Connections between Metabolism and Epigenetic Modification in MDSCs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Metabolism of MDSCs
2.1. Glucose Metabolism
2.1.1. Lactate—An Important Metabolite in MDSCs
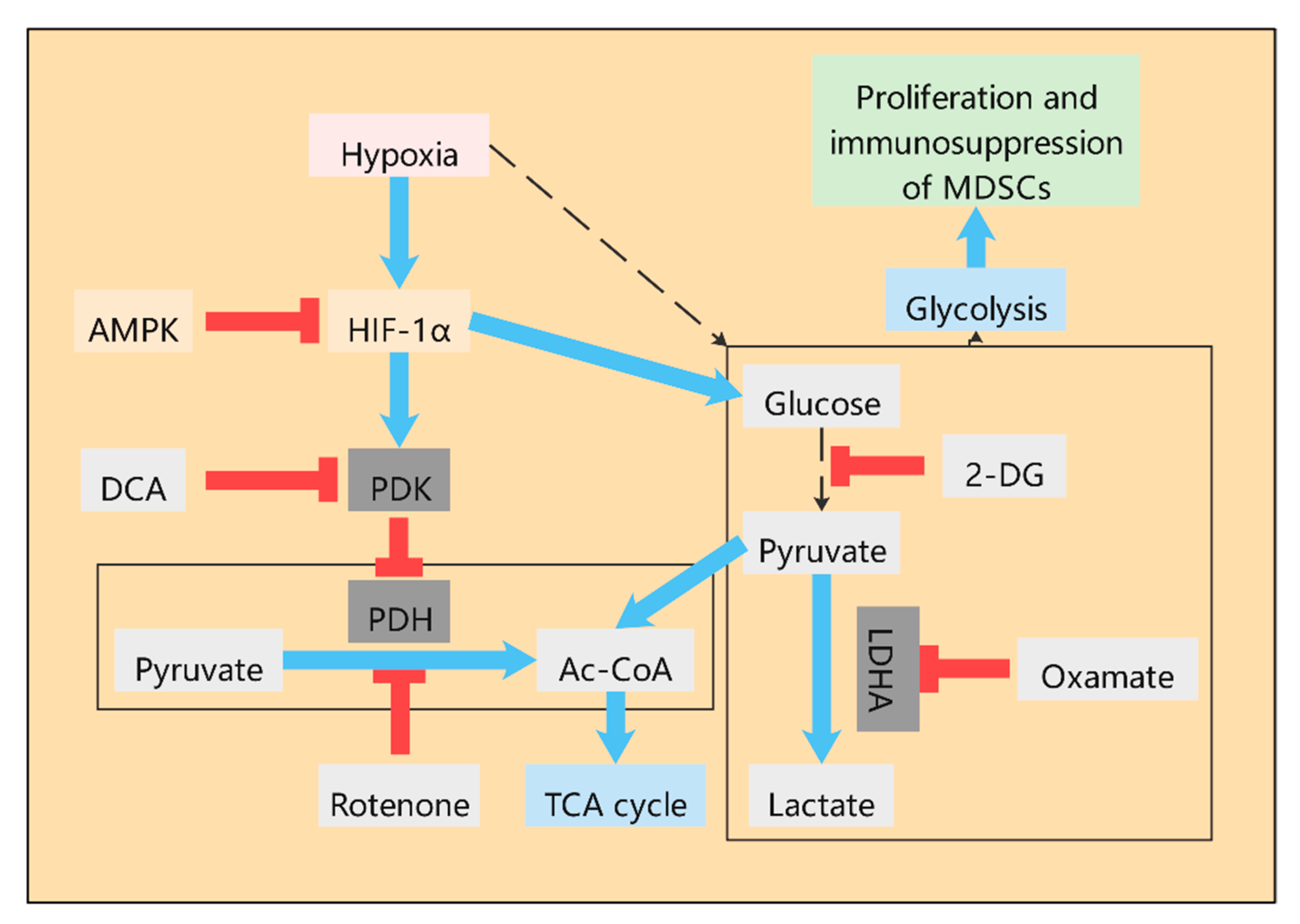
2.1.2. The Role of HIF-1α in the Glycolysis of MDSCs
2.1.3. The Role of AMPK in the Glycolysis of MDSCs
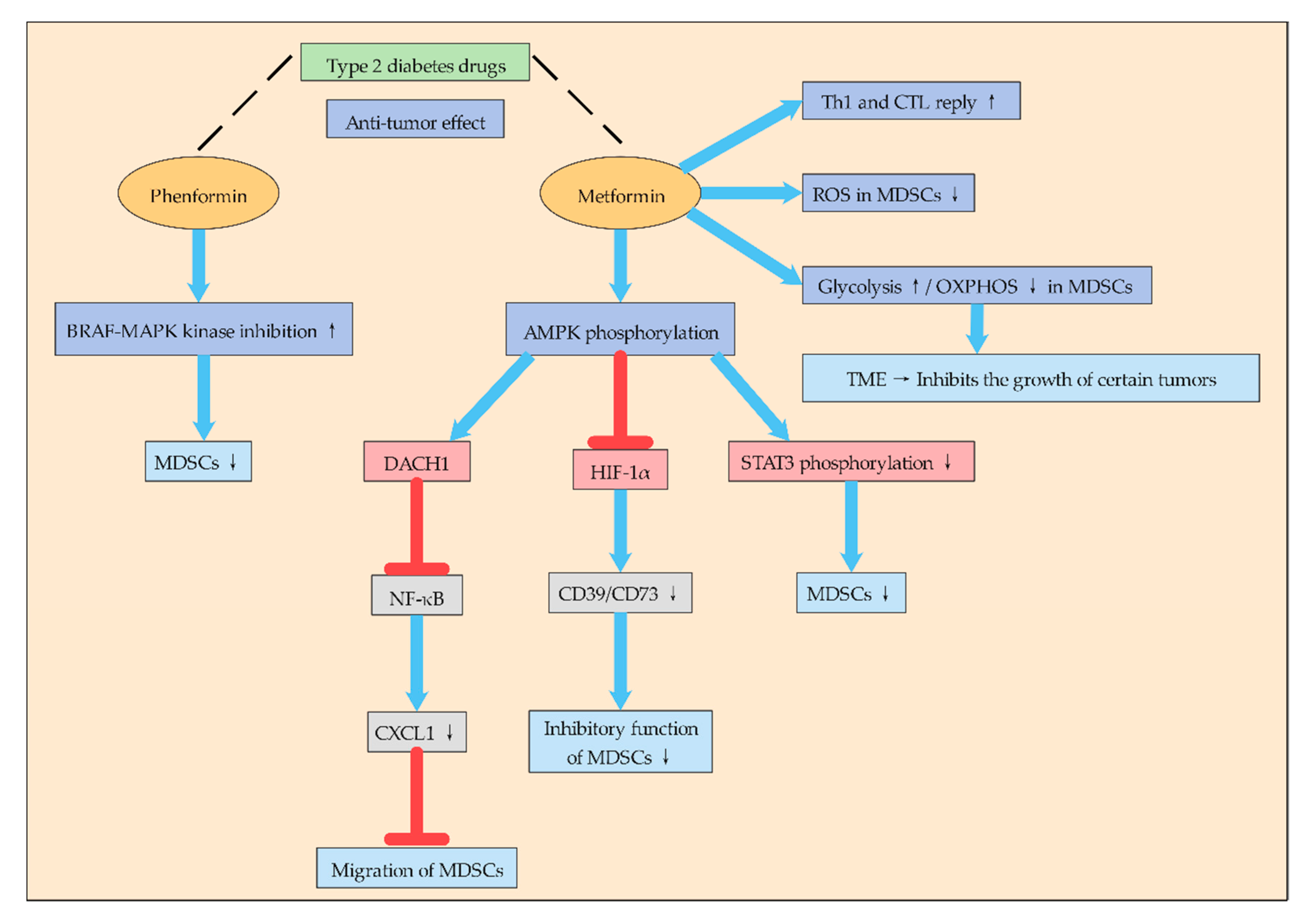
2.1.4. Type 2 Diabetes Drugs May Inhibit MDSCs by Affecting Glycolysis
2.2. Lipid Metabolism
2.3. Amino Acid Metabolism
2.4. A New-Type Metabolite of MDSCs
3. Epigenetic Modification of MDSCs
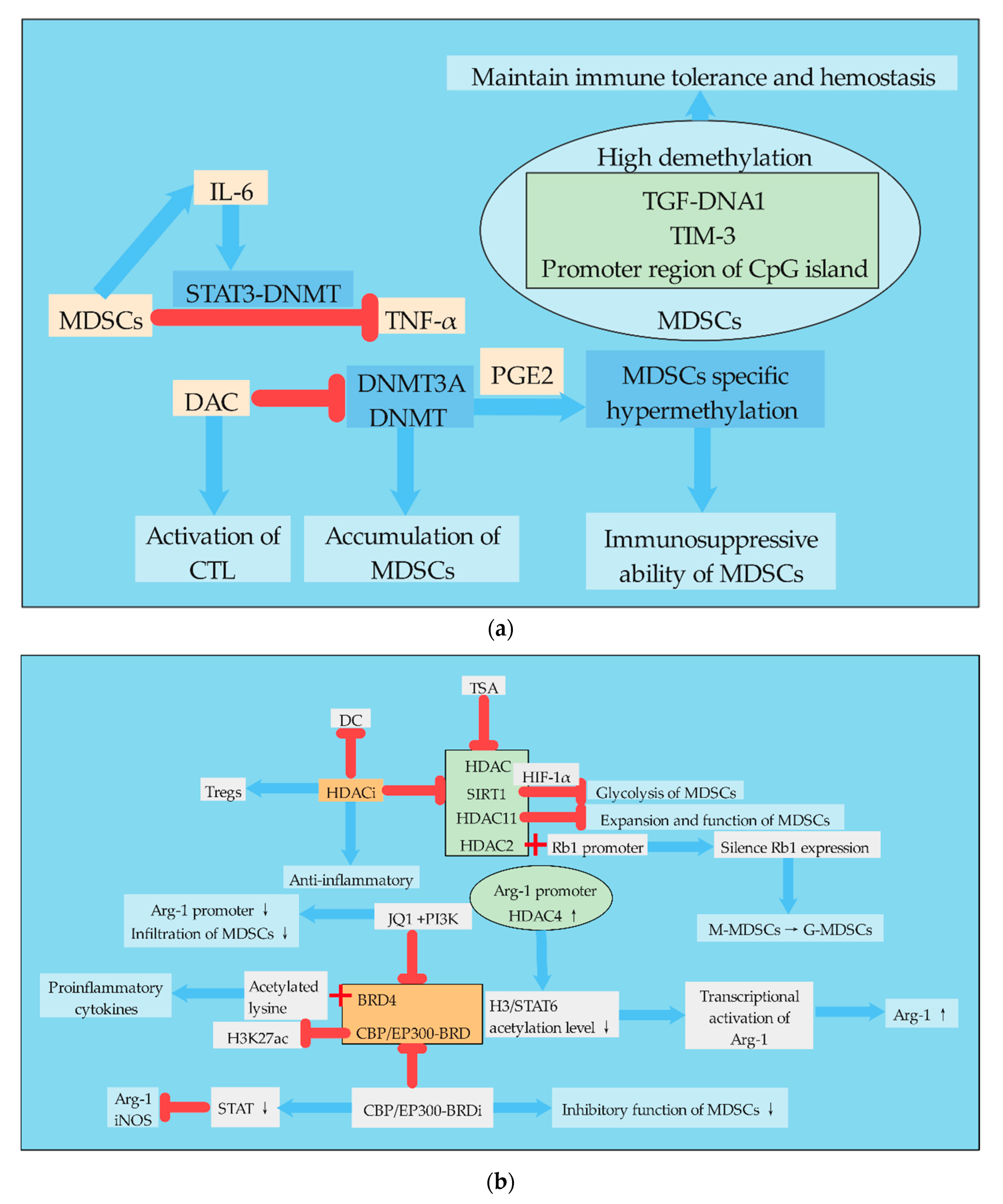
3.1. DNA Methylation
3.2. Histone Acetylation
3.3. Histone Lactylation
4. Connections between Metabolism and Epigenetic Modification in MDSCs
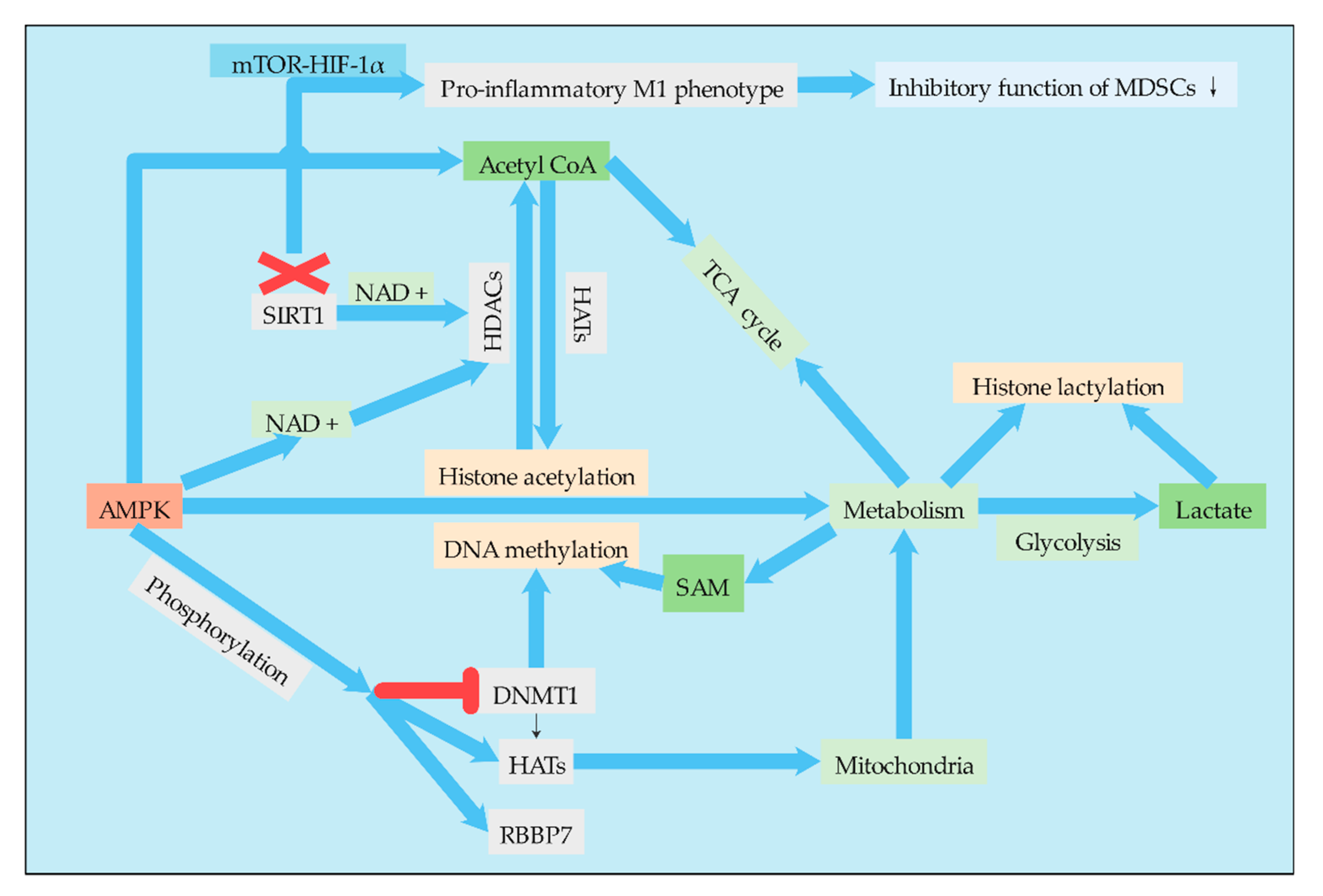
4.1. AMPK and HIF-1α Mediate the Association between Epigenetic Modification and Metabolism
4.1.1. AMPK
4.1.2. HIF-1α
4.2. Limitations
5. Conclusions and Prospect
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MDSCs | Myeloid-derived suppressor cells |
| TME | tumor microenvironment |
| DCs | dendritic cells |
| Arg-1 | Argininase-1 |
| iNOS | inducible nitric oxide synthase |
| ROS | reactive oxygen species |
| Tregs | regulatory T cells |
| TAMs | tumor-associated macrophages |
| PPP | pentose phosphate pathway |
| OXPHOS | oxidative phosphorylation |
| PEP | phosphoenol pyruvate |
| 2-DG | 2-deoxyglucose |
| PDH | Pyruvate dehydrogenase |
| PDK | pyruvate dehydrogenase kinase |
| DCA | dichloroacetic acid |
| NDV | Newcastle disease virus |
| STAT | signal transducer and activator of transcription |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| TNBC | triple negative breast cancer |
| LDHA | lactate dehydrogenase A |
| HIF-1α | hypoxia inducible factor-1α |
| ENTPD2/CD39L1 | ectonucleoside triphosphate diphosphohydrolase 2 |
| Glut-1 | glucose transporter-1 |
| GR | glucocorticoid receptor |
| RPM | Rapamycin |
| PI3K | phosphatidylinositol 3-kinase |
| AKT | serine-threonine protein kinase |
| MENK | ethionine enkephalin |
| AMPK | adenosine 5′-monophosphate (AMP)-activated protein kinase |
| AR | androgen receptors |
| DACH1 | Dachshund homologue 1 |
| FAO | fatty acid oxidation |
| GMP | Granulocytes/macrophage progenitor cells |
