, M.; Jeong, S.-W.; Song, Y.-M.; Lee, S.-T.; Kim, J.-Y.; Lee, S.K.; Roh, J.-K. Continuous cytosine-b-D-arabinofuranoside infusion reduces ectopic granule cells in adult rat hippocampus with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Eur. J. Neurosci. 2004, 19, 3219–3226. [Google Scholar] [CrossRef]
- Jung, K.H.; Chu, K.; Lee, S.T.; Kim, J.; Sinn, D.I.; Kim, J.M.; Park, D.K.; Lee, J.J.; Kim, S.U.; Kim, M.; et al. Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol. Dis. 2006, 23, 237–246. [Google Scholar] [CrossRef]
- Cho, K.-O.; Lybrand, Z.R.; Ito, N.; Brulet, R.; Tafacory, F.; Zhang, L.; Good, L.; Ure, K.; Kernie, S.G.; Birnbaum, S.G.; et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun. 2015, 6, 6606. [Google Scholar] [CrossRef]
- Wang, R.; Tian, S.; Yang, X.; Liu, J.; Wang, Y.; Sun, K. Celecoxib-induced inhibition of neurogenesis in fetal frontal cortex is attenuated by curcumin via Wnt/β-catenin pathway. Life Sci. 2017, 185, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Juliandi, B.; Tanemura, K.; Igarashi, K.; Tominaga, T.; Furukawa, Y.; Otsuka, M.; Moriyama, N.; Ikegami, D.; Abematsu, M.; Sanosaka, T.; et al. Reduced Adult Hippocampal Neurogenesis and Cognitive Impairments following Prenatal Treatment of the Antiepileptic Drug Valproic Acid. Stem Cell Rep. 2015, 5, 996–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, S.K.; Seth, B.; Agarwal, S.; Yadav, A.; Karmakar, M.; Gupta, S.K.; Choubey, V.; Sharma, A.; Chaturvedi, R.K. Ethosuximide Induces Hippocampal Neurogenesis and Reverses Cognitive Deficits in an Amyloid-β Toxin-induced Alzheimer Rat Model via the Phosphatidylinositol 3-Kinase (PI3K)/Akt/Wnt/β-Catenin Pathway. J. Biol. Chem. 2015, 290, 28540–28558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikonomidou, C.; Scheer, I.; Wilhelm, T.; Juengling, F.D.; Titze, K.; Stöver, B.; Lehmkuhl, U.; Koch, S.; Kassubek, J. Brain morphology alterations in the basal ganglia and the hypothalamus following prenatal exposure to antiepileptic drugs. Eur. J. Paediatr. Neurol. 2007, 11, 297–301. [Google Scholar] [CrossRef]
- Lutes, J.M. Chapter 44-Developmental Neurotoxicology of Antiepileptic Drugs. In Handbook of Developmental Neurotoxicology, 2nd ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 499–508. [Google Scholar]
- Bromley, R. The treatment of epilepsy in pregnancy: The neurodevelopmental risks associated with exposure to antiepileptic drugs. Reprod. Toxicol. 2016, 64, 203–210. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Turski, L. Antiepileptic drugs and brain development. Epilepsy Res. 2010, 88, 11–22. [Google Scholar] [CrossRef]
- Hosford, B.E.; Rowley, S.; Liska, J.P.; Danzer, S.C. Ablation of peri-insult generated granule cells after epilepsy onset halts disease progression. Sci. Rep. 2017, 7, 18015. [Google Scholar] [CrossRef]
- Beamer, E.H.; Jurado-Arjona, J.; Jimenez-Mateos, E.M.; Morgan, J.; Reschke, C.R.; Kenny, A.; de Leo, G.; Olivos-Oré, L.A.; Arribas-Blázquez, M.; Madden, S.F.; et al. MicroRNA-22 Controls Aberrant Neurogenesis and Changes in Neuronal Morphology After Status Epilepticus. Front. Mol. Neurosci. 2018, 11, 442. [Google Scholar] [CrossRef]
- Jimenez-Mateos, E.M.; Arribas-Blazquez, M.; Sanz-Rodriguez, A.; Concannon, C.; Olivos-Ore, L.A.; Reschke, C.R.; Mooney, C.M.; Mooney, C.; Lugara, E.; Morgan, J.; et al. MicroRNA targeting of the P2X7 purinoceptor opposes a contralateral epileptogenic focus in the hippocampus. Sci. Rep. 2015, 5, 17486. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Mateos, E.M.; Engel, T.; Merino-Serrais, P.; McKiernan, R.C.; Tanaka, K.; Mouri, G.; Sano, T.; O’Tuathaigh, C.; Waddington, J.L.; Prenter, S.; et al. Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat. Med. 2012, 18, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Guo, M.; Meng, D.; Sun, F.; Guan, L.; Cui, Y.; Zhao, Y.; Wang, X.; Gu, X.; Sun, J.; et al. Silencing MicroRNA-134 Alleviates Hippocampal Damage and Occurrence of Spontaneous Seizures After Intraventricular Kainic Acid-Induced Status Epilepticus in Rats. Front. Cell. Neurosci. 2019, 13, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Mateos, E.M.; Engel, T.; Merino-Serrais, P.; Fernaud-Espinosa, I.; Rodriguez-Alvarez, N.; Reynolds, J.; Reschke, C.R.; Conroy, R.M.; McKiernan, R.C.; de Felipe, J.; et al. Antagomirs targeting microRNA-134 increase hippocampal pyramidal neuron spine volume in vivo and protect against pilocarpine-induced status epilepticus. Brain Struct. Funct. 2015, 220, 2387–2399. [Google Scholar] [CrossRef] [PubMed]
- Vangoor, V.R.; Reschke, C.R.; Senthilkumar, K.; Van De Haar, L.L.; De Wit, M.; Giuliani, G.; Broekhoven, M.H.; Morris, G.; Engel, T.; Brennan, G.P.; et al. Antagonizing increased miR-135a levels at the chronic stage of experimental TLE reduces spontaneous recurrent seizures. J. Neurosci. 2019, 39, 5064–5079. [Google Scholar] [CrossRef] [Green Version]
- Pons-Espinal, M.; Gasperini, C.; Marzi, M.J.; Braccia, C.; Armirotti, A.; Pötzsch, A.; Walker, T.L.; Fabel, K.; Nicassio, F.; Kempermann, G.; et al. MiR-135a-5p Is Critical for Exercise-Induced Adult Neurogenesis. Stem Cell Rep. 2019, 12, 1298–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA Approves First-Of-Its Kind Targeted RNA-Based Therapy to Treat a Rare Disease, FDA, (n.d.). Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-its-kind-targeted-rna-based-therapy-treat-rare-disease (accessed on 24 November 2019).
- Mesraoua, B.; Deleu, D.; Kullmann, D.M.; Shetty, A.K.; Boon, P.; Perucca, E.; Mikati, M.A.; Asadi-Pooya, A.A. Novel therapies for epilepsy in the pipeline. Epilepsy Behav. 2019, 97, 282–290. [Google Scholar] [CrossRef]
- Lybrand, Z.R.; Goswami, S.; Hsieh, J. Stem cells: A path towards improved epilepsy therapies. Neuropharmacology 2019, 107781. [Google Scholar] [CrossRef]
- Cunningham, M.; Cho, J.H.; Leung, A.; Savvidis, G.; Ahn, S.; Moon, M.; Lee, P.K.J.; Han, J.J.; Azimi, N.; Kim, K.S.; et al. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell 2014, 15, 559–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhya, D.; Hattiangady, B.; Castro, O.W.; Shuai, B.; Kodali, M.; Attaluri, S.; Bates, A.; Dong, Y.; Zhang, S.-C.; Prockop, D.J.; et al. Human induced pluripotent stem cell-derived MGE cell grafting after status epilepticus attenuates chronic epilepsy and comorbidities via synaptic integration. Proc. Natl. Acad. Sci. USA 2019, 116, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Takahashi, J. Preparing for first human trial of induced pluripotent stem cell-derived cells for Parkinson’s disease: An interview with Jun Takahashi. Regen. Med. 2019, 14, 93–95. [Google Scholar] [CrossRef]
- Upadhya, D.; Shetty, A.K. Promise of extracellular vesicles for diagnosis and treatment of epilepsy. Epilepsy Behav. 2019, 106499. [Google Scholar] [CrossRef] [PubMed]
- Karttunen, J.; Heiskanen, M.; Lipponen, A.; Poulsen, D.; Pitkänen, A. Extracellular Vesicles as Diagnostics and Therapeutics for Structural Epilepsies. Int. J. Mol. Sci. 2019, 20, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Q.; Upadhya, D.; Hattiangady, B.; Kim, D.-K.; Yeon An, S.; Shuai, B.; Prockop, D.J.; Shetty, A.K. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc. Natl. Acad. Sci. USA 2017, 114, E3536–E3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPartland, J.M. Cannabis Systematics at the Levels of Family, Genus, and Species. Cannabis Cannabinoid Res. 2018, 3, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solymosi, K.; Kofalvi, A. Cannabis: A Treasure Trove or Pandora’s Box? Mini Rev. Med. Chem. 2017, 17. [Google Scholar] [CrossRef]
- United Nations Office on Drugs and Crime. World Drug Report 2019 Booklet 5: “Cannabis and Hallucinogens”; S.l., United Nations Publication; United Nations: Vienna, Austria, 2019; ISBN 978-92-1-148314-7. [Google Scholar]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. In Phytocannabinoids; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 103, pp. 103–131. ISBN 978-3-319-45539-6. [Google Scholar]
- Adams, I.B.; Martin, B.R. Cannabis: Pharmacology and toxicology in animals and humans. Addiction 1996, 91, 1585–1614. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Russo, E.; Guy, G.W. A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypotheses 2006, 66, 234–246. [Google Scholar] [CrossRef]
- Fitzpatrick, J.-M.K.; Downer, E.J. Toll-like receptor signalling as a cannabinoid target in Multiple Sclerosis. Neuropharmacology 2017, 113, 618–626. [Google Scholar] [CrossRef]
- Rice, J.; Cameron, M. Cannabinoids for Treatment of MS Symptoms: State of the Evidence. Curr. Neurol. Neurosci. Rep. 2018, 18. [Google Scholar] [CrossRef]
