Antioxidant Amelioration of Riboflavin Transporter Deficiency in Motoneurons Derived from Patient-Specific Induced Pluripotent Stem Cells

 , ,
, ,  , ,
, ,  , and
, and 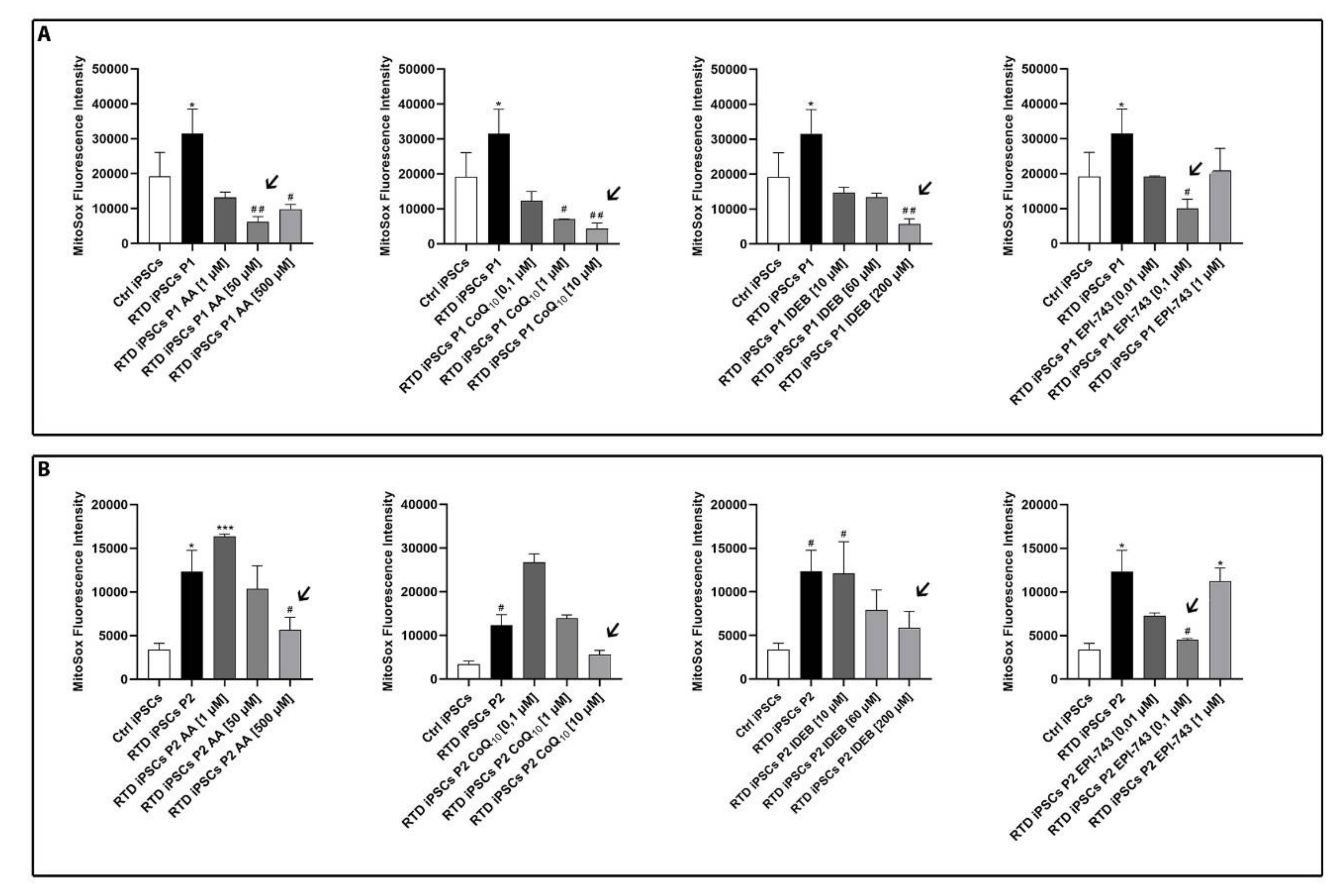{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
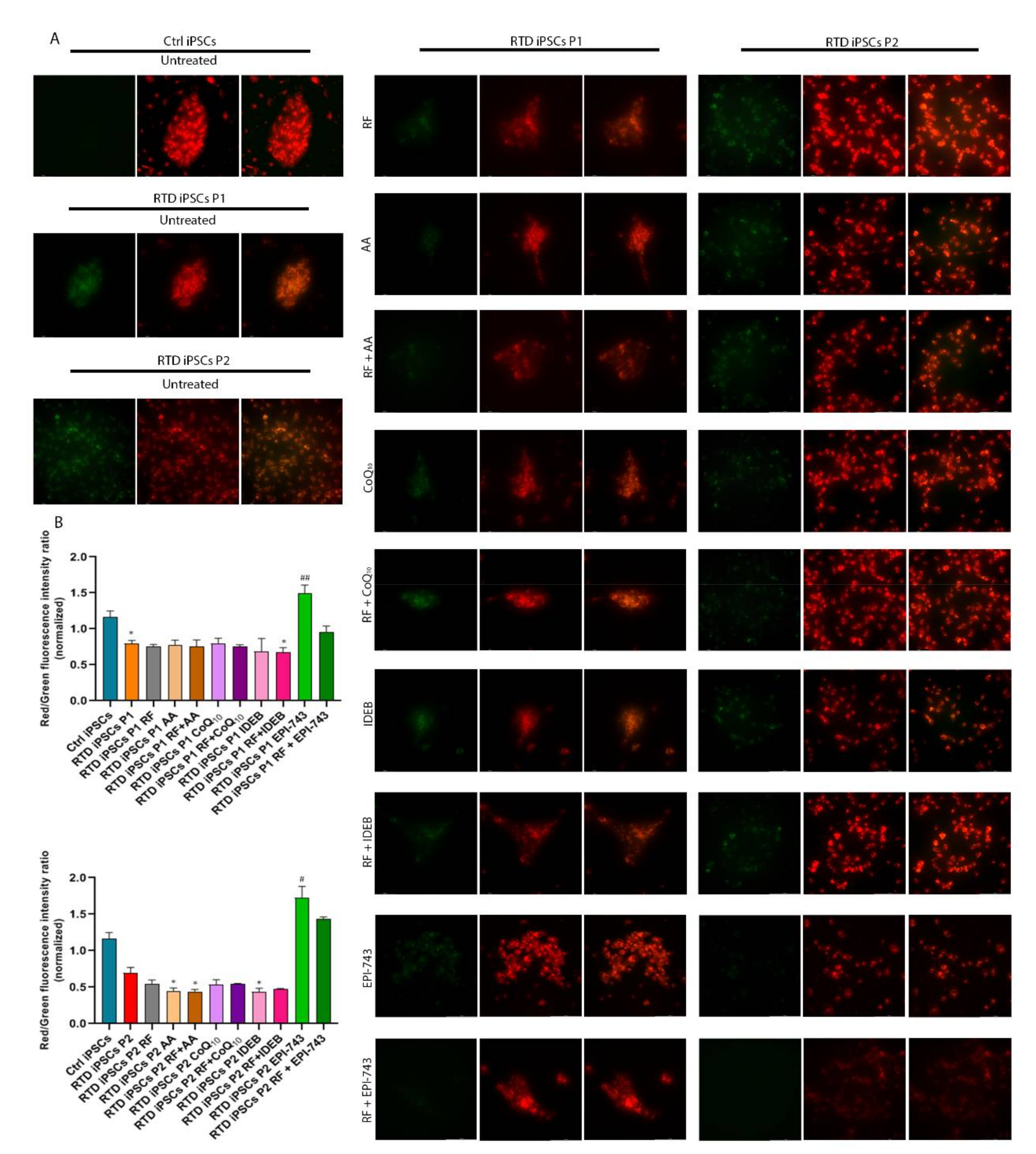
2.1. Antioxidant Treatment Restores Redox Status of RTD MNs
2.2. EPI-743 Treatment Is Able to Reduce the Levels of Oxidized Lipids in RTD iPSCs
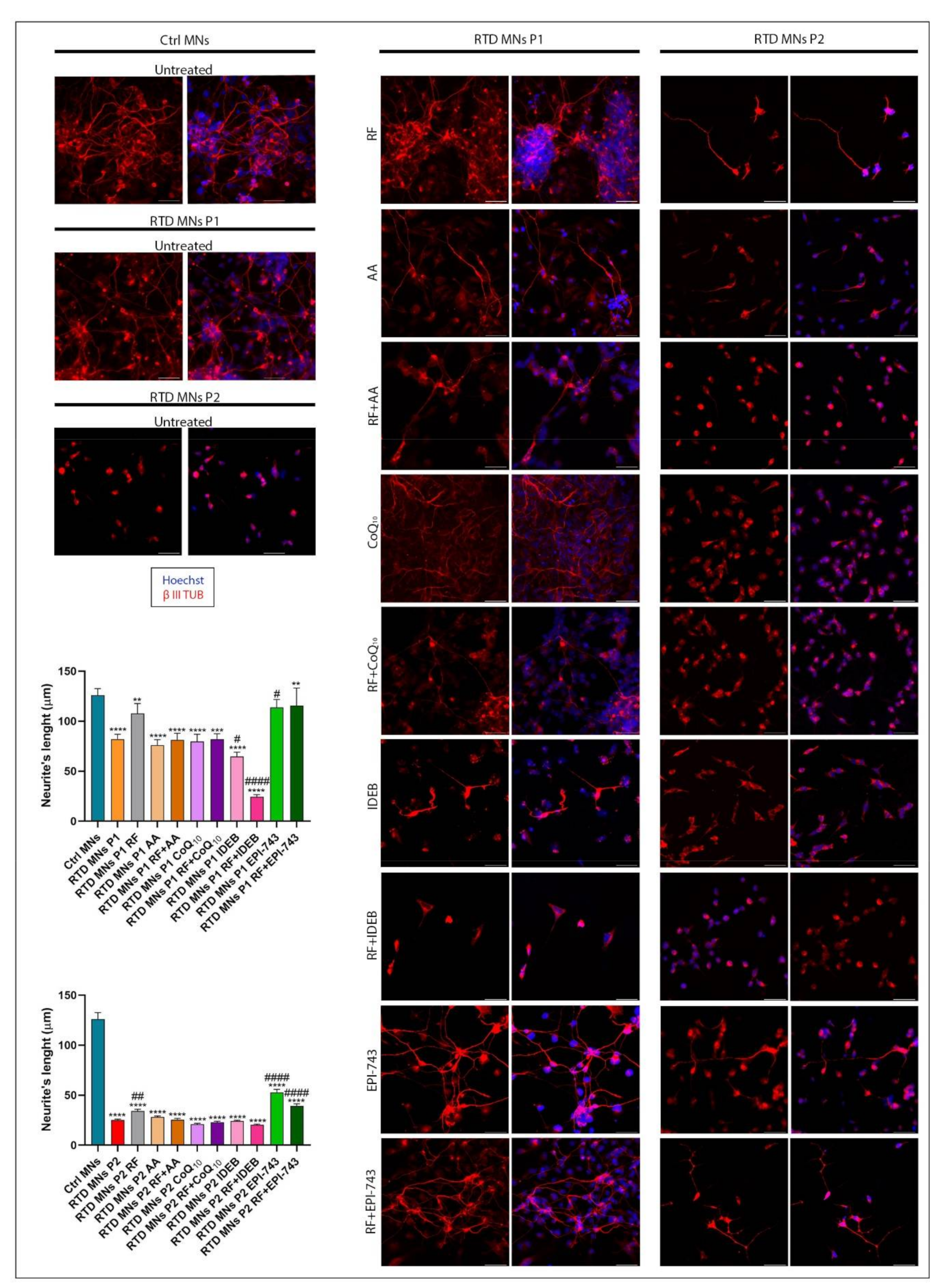
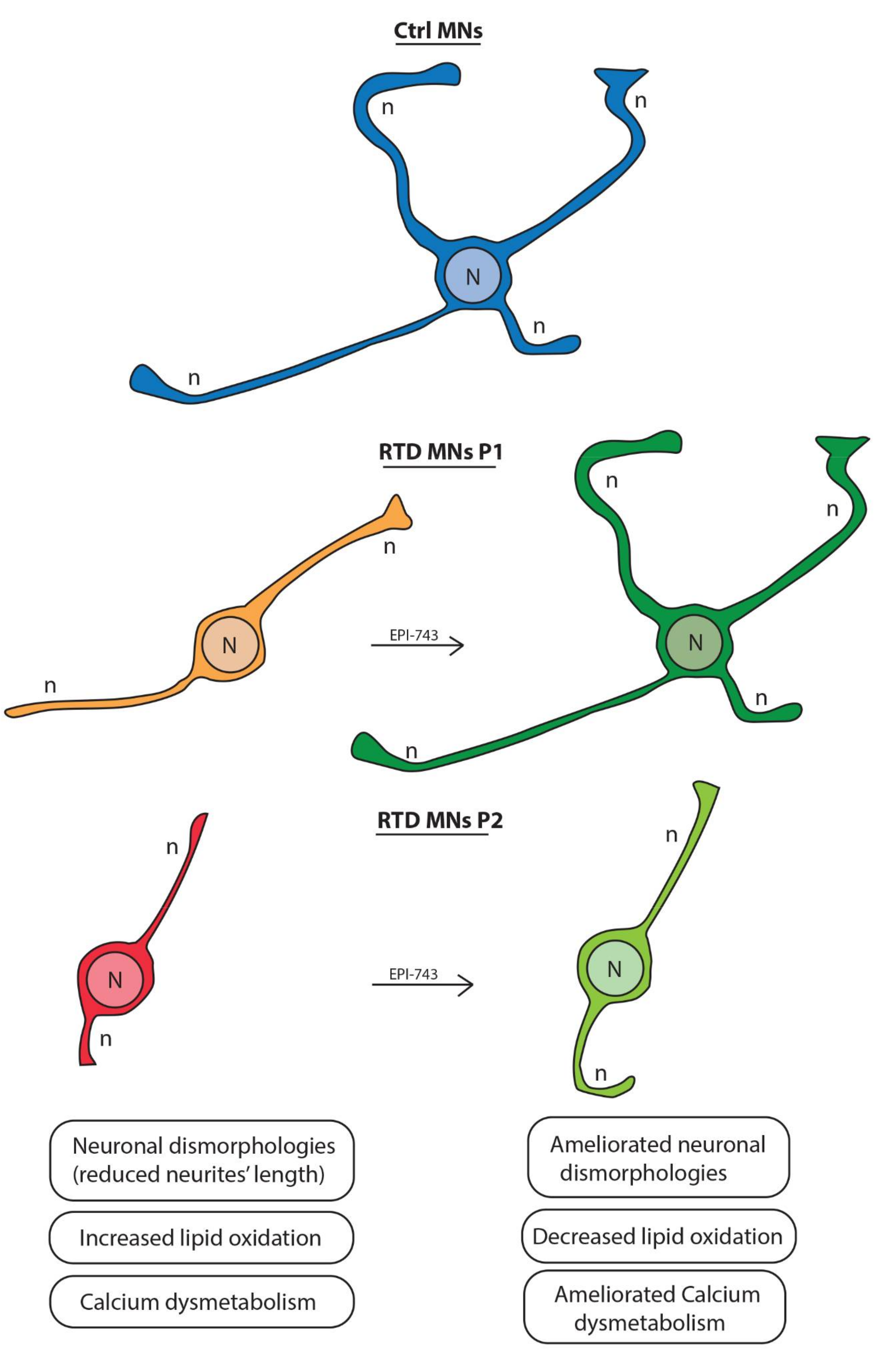
2.3. Morphological Analyses Show that EPI-743 Ameliorates the RTD Phenotype
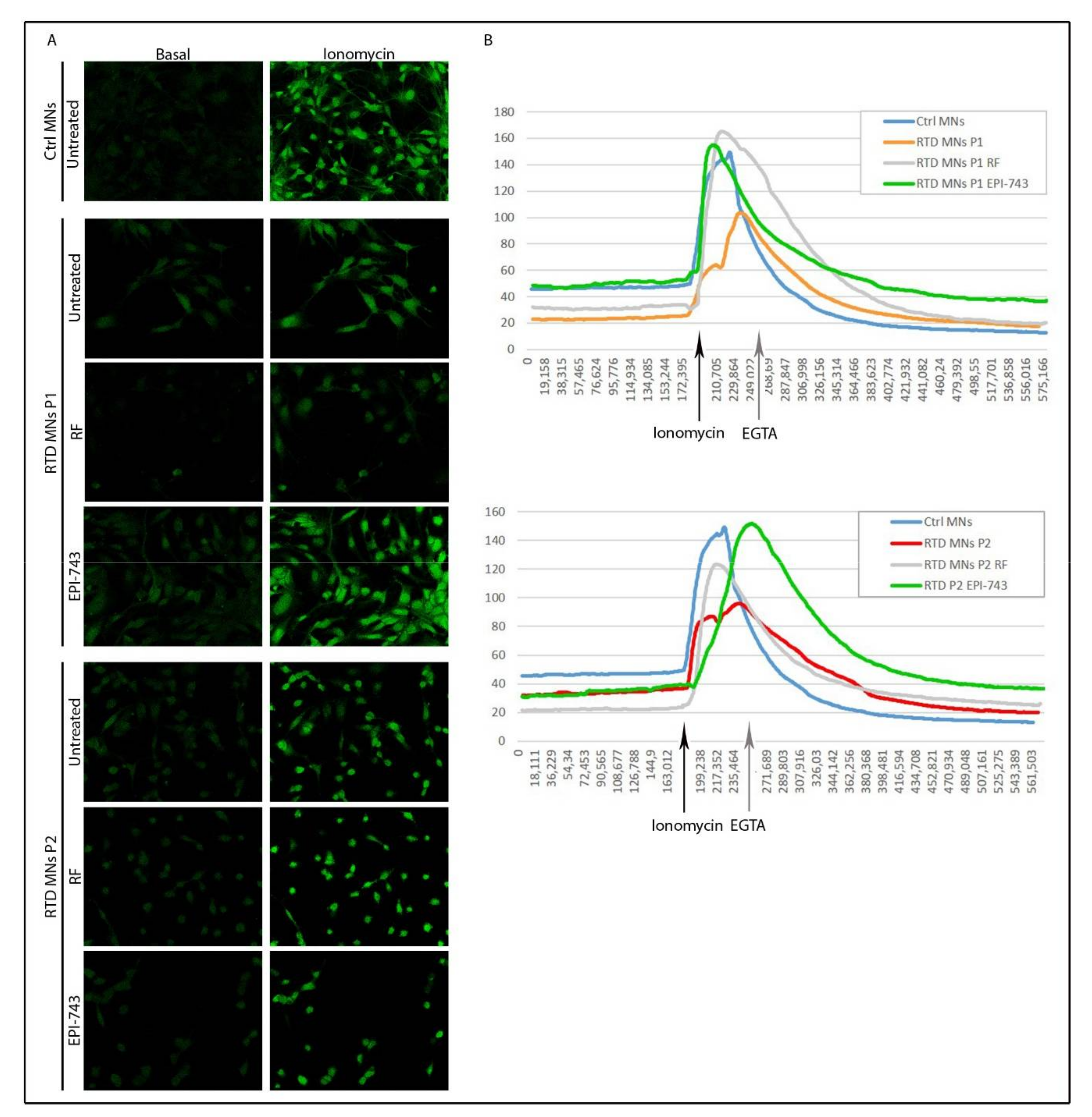
2.4. EPI-743 Has a Beneficial Effect on the Intracellular Calcium Levels in RTD MNs
3. Discussion
4. Materials and Methods
4.1. Derivation of iPSCs:
4.2. Maintenance of iPSCs
4.3. Drug Treatments
4.4. BODIPY Staining
4.5. Differentiation of iPSCs into Motor Neurons
4.6. MitoSOX Red Assay
4.7. Immunofluorescence
4.8. Morphometric Analysis
4.9. Confocal Microscopy
4.10. Calcium Imaging
4.11. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Ascorbic acid |
| AD | Alzheimer disease |
| ATP | Adenosine triphosphate |
| BVVL | Brown-Vialetto-Van Laere syndrome |
| β-III-Tub | β III tubulin |
| Ca2+ | Intracellular calcium |
| COQ10 | Coenzyme Q10 |
| GSH | Glutathione System |
| HD | Huntington’s disease |
| IDEB | Idebenone |
| LHON | Leber hereditary optic neuropathy |
| MNs | Motoneurons |
| ND | Neurodegenerative disease |
| PD | Parkinson’s disease |
| RF | Riboflavin |
| RFTV | Riboflavin transporter |
| ROS | Reactive oxygen species |
| RTD | Riboflavin transporter deficiency |
References
- Zeevalk, G.D.; Bernard, L.P.; Song, C.; Gluck, M.; Ehrhart, J. Mitochondrial inhibition and oxidative stress: Reciprocating players in neurodegeneration. Antioxid. Redox Sign. 2005, 7, 1117–1139. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharm. Exp. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Barja, G. Mitochondrial oxygen radical generation and leak: Sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 1999, 31, 347–366. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.; Zamboni, P.; Mahajan, R. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. CN 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mischley, L.K.; Allen, J.; Bradley, R. Coenzyme Q10 deficiency in patients with parkinson’s disease. J. Neurol. Sci. 2012, 318, 72–75. [Google Scholar] [CrossRef] [Green Version]
- Shults, C. Coenzyme Q10 in neurodegenerative diseases. CMC 2003, 10, 1917–1921. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Calsolaro, V.; Choub, A.; Siciliano, G. Coenzyme Q10 and neurological diseases. Pharmaceuticals 2009, 2, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.; Matson, S.; Matson, W.R.; Cormier, K.; Del Signore, S.J.; Hagerty, S.W.; Stack, E.C.; Ryu, H.; Ferrante, R.J. Dose ranging and efficacy study of high-dose Coenzyme Q10 formulations in Huntington’s disease mice. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 616–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q10 supplementation in aging and disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Carelli, V.; La Morgia, C.; Valentino, M.L.; Rizzo, G.; Carbonelli, M.; De Negri, A.M.; Sadun, F.; Carta, A.; Guerriero, S.; Simonelli, F.; et al. Idebenone treatment in Leber’s Hereditary Optic Neuropathy. Brain 2011, 134, e188. [Google Scholar] [CrossRef]
- Haginoya, K.; Miyabayashi, S.; Kikuchi, M.; Kojima, A.; Yamamoto, K.; Omura, K.; Uematsu, M.; Hino-Fukuyo, N.; Tanaka, S.; Tsuchiya, S. Efficacy of Idebenone for respiratory failure in a patient with leigh syndrome: A long-term follow-up study. J. Neurol. Sci. 2009, 278, 112–114. [Google Scholar] [CrossRef]
- Meier, T.; Buyse, G. Idebenone: An emerging therapy for Friedreich Ataxia. J. Neurol. 2009, 256, 25–30. [Google Scholar] [CrossRef]
- Rötig, A.; Sidi, D.; Munnich, A.; Rustin, P. Molecular insights into Friedreich’s Ataxia and antioxidant-based therapies. Trends. Mol. Med. 2002, 8, 221–224. [Google Scholar] [CrossRef]