| DNMTs | DNA methyltransferases |
| DAC | desitabine |
| MAPK | mitogen-activated protein kinase |
| HDAC | histone deacetylase |
| HAT | histone acetyl transferase |
| HDACi | histone deacetylase inhibitors |
| BRD | bromine domain |
| H3K27ac | H3K27 acetylation |
| KATS | lysine acetyltransferase |
| RBBP7 | retinoblastoma binding protein 7 |
References
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Pang, B.; Lin, G.; Zhen, Y.; Yi, H. Energy metabolism manipulates the fate and function of tumour myeloid-derived suppressor cells. Br. J. Cancer 2020, 122, 23–29. [Google Scholar] [CrossRef]
- Tesi, R.J. MDSC; the Most Important Cell You Have Never Heard of. Trends Pharm. Sci. 2019, 40, 4–7. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Myeloid-derived suppressor cells (MDSC): An important partner in cellular/tissue senescence. Biogerontology 2018, 19, 325–339. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. The role of myeloid-derived suppressor cells (MDSC) in the inflammaging process. Ageing Res. Rev. 2018, 48, 1–10. [Google Scholar] [CrossRef]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Beury, D.W.; Parker, K.H.; Horn, L.A. Survival of the fittest: How myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol. Immunother. CII 2020, 69, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Goffaux, G.; Hammami, I.; Jolicoeur, M. A Dynamic Metabolic Flux Analysis of Myeloid-Derived Suppressor Cells Confirms Immunosuppression-Related Metabolic Plasticity. Sci. Rep. 2017, 7, 9850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, S.L.; Chen, W.W.; Su, Y.C.; Su, Y.W.; Chuang, T.H.; Hsu, S.C.; Huang, L.R. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017, 8, e2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Chang, Y.; Lu, W.; Xu, H.; Wang, S.; Ma, J. Lewis tumor cell conditioned medium enhances immunosuppressive function of mouse myeloid-derived suppressor cells by regulating glycolytic pathway. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi= Chin. J. Cell. Mol. Immunol. 2019, 35, 491–497. [Google Scholar]
- Wu, T.; Zhao, Y.; Wang, H.; Li, Y.; Shao, L.; Wang, R.; Lu, J.; Yang, Z.; Wang, J.; Zhao, Y. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci. Rep. 2016, 6, 20250. [Google Scholar] [CrossRef] [Green Version]