- Maa, E.; Figi, P. The case for medical marijuana in epilepsy. Epilepsia 2014, 55, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Fagan, S.G.; Campbell, V.A. The influence of cannabinoids on generic traits of neurodegeneration. Br. J. Pharmacol. 2014, 171, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, Í.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.I.; Canela, E.I.; Saura, J.; Lanciego, J.L.; et al. Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. Role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levodopa-induced dyskinesia. BrainBehav. Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Carter, G.T.; Javaher, S.P.; Nguyen, M.H.; Garret, S.; Carlini, B.H. Re-branding cannabis: The next generation of chronic pain medicine? Pain Manag. 2015, 5, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamplona, F.A.; da Silva, L.R.; Coan, A.C. Potential Clinical Benefits of CBD-Rich Cannabis Extracts Over Purified CBD in Treatment-Resistant Epilepsy: Observational Data Meta-analysis. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, D.; Sánchez-Robles, E.M.; García, M.M.; Goicoechea, C. Chronic pain and cannabinoids. Great expectations or a christmas carol. Biochem. Pharmacol. 2018, 157, 33–42. [Google Scholar] [CrossRef]
- Venderová, K.; Růžička, E.; Voříšek, V.; Višňovský, P. Survey on cannabis use in Parkinson’s disease: Subjective improvement of motor symptoms. Mov. Disord. 2004, 19, 1102–1106. [Google Scholar] [CrossRef]
- Woodward, M.R.; Harper, D.G.; Stolyar, A.; Forester, B.P.; Ellison, J.M. Dronabinol for the Treatment of Agitation and Aggressive Behavior in Acutely Hospitalized Severely Demented Patients with Noncognitive Behavioral Symptoms. Am. J. Geriatr. Psychiatry 2014, 22, 415–419. [Google Scholar] [CrossRef]
- Gado, F.; Digiacomo, M.; Macchia, M.; Bertini, S.; Manera, C. Traditional Uses of Cannabinoids and New Perspectives in the Treatment of Multiple Sclerosis. Medicines 2018, 5, 91. [Google Scholar] [CrossRef] [Green Version]
- Bricker, J.B.; Russo, J.; Stein, M.B.; Sherbourne, C.; Craske, M.; Schraufnagel, T.J.; Roy-Byrne, P. Does occasional cannabis use impact anxiety and depression treatment outcomes?: Results from a randomized effectiveness trial. Depress. Anxiety 2007, 24, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.S.; Kamil, S.; Shah, M.R.; Bhimanadham, N.N.; Imran, S. Pros and Cons of Marijuana in Treatment of Parkinson’s Disease. Cureus 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, D.; Lippmann, S. Marijuana for Parkinson’s Disease? Innov. Clin. Neurosci. 2019, 16, 33–34. [Google Scholar] [PubMed]
- Kaur, R.; Ambwani, S.; Singh, S. Endocannabinoid System: A Multi-Facet Therapeutic Target. Curr. Clin. Pharmacol. 2016, 11, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petro, D.J.; Ellenberger, C. Treatment of Human Spasticity with Δ9-Tetrahydrocannabinol. J. Clin. Pharmacol. 1981, 21, 413S–416S. [Google Scholar] [CrossRef]
- Verty, A.N.; Evetts, M.J.; Crouch, G.J.; McGregor, I.S.; Stefanidis, A.; Oldfield, B.J. The Cannabinoid Receptor Agonist THC Attenuates Weight Loss in a Rodent Model of Activity-Based Anorexia. Neuropsychopharmacology 2011, 36, 1349–1358. [Google Scholar] [CrossRef] [Green Version]
- Ekert, H.; Waters, K.D.; Jurk, I.H.; Mobilia, J.; Loughnan, P. Amelioration Of Cancer Chemotherapy-Induced Nausea and Vomiting by Delta-9-Tetrahydro-Cannabinol. Med. J. Aust. 1979, 2, 657–659. [Google Scholar] [CrossRef]
- Haney, M.; Gunderson, E.W.; Rabkin, J.; Hart, C.L.; Vosburg, S.K.; Comer, S.D.; Foltin, R.W. Dronabinol and Marijuana in HIV-Positive Marijuana Smokers: Caloric Intake, Mood, and Sleep. J. Acquir. Immune Defic. Syndr. 2007, 45, 545–554. [Google Scholar] [CrossRef] [Green Version]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. In Phytocannabinoids; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 103, pp. 1–36. ISBN 978-3-319-45539-6. [Google Scholar]
- Abush, H.; Akirav, I. Short- and long-term cognitive effects of chronic cannabinoids administration in late-adolescence rats. PLoS ONE 2012, 7, e31731. [Google Scholar] [CrossRef]
- Borgelt, L.M.; Franson, K.L.; Nussbaum, A.M.; Wang, G.S. The Pharmacologic and Clinical Effects of Medical Cannabis. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2013, 33, 195–209. [Google Scholar] [CrossRef]
- Andréasson, S.; Engström, A.; Allebeck, P.; Rydberg, U. Cannabis and Schizophrenia: A Longitudinal Study of Swedish Conscripts. Lancet 1987, 330, 1483–1486. [Google Scholar] [CrossRef]
- Hall, W.; Degenhardt, L. Adverse health effects of non-medical cannabis use. Lancet 2009, 374, 9. [Google Scholar] [CrossRef]
- Khan, M.A.; Akella, S. Cannabis-Induced Bipolar Disorder with Psychotic Features: A Case Report. Psychiatry Edgmont 2009, 6, 44–48. [Google Scholar] [PubMed]
- Lichtman, A.H.; Martin, B.R. Cannabinoid Tolerance and Dependence. In Cannabinoids; Pertwee, R.G., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 168, pp. 691–717. ISBN 978-3-540-22565-2. [Google Scholar]
- Salzet, M.; Stefano, G.B. The endocannabinoid system in invertebrates. Prostaglandins Leukot. Essent. Fat. Acids Plefa 2002, 66, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Elphick, M.R.; Egertova, M. The neurobiology and evolution of cannabinoid signalling. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2001, 356, 381–408. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M. Missing Pieces to the Endocannabinoid Puzzle. Trends Mol. Med. 2020, 26, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J. Transport of endocannabinoids across the plasma membrane and within the cell. Febs J. 2013, 280, 1895–1904. [Google Scholar] [CrossRef]
- Maccarrone, M. The Endocannabinoid System and its Manifold Central Actions. In Handbook of Neurochemistry and Molecular Neurobiology: Neural Lipids; Lajtha, A., Tettamanti, G., Goracci, G., Eds.; Springer: Boston, MA, USA, 2009; pp. 385–405. ISBN 978-0-387-30378-9. [Google Scholar]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Hillard, C.J. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000, 61, 3–18. [Google Scholar] [CrossRef]
- Sugiura, T.; Kishimoto, S.; Oka, S.; Gokoh, M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog. Lipid Res. 2006, 45, 405–446. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-Mediated Control of Synaptic Transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [Green Version]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid Signaling and Synaptic Function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storozhuk, M.V.; Zholos, A.V. TRP Channels as Novel Targets for Endogenous Ligands: Focus on Endocannabinoids and Nociceptive Signalling. Curr. Neuropharmacol. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Marzo, V.D.; Bifulco, M.; Petrocellis, L.D. The endocannabinoid system and its therapeutic exploitation. Nat. Rev. Drug Discov. 2004, 3, 771–784. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabinoids and the gastrointestinal tract. Gut 2001, 48, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Dey, S.K.; Maccarrone, M. Jekyll and Hyde: Two Faces of Cannabinoid Signaling in Male and Female Fertility. Endocr. Rev. 2006, 27, 427–448. [Google Scholar] [CrossRef] [Green Version]
- Montecucco, F.; Di Marzo, V. At the heart of the matter: The endocannabinoid system in cardiovascular function and dysfunction. Trends Pharmacol. Sci. 2012, 33, 331–340. [Google Scholar] [CrossRef]
- Silvestri, C.; Di Marzo, V. The Endocannabinoid System in Energy Homeostasis and the Etiopathology of Metabolic Disorders. Cell Metab. 2013, 17, 475–490. [Google Scholar] [CrossRef] [Green Version]
- Henry, R.J.; Kerr, D.M.; Finn, D.P.; Roche, M. For whom the endocannabinoid tolls: Modulation of innate immune function and implications for psychiatric disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, T.; Karthaus, N.; Kim, B.-S.; Beier, J.P. The endocannabinoid receptors CB1 and CB2 affect the regenerative potential of adipose tissue MSCs. Exp. Cell Res. 2020, 389, 111881. [Google Scholar] [CrossRef] [PubMed]
- Mallipeddi, S.; Janero, D.R.; Zvonok, N.; Makriyannis, A. Functional selectivity at G-protein coupled receptors: Advancing cannabinoid receptors as drug targets. Biochem. Pharmacol. 2017, 128, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles Through Complex Pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, A.C.; Abood, M.E. CB 1 and CB 2 Receptor Pharmacology. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 80, pp. 169–206. ISBN 978-0-12-811232-8. [Google Scholar]
- Howlett, A.C.; Breivogel, C.S.; Childers, S.R.; Deadwyler, S.A.; Hampson, R.E.; Porrino, L.J. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology 2004, 47, 345–358. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Regehr, W.G. Cerebellar Depolarization-Induced Suppression of Inhibition Is Mediated by Endogenous Cannabinoids. J. Neurosci. 2001, 21, RC174. [Google Scholar] [CrossRef]