- Yamada, K.; Tanaka, T.; Han, D.; Senzaki, K.; Kameyama, T.; Nabeshima, T. Protective effects of Idebenone and α-Tocopherol on β-Amyloid-(1-42)-induced learning and memory deficits in rats: Implication of oxidative stress in β-amyloid-induced neurotoxicity in vivo: Oxidative stress and β-amyloid-induced memory deficits. Eur. J. Neurosci. 1999, 11, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Mestre, T.; Ferreira, J.; Coelho, M.M.; Rosa, M.; Sampaio, C. Therapeutic interventions for symptomatic treatment in Huntington’s Disease. Cochrane Database Syst Rev. 2009, 3, CD006456. [Google Scholar] [CrossRef] [PubMed]
- Gillis, J.C.; Benfield, P.; McTavish, D. Idebenone: A Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Use in Age-Related Cognitive Disorders. Drugs Aging 1994, 5, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Zs-Nagy, I. Chemistry, toxicology, pharmacology and pharmacokinetics of idebenone: A review. Arch. Gerontol. Geriatr. 1990, 11, 177–186. [Google Scholar] [CrossRef]
- Giorgio, V.; Petronilli, V.; Ghelli, A.; Carelli, V.; Rugolo, M.; Lenaz, G.; Bernardi, P. The effects of idebenone on mitochondrial bioenergetics. BBA Bioenerg. 2012, 1817, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Esposti, M.D.; Ngo, A.; Ghelli, A.; Benelli, B.; Carelli, V.; McLennan, H.; Linnane, A.W. The interaction of Q analogs, particularly hydroxydecyl benzoquinone (idebenone), with the respiratory complexes of heart mitochondria. Arch. Biochem. Biophys. 1996, 330, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.K.; Russell, O.M.; Lightowlers, R.N.; Turnbull, D.M. Potential compounds for the treatment of mitochondrial disease. Br. Med. Bull. 2015, 116, 5–18. [Google Scholar] [CrossRef]
- Kaufman, S. Coenzymes and hydroxylases: Ascorbate and dopamine-beta-hydroxylase; tetrahydropteridines and phenylalanine and tyrosine hydroxylases. Pharm. Rev. 1966, 18, 61–69. [Google Scholar]
- Rebec, G.V.; Christopher Pierce, R. A Vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog. Neurobiol. 1994, 43, 537–565. [Google Scholar] [CrossRef]
- Majewska, M.D.; Bell, J.A.; London, E.D. Regulation of the NMDA receptor by redox phenomena: Inhibitory role of ascorbate. Brain Res. 1990, 537, 328–332. [Google Scholar] [CrossRef]