- Husain, Z.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate and myeloid-derived suppressor cells: Linking metabolism to cancer immunology. Oncoimmunology 2013, 2, e26383. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. (Berl. Ger.) 2019, 97, 1049–1064. [Google Scholar] [CrossRef] [Green Version]
- Kolb, D.; Kolishetti, N.; Surnar, B.; Sarkar, S.; Guin, S.; Shah, A.S.; Dhar, S. Metabolic Modulation of the Tumor Microenvironment Leads to Multiple Checkpoint Inhibition and Immune Cell Infiltration. ACS Nano 2020, 14, 11055–11066. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Li, B.; Chen, A.; Zheng, M.; Xu, T.; Zhang, H.; Dong, J.; Wu, J.; Yu, D.; Wei, J. Targeting aerobic glycolysis by dichloroacetate improves Newcastle disease virus-mediated viro-immunotherapy in hepatocellular carcinoma. Br. J. Cancer 2020, 122, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Liu, G.; Bi, Y.; Shen, B.; Yang, H.; Zhang, Y.; Wang, X.; Liu, H.; Lu, Y.; Liao, J.; Chen, X.; et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1α-dependent glycolysis. Cancer Res. 2014, 74, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, Z.; Bi, Y.; Fu, Z.; Gong, P.; Li, Y.; Yu, Q.; Jia, A.; Wang, J.; Xue, L.; et al. mTOR signaling disruption from myeloid-derived suppressive cells protects against immune-mediated hepatic injury through the HIF1α-dependent glycolytic pathway. J. Leukoc. Biol. 2016, 100, 1349–1362. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Multhoff, G. Hypoxia-/HIF-1α-Driven Factors of the Tumor Microenvironment Impeding Antitumor Immune Responses and Promoting Malignant Progression. Adv. Exp. Med. Biol. 2018, 1072, 171–175. [Google Scholar] [CrossRef]
- Ou, X.; Lv, W. Metabolic changes and interaction of tumor cell, myeloid-derived suppressor cell and T cell in hypoxic microenvironment. Future Oncol. (Lond. Engl.) 2020, 16, 383–393. [Google Scholar] [CrossRef]
- Yu, Q.; Dong, L.; Li, Y.; Liu, G. SIRT1 and HIF1α signaling in metabolism and immune responses. Cancer Lett. 2018, 418, 20–26. [Google Scholar] [CrossRef]