- Vendel, E.; de Lange, E.C.M. Functions of the CB1 and CB2 Receptors in Neuroprotection at the Level of the Blood–Brain Barrier. Neuromol. Med. 2014, 16, 620–642. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef]
- Cassano, T.; Calcagnini, S.; Pace, L.; De Marco, F.; Romano, A.; Gaetani, S. Cannabinoid Receptor 2 Signaling in Neurodegenerative Disorders: From Pathogenesis to a Promising Therapeutic Target. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; Stella, N.; Zimmer, A. Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci. 2015, 16, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Onaivi, E.S.; Ishiguro, H.; Gu, S.; Liu, Q.-R. CNS effects of CB2 cannabinoid receptors: Beyond neuro-immuno-cannabinoid activity. J. Psychopharmacol. 2012, 26, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Ruiz, J.; Moro, M.A.; Martínez-Orgado, J. Cannabinoids in Neurodegenerative Disorders and Stroke/Brain Trauma: From Preclinical Models to Clinical Applications. Neurotherapeutics 2015, 12, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fride, E. Endocannabinoids in the central nervous system–an overview. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, C.J.; Xi, Z.-X. Progress in brain cannabinoid CB2 receptor research: From genes to behavior. Neurosci. Biobehav. Rev. 2019, 98, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147, S163–S171. [Google Scholar] [CrossRef] [Green Version]
- Petitet, F.; Jeantaud, B.; Reibaud, M.; Imperato, A.; Dubroeucq, M.-C. Complex pharmacology of natural cannabinoids: Evidence for partial agonist activity of Δ9-tetrahydrocannabinol and antagonist activity of cannabidiol on rat brain cannabinoid receptors. Life Sci. 1998, 63, PL1–PL6. [Google Scholar] [CrossRef]
- Strougo, A.; Zuurman, L.; Roy, C.; Pinquier, J.; van Gerven, J.; Cohen, A.; Schoemaker, R. Modelling of the concentration—Effect relationship of THC on central nervous system parameters and heart rate—Insight into its mechanisms of action and a tool for clinical research and development of cannabinoids. J. Psychopharmacol. 2008, 22, 717–726. [Google Scholar] [CrossRef]
- Huestis, M.A.; Boyd, S.J.; Heishman, S.J.; Preston, K.L.; Bonnet, D.; Le Fur, G.; Gorelick, D.A. Single and multiple doses of rimonabant antagonize acute effects of smoked cannabis in male cannabis users. Psychopharmacology 2007, 194, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Qin, N.; Neeper, M.P.; Liu, Y.; Hutchinson, T.L.; Lubin, M.L.; Flores, C.M. TRPV2 Is Activated by Cannabidiol and Mediates CGRP Release in Cultured Rat Dorsal Root Ganglion Neurons. J. Neurosci. 2008, 28, 6231–6238. [Google Scholar] [CrossRef] [Green Version]
- Morales, P.; Reggio, P.H. CBD: A New Hope? ACS Med. Chem. Lett. 2019, 10, 694–695. [Google Scholar] [CrossRef] [Green Version]
- Maccarrone, M.; Maldonado, R.; Casas, M.; Henze, T.; Centonze, D. Cannabinoids therapeutic use: What is our current understanding following the introduction of THC, THC:CBD oromucosal spray and others? Expert Rev. Clin. Pharmacol. 2017, 10, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Rosenthaler, S.; Pöhn, B.; Kolmanz, C.; Nguyen Huu, C.; Krewenka, C.; Huber, A.; Kranner, B.; Rausch, W.D.; Moldzio, R. Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicol. Teratol. 2014, 46, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanuš, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Ibeas Bih, C.; Chen, T.; Nunn, A.V.W.; Bazelot, M.; Dallas, M.; Whalley, B.J. Molecular Targets of Cannabidiol in Neurological Disorders. Neurotherapeutics 2015, 12, 699–730. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro: Cannabinoid antagonism by cannabidiol. Br. J. Pharmacol. 2009, 150, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shabat, S.; Fride, E.; Sheskin, T.; Tamiri, T.; Rhee, M.-H.; Vogel, Z.; Bisogno, T.; De Petrocellis, L.; Di Marzo, V.; Mechoulam, R. An entourage effect: Inactive endogenous fatty acid glycerol esters enhance 2-arachidonoyl-glycerol cannabinoid activity. Eur. J. Pharmacol. 1998, 353, 23–31. [Google Scholar] [CrossRef]
- Cogan, P.S. The ‘entourage effect’ or ‘hodge-podge hashish’: The questionable rebranding, marketing, and expectations of cannabis polypharmacy. Expert Rev. Clin. Pharmacol. 2020, 13, 835–845. [Google Scholar] [CrossRef]
- Lerner, R.; Post, J.; Loch, S.; Lutz, B.; Bindila, L. Targeting brain and peripheral plasticity of the lipidome in acute kainic acid-induced epileptic seizures in mice via quantitative mass spectrometry. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 255–267. [Google Scholar] [CrossRef]
- Shubina, L.; Aliev, R.; Kitchigina, V. Attenuation of kainic acid-induced status epilepticus by inhibition of endocannabinoid transport and degradation in guinea pigs. Epilepsy Res. 2015, 111, 33–44. [Google Scholar] [CrossRef]
- Suleymanova, E.M.; Borisova, M.A.; Vinogradova, L.V. Early endocannabinoid system activation attenuates behavioral impairments induced by initial impact but does not prevent epileptogenesis in lithium–pilocarpine status epilepticus model. Epilepsy Behav. 2019, 92, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, Y.; Yamazaki, M.; Uchigashima, M.; Kobayashi, K.; Watanabe, M.; Sakimura, K.; Kano, M. Crucial Roles of the Endocannabinoid 2-Arachidonoylglycerol in the Suppression of Epileptic Seizures. Cell Rep. 2016, 16, 1405–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shubina, L.; Aliev, R.; Kitchigina, V. Endocannabinoid-dependent protection against kainic acid-induced long-term alteration of brain oscillations in guinea pigs. Brain Res. 2017, 1661, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutiérrez, S.O.; van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Rüden, E.L.; Jafari, M.; Bogdanovic, R.M.; Wotjak, C.T.; Potschka, H. Analysis in conditional cannabinoid 1 receptor-knockout mice reveals neuronal subpopulation-specific effects on epileptogenesis in the kindling paradigm. Neurobiol. Dis. 2015, 73, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Guggenhuber, S.; Monory, K.; Lutz, B.; Klugmann, M. AAV Vector-Mediated Overexpression of CB1 Cannabinoid Receptor in Pyramidal Neurons of the Hippocampus Protects against Seizure-Induced Excitoxicity. PLoS ONE 2010, 5, e15707. [Google Scholar] [CrossRef]
- Ludányi, A.; Eross, L.; Czirják, S.; Vajda, J.; Halász, P.; Watanabe, M.; Palkovits, M.; Maglóczky, Z.; Freund, T.F.; Katona, I. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci. 2008, 28, 2976–2990. [Google Scholar] [CrossRef] [Green Version]
- Racine, R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Phelan, K.D.; Shwe, U.T.; Williams, D.K.; Greenfield, L.J.; Zheng, F. Pilocarpine-induced status epilepticus in mice: A comparison of spectral analysis of electroencephalogram and behavioral grading using the Racine scale. Epilepsy Res. 2015, 117, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Maglóczky, Z.; Tóth, K.; Karlócai, R.; Nagy, S.; Erőss, L.; Czirják, S.; Vajda, J.; Rásonyi, G.; Kelemen, A.; Juhos, V.; et al. Dynamic changes of CB1-receptor expression in hippocampi of epileptic mice and humans: CB1-R Expressing GABAergic Fibers in TLE. Epilepsia 2010, 51, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Karlócai, M.R.; Tóth, K.; Watanabe, M.; Ledent, C.; Juhász, G.; Freund, T.F.; Maglóczky, Z. Redistribution of CB1 Cannabinoid Receptors in the Acute and Chronic Phases of Pilocarpine-Induced Epilepsy. PLoS ONE 2011, 6, e27196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffin, K.; Van Paesschen, W.; Van Laere, K. In vivo activation of endocannabinoid system in temporal lobe epilepsy with hippocampal sclerosis. Brain 2011, 134, 1033–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Ratzliff, A.; Hilgenberg, L.; Gulyás, A.; Freund, T.F.; Smith, M.; Dinh, T.P.; Piomelli, D.; Mackie, K.; Soltesz, I. Long-Term Plasticity of Endocannabinoid Signaling Induced by Developmental Febrile Seizures. Neuron 2003, 39, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Neu, A.; Howard, A.L.; Foldy, C.; Echegoyen, J.; Hilgenberg, L.; Smith, M.; Mackie, K.; Soltesz, I. Prevention of Plasticity of Endocannabinoid Signaling Inhibits Persistent Limbic Hyperexcitability Caused by Developmental Seizures. J. Neurosci. 2007, 27, 46–58. [Google Scholar] [CrossRef] [Green Version]
- McKallip, R.J.; Lombard, C.; Martin, B.R.; Nagarkatti, M.; Nagarkatti, P.S. Δ9-Tetrahydrocannabinol-Induced Apoptosis in the Thymus and Spleen as a Mechanism of Immunosuppression In Vitro and In Vivo. J. Pharmacol. Exp. Ther. 2002, 302, 451–465. [Google Scholar] [CrossRef]
- McKallip, R.J.; Lombard, C.; Fisher, M.; Martin, B.R.; Ryu, S.; Grant, S.; Nagarkatti, P.S.; Nagarkatti, M. Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease. Blood 2002, 100, 627–634. [Google Scholar] [CrossRef]