- Pastore, A.; Petrillo, S.; Tozzi, G.; Carrozzo, R.; Martinelli, D.; Dionisi-Vici, C.; Di Giovamberardino, G.; Ceravolo, F.; Klein, M.B.; Miller, G.; et al. Glutathione: A redox signature in monitoring epi-743 therapy in children with mitochondrial encephalomyopathies. Mol. Genet. Metab. 2013, 109, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P.; Sheena, Y.; Land, J.M.; Heales, S.J.R. Glutathione deficiency in patients with mitochondrial disease: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2005, 28, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 reverses the progression of the pediatric mitochondrial disease—Genetically defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar] [CrossRef]
- Kahn-Kirby, A.H.; Amagata, A.; Maeder, C.I.; Mei, J.J.; Sideris, S.; Kosaka, Y.; Hinman, A.; Malone, S.A.; Bruegger, J.J.; Wang, L.; et al. Targeting ferroptosis: A novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy. PLoS ONE 2019, 14, e0214250. [Google Scholar] [CrossRef] [Green Version]
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-Blind, randomized and controlled trial of EPI-743 in Friedreich’s Ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef]
- Jaeger, B.; Bosch, A.M. Clinical presentation and outcome of Riboflavin Transporter Deficiency: Mini review after five years of experience. J. Inherit. Metab. Dis. 2016, 39, 559–564. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, B.; Bosch, A.M.; Houlden, H. An update on the genetics, clinical presentation, and pathomechanisms of human Riboflavin Transporter Deficiency. J. Inherit. Metab. Dis. 2019, 42, 598–607. [Google Scholar] [CrossRef]
- Koy, A.; Pillekamp, F.; Hoehn, T.; Waterham, H.; Klee, D.; Mayatepek, E.; Assmann, B. Brown-Vialetto-Van Laere Syndrome: A riboflavin-unresponsive patient with a novel mutation in the C20orf54 gene. Pediatr. Neurol. 2012, 46, 407–409. [Google Scholar] [CrossRef]