- Chiu, D.K.; Tse, A.P.; Xu, I.M.; Di Cui, J.; Lai, R.K.; Li, L.L.; Koh, H.Y.; Tsang, F.H.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Elpek, K.G.; Cremasco, V.; Shen, H.; Harvey, C.J.; Wucherpfennig, K.W.; Goldstein, D.R.; Monach, P.A.; Turley, S.J. The tumor microenvironment shapes lineage, transcriptional, and functional diversity of infiltrating myeloid cells. Cancer Immunol. Res. 2014, 2, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Liu, H.; Bi, Y.; Yang, H.; Li, Y.; Wang, J.; Zhang, Z.; Wang, Y.; Li, C.; Jia, A.; et al. Glucocorticoid receptor promotes the function of myeloid-derived suppressor cells by suppressing HIF1α-dependent glycolysis. Cell. Mol. Immunol. 2018, 15, 618–629. [Google Scholar] [CrossRef] [PubMed]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Yang, J.; Luo, F.; Qian, J.; Liu, R.; Zhang, D.; Yu, H.; Chu, Y. mTOR-mediated glycolysis contributes to the enhanced suppressive function of murine tumor-infiltrating monocytic myeloid-derived suppressor cells. Cancer Immunol. Immunother. CII 2018, 67, 1355–1364. [Google Scholar] [CrossRef]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Tuo, Y.; Zhang, Z.; Tian, C.; Hu, Q.; Xie, R.; Yang, J.; Zhou, H.; Lu, L.; Xiang, M. Anti-inflammatory and metabolic reprogramming effects of MENK produce antitumor response in CT26 tumor-bearing mice. J. Leukoc. Biol. 2020, 108, 215–228. [Google Scholar] [CrossRef]
- Trillo-Tinoco, J.; Sierra, R.A.; Mohamed, E.; Cao, Y.; de Mingo-Pulido, Á.; Gilvary, D.L.; Anadon, C.M.; Costich, T.L.; Wei, S.; Flores, E.R.; et al. AMPK Alpha-1 Intrinsically Regulates the Function and Differentiation of Tumor Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 5034–5047. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK—sensing energy while talking to other signaling pathways. Cell Metab. 2014, 20, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Hammami, I.; Chen, J.; Murschel, F.; Bronte, V.; De Crescenzo, G.; Jolicoeur, M. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol. 2012, 13, 18. [Google Scholar] [CrossRef] [Green Version]
- Consiglio, C.R.; Udartseva, O.; Ramsey, K.D.; Bush, C.; Gollnick, S.O. Enzalutamide, an Androgen Receptor Antagonist, Enhances Myeloid Cell-Mediated Immune Suppression and Tumor Progression. Cancer Immunol. Res. 2020, 8, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Yin, K.; Tang, X.; Tian, J.; Zhang, Y.; Ma, J.; Xu, H.; Xu, Q.; Wang, S. Metformin inhibits the function of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Biomed. Pharmacother. 