- Godhwani, N.; Bahna, S.L. Immune Dysregulation in Epilepsy. In Neuroinflammation; Elsevier: Amsterdam, The Netherlands, 2018; pp. 217–231. ISBN 978-0-12-811709-5. [Google Scholar]
- Filloux, F.M. Cannabinoids for pediatric epilepsy? Up in smoke or real science? Transl. Pediatrics 2015, 4, 12. [Google Scholar]
- Shapiro, L.; Wong, J.C.; Escayg, A. Reduced cannabinoid 2 receptor activity increases susceptibility to induced seizures in mice. Epilepsia 2019. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, H. The spatiotemporal expression changes of CB2R in the hippocampus of rats following pilocarpine-induced status epilepticus. Epilepsy Res. 2018, 148, 8–16. [Google Scholar] [CrossRef]
- Romigi, A.; Bari, M.; Placidi, F.; Marciani, M.G.; Malaponti, M.; Torelli, F.; Izzi, F.; Prosperetti, C.; Zannino, S.; Corte, F.; et al. Cerebrospinal fluid levels of the endocannabinoid anandamide are reduced in patients with untreated newly diagnosed temporal lobe epilepsy. Epilepsia 2010, 51, 768–772. [Google Scholar] [CrossRef]
- Bhaskaran, M.D.; Smith, B.N. Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Exp. Neurol. 2010, 223, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffarzadeh, F.; Eslamizade, M.J.; Mousavi, S.M.M.; Abraki, S.B.; Hadjighassem, M.R.; Gorji, A. TRPV1 receptors augment basal synaptic transmission in CA1 and CA3 pyramidal neurons in epilepsy. Neuroscience 2016, 314, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Carletti, F.; Gambino, G.; Rizzo, V.; Ferraro, G.; Sardo, P. Involvement of TRPV1 channels in the activity of the cannabinoid WIN 55,212-2 in an acute rat model of temporal lobe epilepsy. Epilepsy Res. 2016, 122, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.-J.; Guo, W.; Zheng, D.-H.; Zhang, C.-Q.; Li, S.; Liu, S.-Y.; Yin, Q.; Yang, H.; Shu, H.-F. Increased Expression of TRPV1 in the Cortex and Hippocampus from Patients with Mesial Temporal Lobe Epilepsy. J. Mol. Neurosci. 2013, 49, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; de Petrocellis, L.; Pryce, G.; Baker, D.; Guglielmotti, V.; Di Marzo, V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 2006, 139, 1405–1415. [Google Scholar] [CrossRef]
- Xapelli, S.; Agasse, F.; Sardà-Arroyo, L.; Bernardino, L.; Santos, T.; Ribeiro, F.F.; Valero, J.; Bragança, J.; Schitine, C.; de Melo Reis, R.A.; et al. Activation of Type 1 Cannabinoid Receptor (CB1R) Promotes Neurogenesis in Murine Subventricular Zone Cell Cultures. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Compagnucci, C.; Di Siena, S.; Bustamante, M.B.; Di Giacomo, D.; Di Tommaso, M.; Maccarrone, M.; Grimaldi, P.; Sette, C. Type-1 (CB1) Cannabinoid Receptor Promotes Neuronal Differentiation and Maturation of Neural Stem Cells. PLoS ONE 2013, 8, e54271. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, T.; Maroso, M.; Beer, A.; Baddenhausen, S.; Ludewig, S.; Fan, W.; Vennin, C.; Loch, S.; Berninger, B.; Hofmann, C.; et al. Neural stem cell lineage-specific cannabinoid type-1 receptor regulates neurogenesis and plasticity in the adult mouse hippocampus. Cereb. Cortex 2018, 28, 4454–4471. [Google Scholar] [CrossRef]
- Palazuelos, J.; Aguado, T.; Egia, A.; Mechoulam, R.; Guzmán, M.; Galve-Roperh, I. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J. 2006, 20, 2405–2407. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, M.B.; Suetterlin, P.; Yip, P.; Molina-Holgado, F.; Walker, D.J.; Oudin, M.J.; Zentar, M.P.; Pollard, S.; Yáñez-Muñoz, R.J.; Williams, G.; et al. A diacylglycerol lipase-CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age-dependent manner. Mol. Cell. Neurosci. 2008, 38, 526–536. [Google Scholar] [CrossRef]
- Aguado, T.; Monory, K.; Palazuelos, J.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system drives neural progenitor proliferation. FASEB J. 2005, 19, 1704–1706. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T. The Endocannabinoid System Promotes Astroglial Differentiation by Acting on Neural Progenitor Cells. J. Neurosci. 2006, 26, 1551–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galve-Roperh, I.; Chiurchiù, V.; Díaz-Alonso, J.; Bari, M.; Guzmán, M.; Maccarrone, M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog. Lipid Res. 2013, 52, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Falenta, K.; Lalli, G. Endocannabinoid signalling in neuronal migration. Int. J. Biochem. Cell Biol. 2014, 47, 104–108. [Google Scholar] [CrossRef]
- Duff, G.; Argaw, A.; Cecyre, B.; Cherif, H.; Tea, N.; Zabouri, N.; Casanova, C.; Ptito, M.; Bouchard, J.-F. Cannabinoid Receptor CB2 Modulates Axon Guidance. PLoS ONE 2013, 8, e70849. [Google Scholar] [CrossRef] [Green Version]
- Gómez, M.; Hernández, M.; Fernández-Ruiz, J. Cannabinoid signaling system: Does it play a function in cell proliferation and migration, neuritic elongation and guidance and synaptogenesis during brain ontogenesis? Cell Adhes. Migr. 2008, 2, 246–248. [Google Scholar] [CrossRef] [Green Version]
- Harkany, T.; Mackie, K.; Doherty, P. Wiring and firing neuronal networks: Endocannabinoids take center stage. Curr. Opin. Neurobiol. 2008, 18, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Campos, A.C.; Paraíso-Luna, J.; Fogaça, M.V.; Guimarães, F.S.; Galve-Roperh, I. Cannabinoids as Regulators of Neural Development and Adult Neurogenesis. In Lipidomics of Stem Cells; Pébay, A., Wong, R.C.B., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 117–136. ISBN 978-3-319-49342-8. [Google Scholar]
- Begbie, J.; Doherty, P.; Graham, A. Cannabinoid receptor, CB1, expression follows neuronal differentiation in the early chick embryo. J. Anat. 2004, 205, 213–218. [Google Scholar] [CrossRef]
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabas, K.; Ballester Rosado, C.J.; Nguyen, L.; Monory, K.; Marsicano, G.; Di Marzo, V.; Hurd, Y.L.; et al. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc. Natl. Acad. Sci. USA 2008, 105, 8760–8765. [Google Scholar] [CrossRef] [Green Version]
- Berghuis, P.; Dobszay, M.B.; Wang, X.; Spano, S.; Ledda, F.; Sousa, K.M.; Schulte, G.; Ernfors, P.; Mackie, K.; Paratcha, G.; et al. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 19115–19120. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.; Chambers, D.; Hobbs, C.; Doherty, P.; Graham, A. The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol. Cell. Neurosci. 2008, 38, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Ciampo, L.A.D.; Del Ciampo, I.R.L. Cannabis “in utero”: The fetus as a compulsive consumer. Int. J. Med Sci. Clin. Invent. 2019, 6, 4565–4569. [Google Scholar] [CrossRef] [Green Version]
- Huizink, A.C. Prenatal cannabis exposure and infant outcomes: Overview of studies. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 52, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Jew, C.P.; Lu, H.-C. Lasting impacts of prenatal cannabis exposure and the role of endogenous cannabinoids in the developing brain. Future Neurol. 2011, 6, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Corsi, D.J.; Walsh, L.; Weiss, D.; Hsu, H.; El-Chaar, D.; Hawken, S.; Fell, D.B.; Walker, M. Association Between Self-reported Prenatal Cannabis Use and Maternal, Perinatal, and Neonatal Outcomes. JAMA 2019, 322, 145. [Google Scholar] [CrossRef]
- Warshak, C.R.; Regan, J.; Moore, B.; Magner, K.; Kritzer, S.; Van Hook, J. Association between marijuana use and adverse obstetrical and neonatal outcomes. J. Perinatol. 2015, 35, 991–995. [Google Scholar] [CrossRef]
- Goldschmidt, L.; Day, N.L.; Richardson, G.A. Effects of prenatal marijuana exposure on child behavior problems at age 10. Neurotoxicol. Teratol. 2000, 22, 325–336. [Google Scholar] [CrossRef]
- El Marroun, H.; Bolhuis, K.; Franken, I.H.A.; Jaddoe, V.W.V.; Hillegers, M.H.; Lahey, B.B.; Tiemeier, H. Preconception and prenatal cannabis use and the risk of behavioural and emotional problems in the offspring; a multi-informant prospective longitudinal study. Int. J. Epidemiol. 2019, 48, 287–296. [Google Scholar] [CrossRef]
- Jin, K.; Xie, L.; Kim, S.H.; Parmentier-Batteur, S.; Sun, Y.; Mao, X.O.; Childs, J.; Greenberg, D. A Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol. Pharmacol. 2004, 66, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.S.; Ribeiro, F.F.; Ferreira, F.; Vaz, S.H.; Sebastião, A.M.; Xapelli, S. Interaction between cannabinoid type 1 and type 2 receptors in the modulation of subventricular zone and dentate gyrus neurogenesis. Front. Pharmacol. 2017, 8, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019. [Google Scholar] [CrossRef]
- Ahmad, R.; Postnov, A.; Bormans, G.; Versijpt, J.; Vandenbulcke, M.; van Laere, K. Decreased in vivo availability of the cannabinoid type 2 receptor in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2219–2227. [Google Scholar] [CrossRef] [Green Version]