- Bosch, A.M.; Abeling, N.G.G.M.; IJlst, L.; Knoester, H.; van der Pol, W.L.; Stroomer, A.E.M.; Wanders, R.J.; Visser, G.; Wijburg, F.A.; Duran, M.; et al. Brown-Vialetto-Van Laere and Fazio Londe Syndrome is associated with a riboflavin transporter defect mimicking mild MADD: A new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis. 2011, 34, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Anand, G.; Hasan, N.; Jayapal, S.; Huma, Z.; Ali, T.; Hull, J.; Blair, E.; Mcshane, T.; Jayawant, S. Early Use of high-dose riboflavin in a case of Brown-Vialetto-Van Laere Syndrome: Case report. Dev. Med. Child. Neurol. 2012, 54, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett Syndrome using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andoh-Noda, T.O.; Inouye, M.; Miyake, K.; Kubota, T.; Okano, H.; Akamatsu, W. Modeling Rett Syndrome using Human Induced Pluripotent Stem Cells. CNSNDDT 2016, 15, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Auré, K.; et al. Human IPSC-derived neural progenitors are an effective drug discovery model for neurological MtDNA disorders. Cell Stem Cell 2017, 20, 659–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, F.; Ramirez, A.; Compagnucci, C.; Salani, S.; Melzi, V.; Bordoni, A.; Fortunato, F.; Niceforo, A.; Bresolin, N.; Comi, G.P.; et al. Genome-Wide RNA-Seq of IPSC-derived motor neurons indicates selective cytoskeletal perturbation in Brown–Vialetto Disease that is partially rescued by riboflavin. Sci. Rep. 2017, 7, 46271. [Google Scholar] [CrossRef]
- Colasuonno, F.; Niceforo, A.; Marioli, C.; Fracassi, A.; Stregapede, F.; Massey, K.; Tartaglia, M.; Bertini, E.; Compagnucci, C.; Moreno, S. Mitochondrial and peroxisomal alterations contribute to energy dysmetabolism in Riboflavin Transporter Deficiency. Oxid. Med. Cell Long. 2020, 6821247. [Google Scholar] [CrossRef]
- Niceforo, A.; Marioli, C.; Colasuonno, F.; Petrini, S.; Massey, K.; Tartaglia, M.; Bertini, E.; Moreno, S.; Compagnucci, C. Altered Cytoskeletal Arrangement in Induced Pluripotent Stem Cells (IPSCs) and motor neurons from patients with Riboflavin Transporter Deficiency (RTD). Dis. Model. Mech. Submitted in July 2020.