2019, 120, 109458. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, G.; Lian, J.; Huang, L.; Zhao, Q.; Liu, S.; Zhang, Z.; Chen, X.; Yue, D.; Li, L.; Li, F.; et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology 2018, 7, e1442167. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Eikawa, S.; Nishida, M.; Kunisada, Y.; Yoshida, A.; Fujiwara, T.; Kunisada, T.; Ozaki, T.; Udono, H. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: Implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int. Immunol. 2019, 31, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Rodriguez, P.C.; Ochoa, A.C. Metabolic reprogramming of myeloid-derived suppressor cells (MDSC) in cancer. Oncoimmunology 2016, 5, e1200771. [Google Scholar] [CrossRef] [Green Version]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Adeshakin, A.O.; Xu, M.; Afolabi, L.O.; Zhang, G.; Chen, Y.H.; Wan, X. Lipid Metabolic Pathways Confer the Immunosuppressive Function of Myeloid-Derived Suppressor Cells in Tumor. Front. Immunol. 2019, 10, 1399. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef]
- Mandruzzato, S.; Solito, S.; Falisi, E.; Francescato, S.; Chiarion-Sileni, V.; Mocellin, S.; Zanon, A.; Rossi, C.R.; Nitti, D.; Bronte, V.; et al. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J. Immunol. 2009, 182, 6562–6568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, aay1863. [Google Scholar] [CrossRef]
- LXR Agonism Depletes MDSCs to Promote Antitumor Immunity. Cancer Discov. 2018, 8, 263. [CrossRef] [Green Version]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Ladomersky, E.; Zhai, L.; Lenzen, A.; Lauing, K.L.; Qian, J.; Scholtens, D.M.; Gritsina, G.; Sun, X.; Liu, Y.; Yu, F.; et al. IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival Against Advanced Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2559–2573. [Google Scholar] [CrossRef] [Green Version]
- Hammami, I.; Chen, J.; Bronte, V.; DeCrescenzo, G.; Jolicoeur, M. L-glutamine is a key parameter in the immunosuppression phenomenon. Biochem. Biophys. Res. Commun. 2012, 425, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.H.; Sun, I.H.; Zhao, L.; Leone, R.D.; Sun, I.M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020, 130, 3865–3884. [Google Scholar] [CrossRef] [PubMed]
