Serum-Based Proteomics Profiling in Adult Patients with Cystic Fibrosis

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Clinical Characteristics of Study Subjects
2.2. Mass Spectrometry Identification of Differentially Expressed Proteins on 2D-DIGE
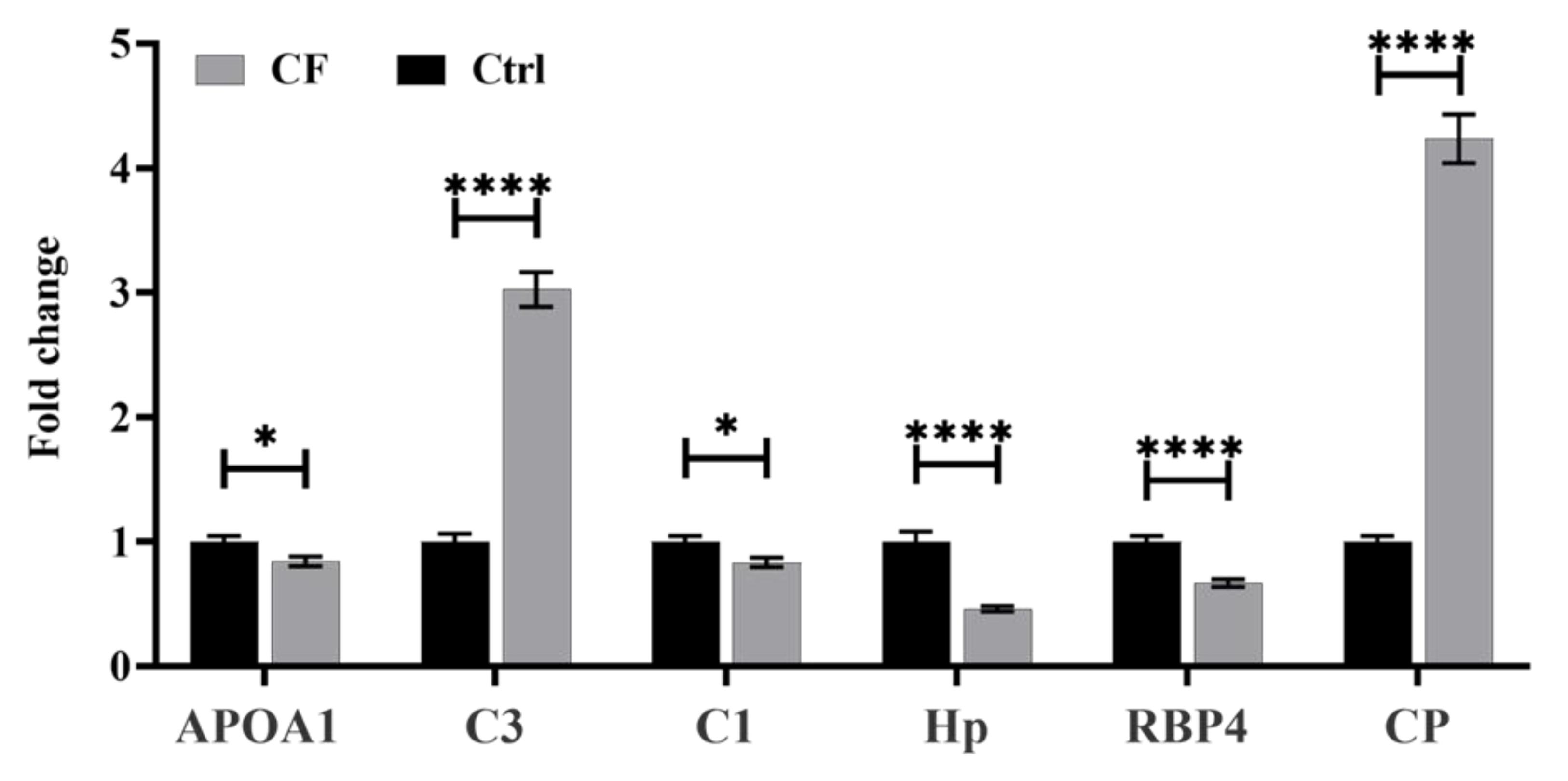
2.3. Methodology Validation Using Multiple Reaction Monitoring (MRM) Mass Spectrometry
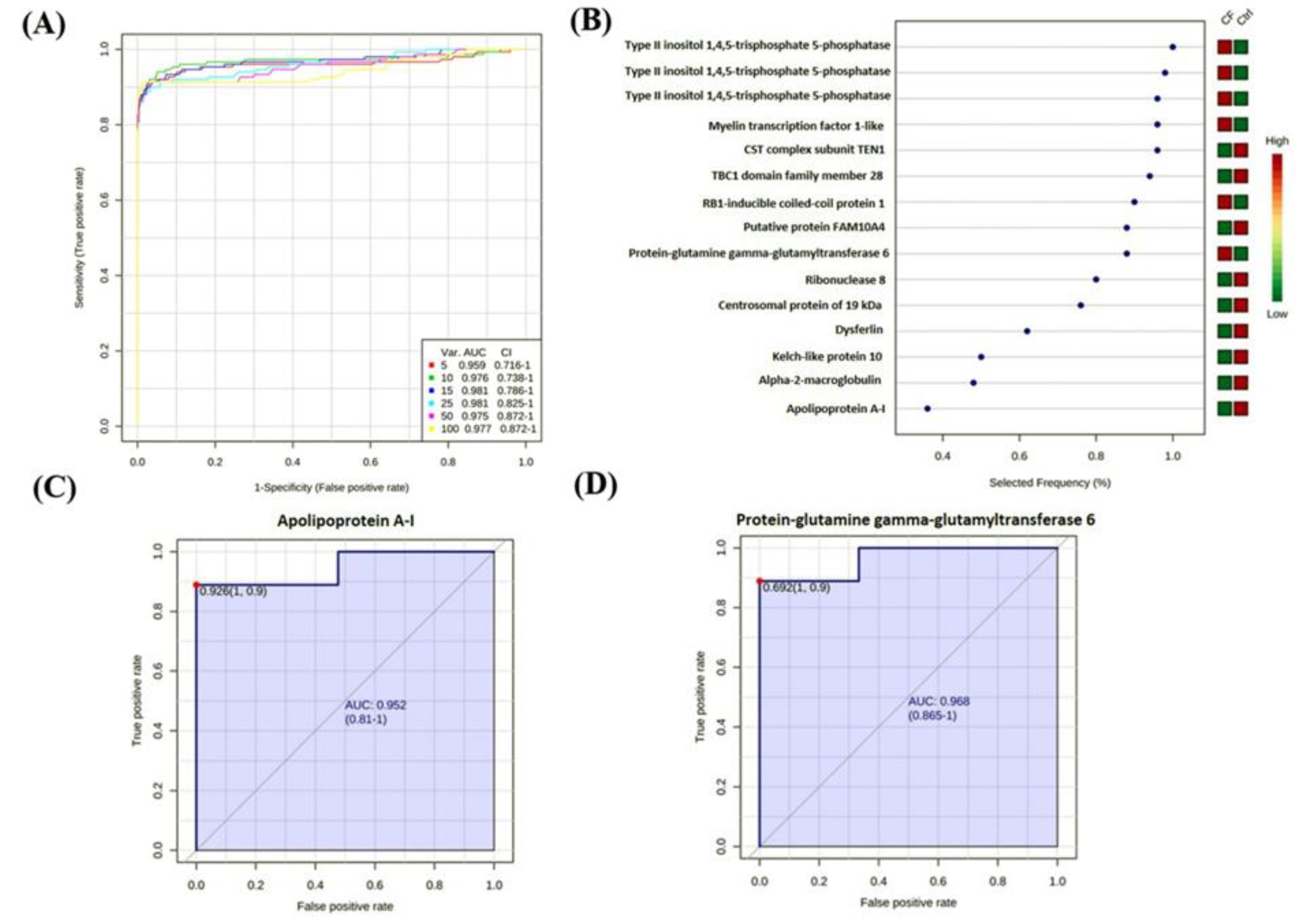
2.4. Principal Component Analysis (PCA) and Cluster Analysis
2.5. Biomarker Evaluation and Analysis
2.6. Interactions of Identified Proteins and Network Connectivity Mapping Asing an Ingenuity Pathway Analysis (IPA)
2.7. Classification of Key Proteins Based On Function
3. Discussion
3.1. Proteases and Antiproteases
3.2. Complement System
3.3. Redox Status and Antioxidants
3.4. Mitochondrial Proteins
3.5. Lipid Metabolism
3.6. Vitamin Transporters
3.7. Network Pathway Analysis
4. Materials and Methods
4.1. Ethical Considerations and Informed Consent
4.2. Study Design and Subjects
4.3. Sample Processing and Protein Extraction
4.4. CyDye Labeling, 2D-DIGE, and Imaging
4.5. Protein Identification by MALDI-TOF MS
4.6. Multiple Reaction Monitoring-Tandem Mass Spectrometry for Validation
4.7. Bioinformatic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| CF | Cystic fibrosis |
| CFRD | CF-related diabetes |
| CFTR | CF transmembrane conductance regulator |
| DBS | Dried blood spot |
| DIG | Differential in-gel electrophoresis |
| ER | Endoplasmic reticulum |
| GGT | Gamma-glutamyl transferase |
| GST | Glutathione S-transferase |
| IPA | Ingenuity pathway analysis |
| IRB | Institutional Review Board |
| IRT | Immunoreactive trypsinogen |
| KFSHRC | King Faisal Specialist Hospital and Research Centre |
| KKUH | King Khalid University Hospital |
| LIPL | Lipoprotein lipase |
| MAPK | Mitogen-activated protein kinase |
| MRM | Multiple reaction monitoring |
| MS | Mass spectrometry |
| NCGT | National Center for Genomic Technology |
| NE | Neutrophil elastase |
| PANTHER | Protein analysis through evolutionary relationships |
| PCA | Principal component analysis |