- Parent, J.M.; Elliott, R.C.; Pleasure, S.J.; Barbaro, N.M.; Lowenstein, D.H. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann. Neurol. 2006, 59, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Marxreiter, F.; Nuber, S.; Kandasamy, M.; Klucken, J.; Aigner, R.; Burgmayer, R.; Couillard-Despres, S.; Riess, O.; Winkler, J.; Winner, B. Changes in adult olfactory bulb neurogenesis in mice expressing the A30P mutant form of alpha-synuclein. Eur. J. Neurosci. 2009, 29, 879–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, V.; Hill, M.P.; Bisogno, T.; Crossman, A.R.; Brotchie, J.M. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB J. 2000, 14, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Micale, V.; Mazzola, C.; Drago, F. Endocannabinoids and neurodegenerative diseases. Pharmacol. Res. 2007, 56, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Galve-Roperh, I.; Aguado, T.; Palazuelos, J.; Guzmán, M. The Endocannabinoid System and Neurogenesis in Health and Disease. Neuroscience 2007, 13, 109–114. [Google Scholar] [CrossRef]
- Oddi, S.; Scipioni, L.; Maccarrone, M. Endocannabinoid system and adult neurogenesis: A focused review. Curr. Opin. Pharmacol. 2020, 50, 25–32. [Google Scholar] [CrossRef]
- Handler, M.; Yang, X.; Shen, J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development 2000, 127, 2593–2606. [Google Scholar]
- Zhang, J.; Ooi, J.; Utami, K.H.; Langley, S.R.; Aning, O.A.; Park, D.S.; Renner, M.; Ma, S.; Cheok, C.F.; Knoblich, J.A.; et al. Expanded huntingtin CAG repeats disrupt the balance between neural progenitor expansion and differentiation in human cerebral organoids. bioRxiv 2019, 850586. [Google Scholar] [CrossRef]
- Sherstnev, V.V.; Kedrov, A.V.; Solov’eva, O.A.; Gruden’, M.A.; Konovalova, E.V.; Kalinin, I.A.; Proshin, A.T. The effects of α-synuclein oligomers on neurogenesis in the hippocampus and the behavior of aged mice. Neurochem. J. 2017, 11, 282–289. [Google Scholar] [CrossRef]
- Navarrete, F.; García-Gutiérrez, M.S.; Aracil-Fernández, A.; Lanciego, J.L.; Manzanares, J. Cannabinoid CB1 and CB2 Receptors, and Monoacylglycerol Lipase Gene Expression Alterations in the Basal Ganglia of Patients with Parkinson’s Disease. Neurotherapeutics 2018, 15, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aso, E.; Andrés-Benito, P.; Ferrer, I. Genetic deletion of CB1 cannabinoid receptors exacerbates the Alzheimer-like symptoms in a transgenic animal model. Biochem. Pharmacol. 2018, 157, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Calvo, A.; Bajo-Grañeras, R.; Maroto, I.B.; Zian, D.; Grabner, G.F.; García-Taboada, E.; Resel, E.; Zechner, R.; Zimmermann, R.; Ortega-Gutiérrez, S.; et al. Astroglial monoacylglycerol lipase controls mutant huntingtin-induced damage of striatal neurons. Neuropharmacology 2019, 150, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, A.A.; Milligan, C.E.; Pharr, E.P.; Howlett, A.C. The Endocannabinoid System and Oligodendrocytes in Health and Disease. Front. Neurosci. 2018, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, R.W.; Oliveira, C.L.; Guimarães, F.S.; Campos, A.C. Cannabinoid signalling in embryonic and adult neurogenesis: Possible implications for psychiatric and neurological disorders. Acta Neuropsychiatr. 2018, 1–16. [Google Scholar] [CrossRef]
- O’Connell, B.K.; Gloss, D.; Devinsky, O. Cannabinoids in treatment-resistant epilepsy: A review. Epilepsy Behav. 2017, 70, 341–348. [Google Scholar] [CrossRef]
- Leo, A.; Russo, E.; Elia, M. Cannabidiol and epilepsy: Rationale and therapeutic potential. Pharmacol. Res. 2016, 107, 85–92. [Google Scholar] [CrossRef]
- Rosenberg, E.C.; Patra, P.H.; Whalley, B.J. Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy-related neuroprotection. Epilepsy Behav. 2017, 70, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Naderi, N.; Ahmad-Molaei, L.; Aziz Ahari, F.; Motamedi, F. Modulation of Anticonvulsant Effects of Cannabinoid Compounds by GABA-A Receptor Agonist in Acute Pentylenetetrazole Model of Seizure in Rat. Neurochem. Res. 2011, 36, 1520–1525. [Google Scholar] [CrossRef]
- Vilela, L.R.; Medeiros, D.C.; Rezende, G.H.S.; de Oliveira, A.C.P.; Moraes, M.F.D.; Moreira, F.A. Effects of cannabinoids and endocannabinoid hydrolysis inhibition on pentylenetetrazole-induced seizure and electroencephalographic activity in rats. Epilepsy Res. 2013, 104, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Mikheeva, I.B.; Shubina, L.; Matveeva, N.; Pavlik, L.L.; Kitchigina, V.F. Fatty acid amide hydrolase inhibitor URB597 may protect against kainic acid–induced damage to hippocampal neurons: Dependence on the degree of injury. Epilepsy Res. 2017, 137, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.S.S.; Umathe, S.N. Involvement of transient receptor potential vanilloid type 1 channels in the pro-convulsant effect of anandamide in pentylenetetrazole-induced seizures. Epilepsy Res. 2012, 100, 113–124. [Google Scholar] [CrossRef]
- Griebel, G.; Pichat, P.; Beeské, S.; Leroy, T.; Redon, N.; Jacquet, A.; Françon, D.; Bert, L.; Even, L.; Lopez-Grancha, M.; et al. Selective blockade of the hydrolysis of the endocannabinoid 2-arachidonoylglycerol impairs learning and memory performance while producing antinociceptive activity in rodents. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, A.V.; Horne, E.A.; Cheah, C.S.; Swinney, K.; Hsu, K.-L.; Cao, J.K.; Marrs, W.R.; Blankman, J.L.; Tu, S.; Cherry, A.E.; et al. ABHD6 Blockade Exerts Antiepileptic Activity in PTZ-Induced Seizures and in Spontaneous Seizures in R6/2 Mice. Neuron 2014, 83, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Andres-Mach, M.; Haratym-Maj, A.; Zagaja, M.; Rola, R.; Maj, M.; Chrościńska-Krawczyk, M.; Luszczki, J.J. ACEA (a highly selective cannabinoid CB1 receptor agonist) stimulates hippocampal neurogenesis in mice treated with antiepileptic drugs. Brain Res. 2015, 1624, 86–94. [Google Scholar] [CrossRef]
- Zagaja, M.; Haratym-Maj, A.; Szewczyk, A.; Rola, R.; Maj, M.; Łuszczki, J.J.; Andres-Mach, M. Levetiracetam combined with ACEA, highly selective cannabinoid CB1 receptor agonist changes neurogenesis in mouse brain. Neurosci. Lett. 2019, 696, 79–86. [Google Scholar] [CrossRef]
- Andres-Mach, M.; Szewczyk, A.; Zagaja, M.; Luszczki, J.; Maj, M.; Rola, R.; Abram, M.; Kaminski, K. Evaluation of the impact of compound C11 a new anticonvulsant candidate on cognitive functions and hippocampal neurogenesis in mouse brain. Neuropharmacology 2020, 163, 107849. [Google Scholar] [CrossRef]
- Andres-Mach, M.; Zagaja, M.; Haratym-Maj, A.; Rola, R.; Maj, M.; Haratym, J.; Dudra-Jastrzębska, M.; Łuszczki, J. A Long-Term Treatment with Arachidonyl-2′-Chloroethylamide Combined with Valproate Increases Neurogenesis in a Mouse Pilocarpine Model of Epilepsy. Int. J. Mol. Sci. 2017, 18, 900. [Google Scholar] [CrossRef]
- Zagaja, M.; Szewczyk, A.; Haratym-Maj, A.; Rola, R.; Maj, M.; Łuszczki, J.J.; Andres-Mach, M. Increased neurogenesis after ACEA and levetiracetam treatment in mouse pilocarpine model of epilepsy. J. Pre Clin. Clin. Res. 2017, 11, 136–141. [Google Scholar] [CrossRef]
- Andres-Mach, M.; Zolkowska, D.; Barcicka-Klosowska, B.; Haratym-Maj, A.; Florek-Luszczki, M.; Luszczki, J.J. Effect of ACEA—A selective cannabinoid CB1 receptor agonist on the protective action of different antiepileptic drugs in the mouse pentylenetetrazole-induced seizure model. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2012, 39, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Cannon, J.R.; Greenamyre, J.T. Post-status epilepticus treatment with the cannabinoid agonist WIN 55,212-2 prevents chronic epileptic hippocampal damage in rats. Neurobiol. Dis. 2015, 73, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Colangeli, R.; Di Giovanni, G. WIN 55,212-2 Reverted Pilocarpine-Induced Status Epilepticus Early Changes of the Interaction among 5-HT2C/NMDA/CB1/Receptors in the Rat Hippocampus. ACS Chem. Neurosci. 2019, 10, 3296–3306. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.J.; Wiley, J.L.; Martin, B.R.; DeLorenzo, R.J. Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur. J. Pharmacol. 2001, 428, 51–57. [Google Scholar] [CrossRef]
- Naderi, N.; Aziz Ahari, F.; Shafaghi, B.; Hosseini Najarkolaei, A.; Motamedi, F. Evaluation of interactions between cannabinoid compounds and diazepam in electroshock-induced seizure model in mice. J. Neural Transm. 2008, 115, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Luszczki, J.J.; Misiuta-Krzesinska, M.; Florek, M.; Tutka, P.; Czuczwar, S.J. Synthetic cannabinoid WIN 55,212-2 mesylate enhances the protective action of four classical antiepileptic drugs against maximal electroshock-induced seizures in mice. Pharmacol. Biochem. Behav. 2011, 98, 261–267. [Google Scholar] [CrossRef]