- Compagnucci, C.; Di Siena, S.; Bustamante, M.B.; Di Giacomo, D.; Di Tommaso, M.; Maccarrone, M.; Grimaldi, P.; Sette, C. Type-1 (CB1) cannabinoid receptor promotes neuronal differentiation and maturation of neural stem cells. PLoS ONE 2013, 8, e54271. [Google Scholar] [CrossRef] [Green Version]
- Corti, S.; Nizzardo, M.; Simone, C.; Falcone, M.; Donadoni, C.; Salani, S.; Rizzo, F.; Nardini, M.; Riboldi, G.; Magri, F.; et al. Direct reprogramming of human astrocytes into neural stem cells and neurons. Exp. Cell Res. 2012, 318, 1528–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, H.C.; Cleveland, D.W. Differential utilization of beta-tubulin isotypes in differentiating neurites. J. Cell Biol. 1989, 109, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B. Calcium signalling in health and disease. Semin. Cell Dev. Biol. 2019, 94, 1–2. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Petrillo, S.; Bertini, E.S.; Piemonte, F. Oxidative stress in DNA repeat expansion disorders: A focus on NRF2 signaling involvement. Biomolecules 2020, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 signaling network defines clinical biomarkers and therapeutic opportunity in Friedreich’s Ataxia. IJMS 2020, 21, 916. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Hamilton, R.; Jernigan, A.; Zhang, Y.; Sabia, M.; Rahman, M.M.; Li, Y.; Wei, R.; Chaudhuri, A.; Van Remmen, H. Genetic ablation of 12/15-Lipoxygenase but not 5-Lipoxygenase protects against denervation-induced muscle atrophy. Free Radic. Biol. Med. 2014, 67, 30–40. [Google Scholar] [CrossRef]
- Habouri, L.; El Mansouri, F.E.; Ouhaddi, Y.; Lussier, B.; Pelletier, J.-P.; Martel-Pelletier, J.; Benderdour, M.; Fahmi, H. Deletion of 12/15-Lipoxygenase accelerates the development of aging-associated and instability-induced osteoarthritis. Osteoarthr. Cart. 2017, 25, 1719–1728. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the treatment of Friedreich’s Ataxia: A comparison among drugs. IJMS 2019, 20, 5211. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 induction re-establishes a proper neuronal differentiation program in Friedreich’s Ataxia neural stem cells. Front. Cell. Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Yan, X.; Wintergerst, K.A.; Cai, L.; Keller, B.B.; Tan, Y. Nrf2: Redox and metabolic regulator of stem cell state and function. Trends. Mol. Med. 2020, 26, 185–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochmuth, C.E.; Biteau, B.; Bohmann, D.; Jasper, H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in drosophila. Cell Stem Cell 2011, 8, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, A.A.; Singh, A.; Matsui, W.; Biswal, S. The redox-sensitive transcription factor Nrf2 regulates murine hematopoietic stem cell survival independently of ROS levels. Blood 2011, 118, 6572–6579. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.; Wang, Y.; Kim, H.-S.; Lalli, M.A.; Kosik, K.S. Nrf2, a regulator of the proteasome, controls self-renewal and pluripotency in Human Embryonic Stem Cells: Nrf2-proteasome pathway controls stemness in HESCs. Stem Cells 2014, 32, 2616–2625. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, K.E.; Joy, S.; Delhove, J.M.K.M.; Kotiadis, V.N.; Fernandez, E.; Fitzpatrick, L.M.; Whiteford, J.R.; King, P.J.; Bolanos, J.P.; Duchen, M.R.; et al. NRF2 orchestrates the metabolic shift during Induced Pluripotent Stem Cell reprogramming. Cell Rep. 2016, 14, 1883–1891. [Google Scholar] [CrossRef] [Green Version]
- Brasaemle, D.L.; Rubin, B.; Harten, I.A.; Gruia-Gray, J.; Kimmel, A.R.; Londos, C. Perilipin a increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis. J. Biol. Chem. 2000, 275, 38486–38493. [Google Scholar] [CrossRef] [Green Version]
- Gocze, P.M.; Freeman, D.A. Factors underlying the variability of lipid droplet fluorescence in MA-10 Leydig tumor cells. Cytometry 1994, 17, 151–158. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Drummen, G.P.C.; van Liebergen, L.C.M.; Op den Kamp, J.A.F.; Post, J.A. C11-BODIPY581/591, an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Ciccolella, M.; Corti, S.; Catteruccia, M.; Petrini, S.; Tozzi, G.; Rizza, T.; Carrozzo, R.; Nizzardo, M.; Bordoni, A.; Ronchi, D.; et al. Riboflavin Transporter 3 involvement in infantile Brown-Vialetto-Van Laere Disease: Two novel mutations. J. Med. Genet. 2013, 50, 104–107. [Google Scholar] [CrossRef]