| PMF | Peptide mass fingerprinting |
| ROC | Receiver operating characteristic |
| ROS | Reactive oxygen species |
| TOF | Time-of-flight |
| FDR | The false discovery rate |
References
- Rauniyar, N.; Gupta, V.; Balch, W.E.; Yates, J.R., III. Quantitative proteomic profiling reveals differentially regulated proteins in cystic fibrosis cells. J. Proteome Res. 2014, 13, 4668–4675. [Google Scholar] [CrossRef] [PubMed]
- Kamath, K.S.; Kumar, S.S.; Kaur, J.; Venkatakrishnan, V.; Paulsen, I.T.; Nevalainen, H.; Molloy, M.P. Proteomics of hosts and pathogens in cystic fibrosis. Proteom. Clin. Appl. 2015, 9, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, L.; Corey, M.; Zou, F.; Lee, S.; Cojocaru, A.L.; Taylor, C.; Blackman, S.M.; Stephenson, A.; Sandford, A.J.; et al. Understanding the population structure of North American patients with cystic fibrosis. Clin. Genet. 2011, 79, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Banjar, H.; Kambouris, M.; Meyer, B.; Al-Mehaidib, A.; Mogarri, I. Geographic distribution of cystic fibrosis transmembrane regulator gene mutations in Saudi Arabia. Ann. Trop. Paediatr. 1999, 19, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Puglia, M.; Landi, C.; Gagliardi, A.; Breslin, L.; Armini, A.; Brunetti, J.; Pini, A.; Bianchi, L.; Bini, L. The proteome speciation of an immortalized cystic fibrosis cell line: New perspectives on the pathophysiology of the disease. J. Proteom. 2018, 170, 28–42. [Google Scholar] [CrossRef]
- Hunt, A.; Glasgow, A.; Humphreys, H.; Greene, C.M. Alpha-1 Antitrypsin—A Target for MicroRNA-Based Therapeutic Development for Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 836. [Google Scholar] [CrossRef] [Green Version]
- Balch, W.E.; Yates, J.R. Application of mass spectrometry to study proteomics and interactomics in cystic fibrosis. In Cystic Fibrosis; Springer: Berlin/Heidelberg, Germany, 2011; pp. 227–247. [Google Scholar]
- Peretti, N.; Roy, C.C.; Drouin, E.; Seidman, E.; Brochu, P.; Casimir, G.; Levy, E. Abnormal intracellular lipid processing contributes to fat malabsorption in cystic fibrosis patients. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G609–G615. [Google Scholar] [CrossRef] [Green Version]