- Baumann, T.; Dunkel, A.; Schmid, C.; Schmitt, S.; Hiltensperger, M.; Lohr, K.; Laketa, V.; Donakonda, S.; Ahting, U.; Lorenz-Depiereux, B.; et al. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat. Immunol. 2020, 21, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, Z.H.; Chen, Y.; Jarosz, D.F. Protein-Based Inheritance: Epigenetics beyond the Chromosome. Mol. Cell 2018, 69, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hong, T.; Wang, S.; Mo, J.; Tian, T.; Zhou, X. Epigenetic modification of nucleic acids: From basic studies to medical applications. Chem. Soc. Rev. 2017, 46, 2844–2872. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, J.; Chen, Z.; Cheng, A.S. Understanding the epigenetic regulation of tumours and their microenvironments: Opportunities and problems for epigenetic therapy. J. Pathol. 2017, 241, 10–24. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, S.; Liu, Y.; Yang, C. Epigenetics in myeloid derived suppressor cells: A sheathed sword towards cancer. Oncotarget 2016, 7, 57452–57463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Chen, P.; Leng, Y.; Kang, J. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3-COX2-dependent pathway. Oncogene 2018, 37, 5952–5966. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [Green Version]
- Ginder, G.D.; Williams, D.C., Jr. Corrigendum to “Readers of DNA methylation, the MBD family as potential therapeutic targets” [Pharmacology & Therapeutics 184 (1) (2018) 98-111]. Pharmacol. Ther. 2018, 190, 237–238. [Google Scholar] [CrossRef]
- Koch, A.; Joosten, S.C.; Feng, Z.; de Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef]
- Rodríguez-Ubreva, J.; Català-Moll, F.; Obermajer, N.; Álvarez-Errico, D.; Ramirez, R.N.; Company, C.; Vento-Tormo, R.; Moreno-Bueno, G.; Edwards, R.P.; Mortazavi, A.; et al. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.D.; Lu, C.; Payne, D.; Paschall, A.V.; Klement, J.D.; Redd, P.S.; Ibrahim, M.L.; Yang, D.; Han, Q.; Liu, Z.; et al. Autocrine IL6-Mediated Activation of the STAT3-DNMT Axis Silences the TNFα-RIP1 Necroptosis Pathway to Sustain Survival and Accumulation of Myeloid-Derived Suppressor Cells. Cancer Res. 2020, 80, 3145–3156. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, X.; Zhang, W.; Guo, H.; Cheng, S.; He, Q.; Yang, B.; Ding, L. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm. Sin. B 2020, 10, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Toor, S.M.; Taha, R.Z.; Al-Ali, D.; Sasidharan Nair, V.; Elkord, E. DNA methylation in the promoters of PD-L1, MMP9, ARG1, galectin-9, TIM-3, VISTA and TGF-β genes in HLA-DR(-) myeloid cells, compared with HLA-DR(+) antigen-presenting cells. Epigenetics 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Saleh, R.; Toor, S.M.; Taha, R.Z.; Ahmed, A.A.; Kurer, M.A.; Murshed, K.; Alajez, N.M.; Abu Nada, M.; Elkord, E. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin. Epigenet. 2020, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.A.; Richon, V.M.; Breslow, R.; Rifkind, R.A. Histone deacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol. 2001, 13, 477–483. [Google Scholar] [CrossRef]
- Sahakian, E.; Powers, J.J.; Chen, J.; Deng, S.L.; Cheng, F.; Distler, A.; Woods, D.M.; Rock-Klotz, J.; Sodre, A.L.; Youn, J.I.; et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015, 63, 579–585. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.I.; Kumar, V.; Collazo, M.; Nefedova, Y.; Condamine, T.; Cheng, P.; Villagra, A.; Antonia, S.; McCaffrey, J.C.; Fishman, M.; et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat. Immunol. 2013, 14, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Wei, J.; Zhong, L.; Shi, M.; Zhou, P.; Zuo, S.; Wu, K.; Zhu, M.; Huang, X.; Yu, Y.; et al. Cross talk between histone deacetylase 4 and STAT6 in the transcriptional regulation of arginase 1 during mouse dendritic cell differentiation. Mol. Cell. Biol. 2015, 35, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Rosborough, B.R.; Castellaneta, A.; Natarajan, S.; Thomson, A.W.; Turnquist, H.R. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J. Leukoc. Biol. 2012, 91, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Singh, A.R.; Liu, K.X.; Pham, T.V.; Zulcic, M.; Skola, D.; Chun, H.B.; Glass, C.K.; Morales, G.A.; Garlich, J.R.; et al. SF2523: Dual PI3K/BRD4 Inhibitor Blocks Tumor Immunosuppression and Promotes Adaptive Immune Responses in Cancer. Mol. Cancer 2019, 18, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- De Almeida Nagata, D.E.; Chiang, E.Y.; Jhunjhunwala, S.; Caplazi, P.; Arumugam, V.; Modrusan, Z.; Chan, E.; Merchant, M.; Jin, L.; Arnott, D.; et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 2019, 27, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019, 57, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Rando, O.J. Metabolic Inputs into the Epigenome. Cell Metab. 