- Glaser, T.; Castillo, A.R.G.; Oliveira, Á.; Ulrich, H. Intracellular calcium measurements for functional characterization of neuronal phenotypes. Methods Mol. Biol. 2016, 1341, 245–255. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marioli, C.; Magliocca, V.; Petrini, S.; Niceforo, A.; Borghi, R.; Petrillo, S.; La Rosa, P.; Colasuonno, F.; Persichini, T.; Piemonte, F.; et al. Antioxidant Amelioration of Riboflavin Transporter Deficiency in Motoneurons Derived from Patient-Specific Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 7402. https://doi.org/10.3390/ijms21197402
Marioli C, Magliocca V, Petrini S, Niceforo A, Borghi R, Petrillo S, La Rosa P, Colasuonno F, Persichini T, Piemonte F, et al. Antioxidant Amelioration of Riboflavin Transporter Deficiency in Motoneurons Derived from Patient-Specific Induced Pluripotent Stem Cells. International Journal of Molecular Sciences. 2020; 21(19):7402. https://doi.org/10.3390/ijms21197402
Chicago/Turabian StyleMarioli, Chiara, Valentina Magliocca, Stefania Petrini, Alessia Niceforo, Rossella Borghi, Sara Petrillo, Piergiorgio La Rosa, Fiorella Colasuonno, Tiziana Persichini, Fiorella Piemonte, and et al. 2020. "Antioxidant Amelioration of Riboflavin Transporter Deficiency in Motoneurons Derived from Patient-Specific Induced Pluripotent Stem Cells" International Journal of Molecular Sciences 21, no. 19: 7402. https://doi.org/10.3390/ijms21197402
APA StyleMarioli, C., Magliocca, V., Petrini, S., Niceforo, A., Borghi, R., Petrillo, S., La Rosa, P., Colasuonno, F., Persichini, T., Piemonte, F., Massey, K., Tartaglia, M., Moreno, S., Bertini, E., & Compagnucci, C. (2020). Antioxidant Amelioration of Riboflavin Transporter Deficiency in Motoneurons Derived from Patient-Specific Induced Pluripotent Stem Cells. International Journal of Molecular Sciences, 21(19), 7402. https://doi.org/10.3390/ijms21197402