- Jeanson, L.; Guerrera, I.C.; Papon, J.-F.; Chhuon, C.; Zadigue, P.; Prulière-Escabasse, V.; Amselem, S.; Escudier, E.; Coste, A.; Edelman, A. Proteomic analysis of nasal epithelial cells from cystic fibrosis patients. PLoS ONE 2014, 9, e108671. [Google Scholar] [CrossRef] [Green Version]
- Oberdorf, J.; Carlson, E.J.; Skach, W.R. Uncoupling proteasome peptidase and ATPase activities results in cytosolic release of an ER polytopic protein. J. Cell Sci. 2006, 119, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Okiyoneda, T.; Harada, K.; Takeya, M.; Yamahira, K.; Wada, I.; Shuto, T.; Suico, M.A.; Hashimoto, Y.; Kai, H. Delta F508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol. Biol. Cell 2004, 15, 563–574. [Google Scholar] [CrossRef]
- Pattison, S.H.; Elborn, J.S. Protein biomarkers in cystic fibrosis research: Where next? Genome medicine 2010, 2, 88. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Braag, S.A.; Keeler, A.; Hodges, C.; Drumm, M.; Flotte, T.R. Lack of cystic fibrosis transmembrane conductance regulator in CD3+ lymphocytes leads to aberrant cytokine secretion and hyperinflammatory adaptive immune responses. Am. J. Respir. Cell Mol. Biol. 2011, 44, 922–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloane, A.J.; Lindner, R.A.; Prasad, S.S.; Sebastian, L.T.; Pedersen, S.K.; Robinson, M.; Bye, P.T.; Nielson, D.W.; Harry, J.L. Proteomic analysis of sputum from adults and children with cystic fibrosis and from control subjects. Am. J. Respir. Crit. Care Med. 2005, 172, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Charro, N.; Hood, B.L.; Faria, D.; Pacheco, P.; Azevedo, P.; Lopes, C.; de Almeida, A.B.; Couto, F.M.; Conrads, T.P.; Penque, D. Serum proteomics signature of Cystic Fibrosis patients: A complementary 2-DE and LC–MS/MS approach. J. Proteom. 2011, 74, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Moon, K.