2017, 25, 544–558. [Google Scholar] [CrossRef]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef] [Green Version]
- De Veirman, K.; Menu, E.; Maes, K.; De Beule, N.; De Smedt, E.; Maes, A.; Vlummens, P.; Fostier, K.; Kassambara, A.; Moreaux, J.; et al. Myeloid-derived suppressor cells induce multiple myeloma cell survival by activating the AMPK pathway. Cancer Lett. 2019, 442, 233–241. [Google Scholar] [CrossRef]
- Vancura, A.; Nagar, S.; Kaur, P.; Bu, P.; Bhagwat, M.; Vancurova, I. Reciprocal Regulation of AMPK/SNF1 and Protein Acetylation. Int. J. Mol. Sci. 2018, 19, 3314. [Google Scholar] [CrossRef] [Green Version]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, aaf7478. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [Green Version]
- Katzenelenbogen, Y.; Sheban, F.; Yalin, A.; Yofe, I.; Svetlichnyy, D.; Jaitin, D.A.; Bornstein, C.; Moshe, A.; Keren-Shaul, H.; Cohen, M.; et al. Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 2020, 182, 872–885. [Google Scholar] [CrossRef]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2020, 53, 100715. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, H.; Xu, H.; Wang, S.; Ma, J. Connections between Metabolism and Epigenetic Modification in MDSCs. Int. J. Mol. Sci. 2020, 21, 7356. https://doi.org/10.3390/ijms21197356
Dai H, Xu H, Wang S, Ma J. Connections between Metabolism and Epigenetic Modification in MDSCs. International Journal of Molecular Sciences. 2020; 21(19):7356. https://doi.org/10.3390/ijms21197356
Chicago/Turabian StyleDai, Haiyan, Huaxi Xu, Shengjun Wang, and Jie Ma. 2020. "Connections between Metabolism and Epigenetic Modification in MDSCs" International Journal of Molecular Sciences 21, no. 19: 7356. https://doi.org/10.3390/ijms21197356
APA StyleDai, H., Xu, H., Wang, S., & Ma, J. (2020). Connections between Metabolism and Epigenetic Modification in MDSCs. International Journal of Molecular Sciences, 21(19), 7356. https://doi.org/10.3390/ijms21197356