- Luszczki, J.J.; Wlaz, A.; Karwan, S.; Florek-Luszczki, M.; Czuczwar, S.J. Effects of WIN 55,212-2 mesylate on the anticonvulsant action of lamotrigine, oxcarbazepine, pregabalin and topiramate against maximal electroshock-induced seizures in mice. Eur. J. Pharmacol. 2013, 720, 247–254. [Google Scholar] [CrossRef]
- Naderi, N.; Shafieirad, E.; Lakpoor, D.; Rahimi, A.; Mousavi, Z. Interaction between Cannabinoid Compounds and Capsazepine in Protection against Acute Pentylenetetrazole-induced Seizure in Mice. Iran. J. Pharm. Res. 2015, 14, 115–120. [Google Scholar]
- Bahremand, A.; Shafaroodi, H.; Ghasemi, M.; Nasrabady, S.E.; Gholizadeh, S.; Dehpour, A.R. The cannabinoid anticonvulsant effect on pentylenetetrazole-induced seizure is potentiated by ultra-low dose naltrexone in mice. Epilepsy Res. 2008, 81, 44–51. [Google Scholar] [CrossRef]
- Bahremand, A.; Nasrabady, S.E.; Shafaroodi, H.; Ghasemi, M.; Dehpour, A.R. Involvement of nitrergic system in the anticonvulsant effect of the cannabinoid CB1 agonist ACEA in the pentylenetetrazole-induced seizure in mice. Epilepsy Res. 2009, 84, 110–119. [Google Scholar] [CrossRef]
- Shafaroodi, H.; Samini, M.; Moezi, L.; Homayoun, H.; Sadeghipour, H.; Tavakoli, S.; Hajrasouliha, A.R.; Dehpour, A.R. The interaction of cannabinoids and opioids on pentylenetetrazole-induced seizure threshold in mice. Neuropharmacology 2004, 47, 390–400. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, C.R.; Hoeller, A.A.; Franco, P.L.C.; Martini, A.P.S.; Soares, F.M.S.; Lin, K.; Prediger, R.D.; Whalley, B.J.; Walz, R. The cannabinoid CB2 receptor-specific agonist AM1241 increases pentylenetetrazole-induced seizure severity in Wistar rats. Epilepsy Res. 2016, 127, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Huizenga, M.N.; Wicker, E.; Beck, V.C.; Forcelli, P.A. Anticonvulsant effect of cannabinoid receptor agonists in models of seizures in developing rats. Epilepsia 2017, 58, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Malyshevskaya, O.; Aritake, K.; Kaushik, M.K.; Uchiyama, N.; Cherasse, Y.; Kikura-Hanajiri, R.; Urade, Y. Natural (∆9-THC) and synthetic (JWH-018) cannabinoids induce seizures by acting through the cannabinoid CB1 receptor. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breivogel, C.S.; Wells, J.R.; Jonas, A.; Mistry, A.H.; Gravley, M.L.; Patel, R.M.; Whithorn, B.E.; Brenseke, B.M. Comparison of the Neurotoxic and Seizure-Inducing Effects of Synthetic and Endogenous Cannabinoids with Δ9-Tetrahydrocannabinol. Cannabis Cannabinoid Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, A.F.; Lycas, M.D.; Kaczmarzyk, J.R.; Spivak, C.E.; Baumann, M.H.; Lupica, C.R. Disruption of hippocampal synaptic transmission and long-term potentiation by psychoactive synthetic cannabinoid ‘Spice’ compounds: Comparison with Δ9-tetrahydrocannabinol: Spice and hippocampal function. Addict. Biol. 2017, 22, 390–399. [Google Scholar] [CrossRef] [Green Version]
- De Salas-Quiroga, A.; Díaz-Alonso, J.; García-Rincón, D.; Remmers, F.; Vega, D.; Gómez-Cañas, M.; Lutz, B.; Guzmán, M.; Galve-Roperh, I. Prenatal exposure to cannabinoids evokes long-lasting functional alterations by targeting CB1 receptors on developing cortical neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 13693–13698. [Google Scholar] [CrossRef] [Green Version]
- Beggiato, S.; Borelli, A.C.; Tomasini, M.C.; Morgano, L.; Antonelli, T.; Tanganelli, S.; Cuomo, V.; Ferraro, L. Long-lasting alterations of hippocampal GABAergic neurotransmission in adult rats following perinatal Δ9-THC exposure. Neurobiol. Learn. Mem. 2017, 139, 135–143. [Google Scholar] [CrossRef]
- Castaldo, P.; Magi, S.; Cataldi, M.; Arcangeli, S.; Lariccia, V.; Nasti, A.A.; Ferraro, L.; Tomasini, M.C.; Antonelli, T.; Cassano, T.; et al. Altered regulation of glutamate release and decreased functional activity and expression of GLT1 and GLAST glutamate transporters in the hippocampus of adolescent rats perinatally exposed to Δ9-THC. Pharmacol. Res. 2010, 61, 334–341. [Google Scholar] [CrossRef]
- Hill, A.J.; Mercier, M.S.; Hill, T.D.M.; Glyn, S.E.; Jones, N.A.; Yamasaki, Y.; Futamura, T.; Duncan, M.; Stott, C.G.; Stephens, G.J.; et al. Cannabidivarin is anticonvulsant in mouse and rat. Br. J. Pharmacol. 2012, 167, 1629–1642. [Google Scholar] [CrossRef]
- Whalley, B.; Williams, C.; Stephens, G.; Futamura, T. Use of the Phytocannabinoid Cannabidivarin (CBDV) in the Treatment of Epilepsy. U.S. Patent No. 9,125,859, 8 August 2015. [Google Scholar]
- Henley, B. Characterising the Anti-Convulsant Effects of CBD and CBDV on Layer II of the Medial Entorhinal Cortex of Rat and Human Brain Tissue In Vitro. Ph.D. Thesis, Aston University, Birmingham, UK, 2018; p. 273. [Google Scholar]
- Thompson, M.D.; Ross, R.A.; McIntyre Burnham, W. The anticonvulsant effects of cannabidiol examined in animal seizure models. Clin. Neurophysiol. 2016, 127, e164. [Google Scholar] [CrossRef]
- do val-da Silva, R.A.; Peixoto-Santos, J.E.; Kandratavicius, L.; de Ross, J.B.; Esteves, I.; de Martinis, B.S.; Alves, M.N.R.; Scandiuzzi, R.C.; Hallak, J.E.C.; Zuardi, A.W.; et al. Protective Effects of Cannabidiol against Seizures and Neuronal Death in a Rat Model of Mesial Temporal Lobe Epilepsy. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozela, E.; Juknat, A.; Vogel, Z. Modulation of Astrocyte Activity by Cannabidiol, a Nonpsychoactive Cannabinoid. Int. J. Mol. Sci. 2017, 18, 1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.A.; Shekh-Ahmad, T.; Khalil, A.; Walker, M.C.; Ali, A.B. Cannabidiol exerts antiepileptic effects by restoring hippocampal interneuron functions in a temporal lobe epilepsy model: Antiepileptic/neuroprotective effects of CBD in hippocampus. Br. J. Pharmacol. 2018, 175, 2097–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonca, E.; Darıcı, F. The Effect of Cannabidiol on Ischemia/Reperfusion-Induced Ventricular Arrhythmias: The Role of Adenosine A1 Receptors. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 76–83. [Google Scholar] [CrossRef]
- Stott, C.G.; Nichol, K.; Jones, N.A.; Gray, R.A.; Bazelot, M.; Whalley, B.J. The Proposed Multimodal Mechanism of Action of Cannabidiol (CBD) in Epilepsy: Modulation of Intracellular Calcium and Adenosine-mediated Signalling. Epilepsy Behav. 2019, 101, 106734. [Google Scholar] [CrossRef]
- Aso, E.; Fernández-Dueñas, V.; López-Cano, M.; Taura, J.; Watanabe, M.; Ferrer, I.; Luján, R.; Ciruela, F. Adenosine A2A-Cannabinoid CB1 Receptor Heteromers in the Hippocampus: Cannabidiol Blunts Δ9-Tetrahydrocannabinol-Induced Cognitive Impairment. Mol. Neurobiol. 2019, 56, 5382–5391. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.I.; Auchampach, J.A.; Hillard, C.J.; Zhu, G.; Yousufzai, B.; Mian, S.; Khan, S.; Khalifa, Y. Mediation of Cannabidiol Anti-inflammation in the Retina by Equilibrative Nucleoside Transporter and A2A Adenosine Receptor. Investig. Opthalmol. Vis. Sci. 2008, 49, 5526. [Google Scholar] [CrossRef] [Green Version]
- Sebastião, A.M.; Ribeiro, J.A. Adenosine Receptors and the Central Nervous System. Adenosine Receptors in Health and Disease. In Handbook of Experimental Pharmacology; Wilson, C.N., Mustafa, S.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 471–534. ISBN 978-3-540-89615-9. [Google Scholar]
- Rombo, D.M.; Newton, K.; Nissen, W.; Badurek, S.; Horn, J.M.; Minichiello, L.; Jefferys, J.G.R.; Sebastiao, A.M.; Lamsa, K.P. Synaptic mechanisms of adenosine A2A receptor-mediated hyperexcitability in the hippocampus: Synapse-Specific Modulation Via A2AR. Hippocampus 2015, 25, 566–580. [Google Scholar] [CrossRef]
- Franco, R.; Reyes-Resina, I.; Aguinaga, D.; Lillo, A.; Jiménez, J.; Raïch, I.; Borroto-Escuela, D.O.; Ferreiro-Vera, C.; Canela, E.I.; Sánchez de Medina, V.; et al. Potentiation of cannabinoid signaling in microglia by adenosine A2A receptor antagonists. Glia 2019, 67, 2410–2423. [Google Scholar] [CrossRef]
- Zhao, X.; Liao, Y.; Morgan, S.; Mathur, R.; Feustel, P.; Mazurkiewicz, J.; Qian, J.; Chang, J.; Mathern, G.W.; Adamo, M.A.; et al. Noninflammatory Changes of Microglia Are Sufficient to Cause Epilepsy. Cell Rep. 2018, 22, 2080–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyo, U.B.; Peng, J.; Murugan, M.; Mo, M.; Lalani, A.; Xie, P.; Xu, P.; Margolis, D.J.; Wu, L.-J. Regulation of Physical Microglia–Neuron Interactions by Fractalkine Signaling after Status Epilepticus. Eneuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Basilico, B.; Marrone, M.C.; Ragozzino, D. Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Semin. Cell Dev. Biol. 2019, 94, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron–glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Search of: Cannabinoids Epilepsy-List Results-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?term=cannabinoids+epilepsy&Search=Search (accessed on 1 December 2019).