-M.; Chen, V.; Ng, R.; Foster, L.J.; Tebbutt, S.J.; Quon, B.S. Identification of novel blood biomarkers of treatment response in cystic fibrosis pulmonary exacerbations by label-free quantitative proteomics. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- DeBoer, E.M.; Wagner, B.D.; Popler, J.; Harris, J.K.; Zemanick, E.T.; Accurso, F.J.; Sagel, S.D.; Deterding, R.R. Novel application of aptamer proteomic analysis in cystic fibrosis bronchoalveolar lavage fluid. Proteom. Clin. Appl. 2019, 13, 1800085. [Google Scholar] [CrossRef]
- Pattison, S.H.; Gibson, D.S.; Johnston, E.; Peacock, S.; Rivera, K.; Tunney, M.M.; Pappin, D.J.; Elborn, J.S. Proteomic profile of cystic fibrosis sputum cells in adults chronically infected with Pseudomonas aeruginosa. Eur. Respir. J. 2017, 50, 1601569. [Google Scholar] [CrossRef] [Green Version]
- Banjar, H.H.; Tuleimat, L.; El Seoudi AA, A.; Mogarri, I.; Alhaider, S.; Nizami, I.Y.; AlMaghamsi, T.; Alkaf, S.A.; Moghrabi, N. Genotype patterns for mutations of the cystic fibrosis transmembrane conductance regulator gene: A retrospective descriptive study from Saudi Arabia. Annal. Saudi Med. 2020, 40, 15–24. [Google Scholar] [CrossRef]
- Ramalho, A.S.; Clarke, L.A.; Sousa, M.; Felicio, V.; Barreto, C.; Lopes, C.; Amaral, M.D. Comparative ex vivo, in vitro and in silico analyses of a CFTR splicing mutation: Importance of functional studies to establish disease liability of mutations. J. Cyst. Fibros. 2016, 15, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Al-Qahtani, W.S.; Abduljabbar, M.; AlSuhaibani, E.S.; Abdel Rahman, A.; Aljada, A. Quantification of the lamin A/C transcript variants in cancer cell lines by targeted absolute quantitative proteomics and correlation with mRNA expression. Int. J. Mol. Sci. 2019, 20, 1902. [Google Scholar] [CrossRef] [Green Version]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hair, P.S.; Sass, L.A.; Vazifedan, T.; Shah, T.A.; Krishna, N.K.; Cunnion, K.M. Complement effectors, C5a and C3a, in cystic fibrosis lung fluid correlate with disease severity. PLoS ONE 2017, 12, e0173257. [Google Scholar] [CrossRef] [PubMed]