- Eisenstein, M. The reality behind cannabidiol’s medical hype. Nature 2019, 572, 3. [Google Scholar] [CrossRef] [Green Version]
- Thodeson, D.M.; Brulet, R.; Hsieh, J. Neural stem cells and epilepsy: Functional roles and disease-in-a-dish models. Cell Tissue Res. 2018, 371, 47–54. [Google Scholar] [CrossRef]
- Ferreiros, A.; Beltrán-Carrascal, E.; Restrepo, P.; Pantoja, C.; Castañeda-Cardona, C.; Lasalvia, P.; Van Der Werf, L.; Nariño, D.; Rosselli, D.; Ferreiros, A.; et al. Efficacy of cannabinoids in pharmacoresistant epilepsy: A narrative review of the literature. Iatreia 2020, 33, 167–176. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martín, A.; Almazán, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Hill, C.L.; Leo, A.; Alhusaini, A.; Soubrane, C.; Mazzarella, E.; Russo, E.; Whalley, B.J.; Di Marzo, V.; Stephens, G.J. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: Potential for the treatment of neuronal hyperexcitability. ACS Chem. Neurosci. 2014, 5, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Ruiz, J.; Galve-Roperh, I.; Sagredo, O.; Guzmán, M. Possible therapeutic applications of cannabis in the neuropsychopharmacology field. Eur. Neuropsychopharmacol. 2020. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Aberrant neurogenesis in adult epileptic brain: Compensatory or pathologic. Neurochem. J. 2010, 4, 84–89. [Google Scholar] [CrossRef]






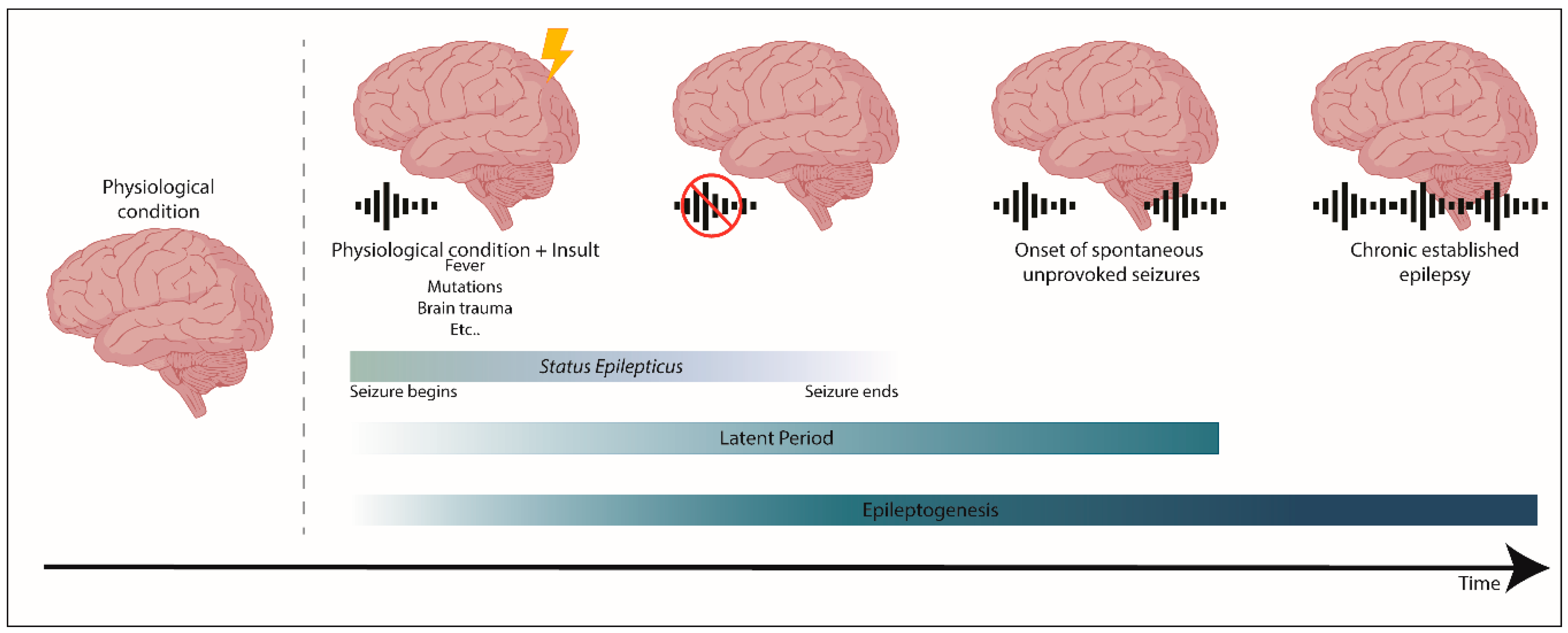{kind=link}
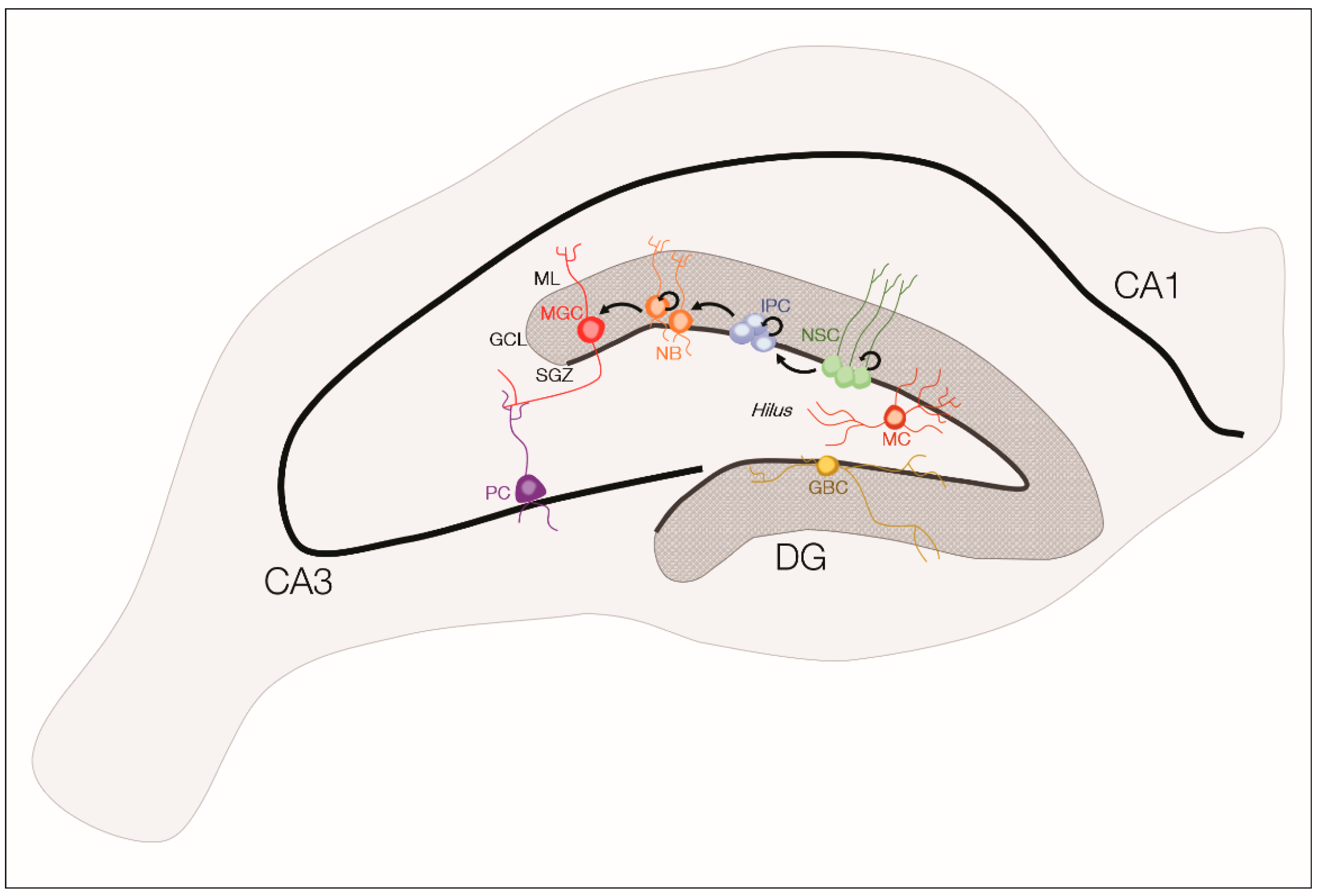{kind=link}
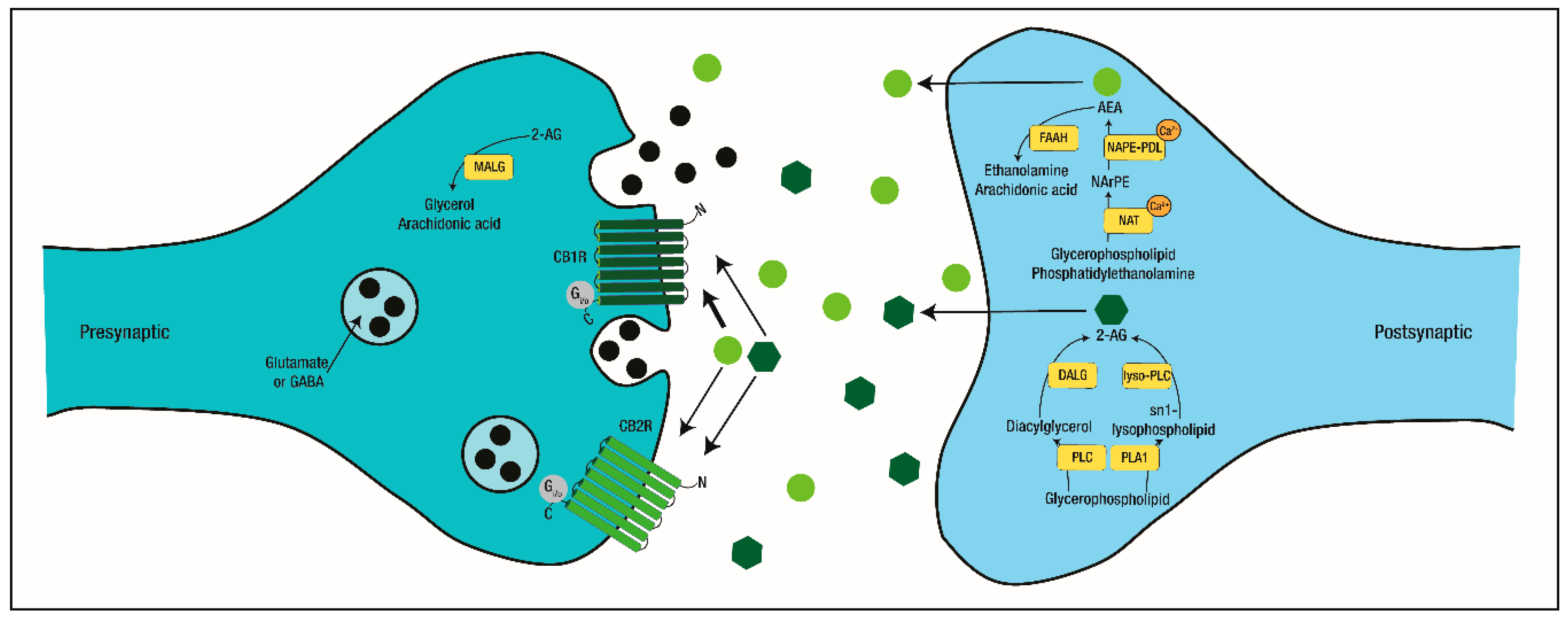{kind=link}
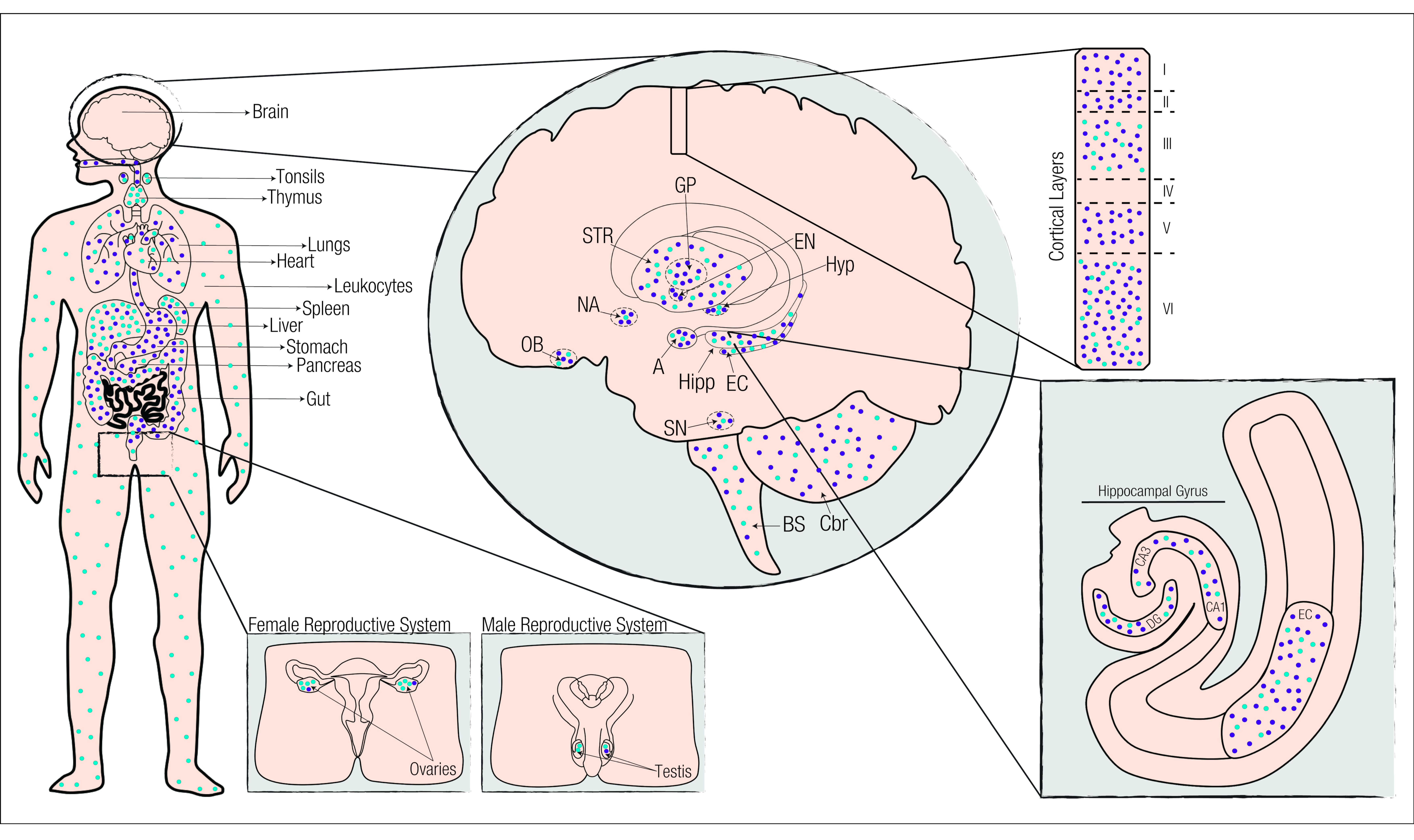{kind=link}
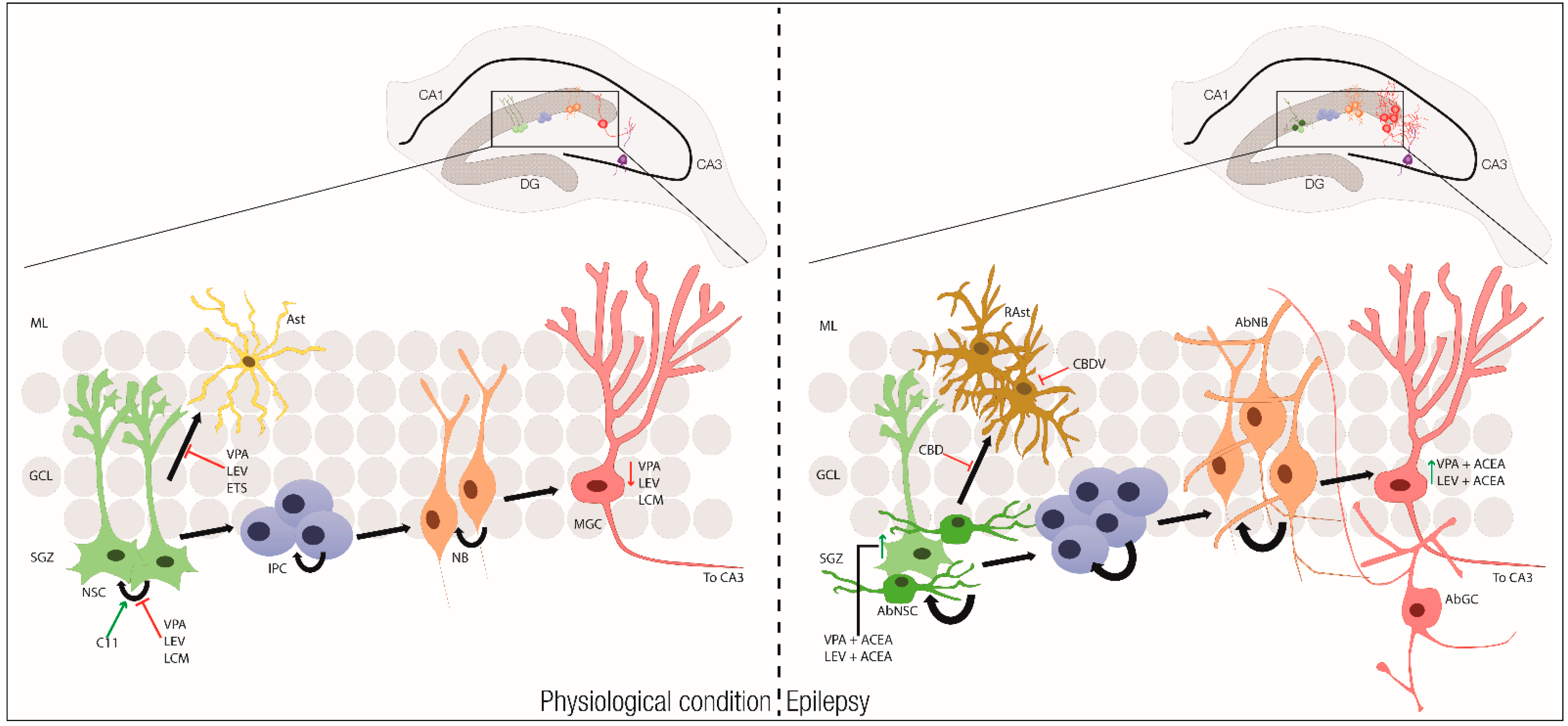{kind=link}
| Neural Stem Cell Therapies for Epilepsy | ||
|---|---|---|
| Modulating miRNA | Grafting Medial Ganglionic Eminence (MGE) Cells | Extracellular Vesicles Delivering |
|
|
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourenço, D.M.; Ribeiro-Rodrigues, L.; Sebastião, A.M.; Diógenes, M.J.; Xapelli, S. Neural Stem Cells and Cannabinoids in the Spotlight as Potential Therapy for Epilepsy. Int. J. Mol. Sci. 2020, 21, 7309. https://doi.org/10.3390/ijms21197309
Lourenço DM, Ribeiro-Rodrigues L, Sebastião AM, Diógenes MJ, Xapelli S. Neural Stem Cells and Cannabinoids in the Spotlight as Potential Therapy for Epilepsy. International Journal of Molecular Sciences. 2020; 21(19):7309. https://doi.org/10.3390/ijms21197309
Chicago/Turabian StyleLourenço, Diogo M., Leonor Ribeiro-Rodrigues, Ana M. Sebastião, Maria J. Diógenes, and Sara Xapelli. 2020. "Neural Stem Cells and Cannabinoids in the Spotlight as Potential Therapy for Epilepsy" International Journal of Molecular Sciences 21, no. 19: 7309. https://doi.org/10.3390/ijms21197309
APA StyleLourenço, D. M., Ribeiro-Rodrigues, L., Sebastião, A. M., Diógenes, M. J., & Xapelli, S. (2020). Neural Stem Cells and Cannabinoids in the Spotlight as Potential Therapy for Epilepsy. International Journal of Molecular Sciences, 21(19), 7309. https://doi.org/10.3390/ijms21197309