- Cater, J.H.; Wilson, M.R.; Wyatt, A.R. Alpha-2-macroglobulin, a hypochlorite-regulated chaperone and immune system modulator. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cserhalmi, M.; Papp, A.; Brandus, B.; Uzonyi, B.; Józsi, M. Regulation of Regulators: Role of the Complement Factor H-Related Proteins. In Seminars in Immunology; Elsevier: Amsterdam, The Netherlands, 2019; p. 101341. [Google Scholar]
- Roum, J.; Buhl, R.; McElvaney, N.; Borok, Z.; Crystal, R. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Groffen, J.; Warburton, D.; Sneddon, T.P. The human gamma-glutamyltransferase gene family. Hum. Genet. 2008, 123, 321–332. [Google Scholar] [CrossRef]
- Bielli, P.; Calabrese, L. Structure to function relationships in ceruloplasmin: A’moonlighting’protein. Cell. Mol. Life Sci. 2002, 59, 1413–1427. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, G.; Zhang, X.; Hüttemann, P.P.; Qiu, Y.; Liu, J.; Mitchell, A.; Lee, I.; Zhang, C.; Lee, J.-S. COX7AR is a stress-inducible mitochondrial COX subunit that promotes breast cancer malignancy. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Boehm, J.; Lee, J.C. p38 MAP kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef]
- Raia, V.; Maiuri, L.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Auricchio, S.; Cimmino, M.; Cavaliere, M.; Nardone, M.; Cesaro, A.; et al. Inhibition of p38 mitogen activated protein kinase controls airway inflammation in cystic fibrosis. Thorax 2005, 60, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Harris, W.T.; Kortyka, S.; Kotha, K.; Ostmann, A.J.; Rezayat, A.; Sridharan, A.; Sanders, Y.; Naren, A.P.; Clancy, J.P. Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS ONE 2014, 9, e106842. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, W.; Abdel Jabar, M.; Masood, A.; Jacob, M.; Nizami, I.; Dasouki, M.; Abdel Rahman, A.M. Dried blood spot-based metabolomic profiling in adults with cystic fibrosis. J. Proteome Res. 2020, 19, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Lin, S.Y.; Yeh, Y.Y.; Hsiao, H.H.; Wu, C.Y.; Chen, S.T.; Wang AH, J. A modified protein precipitation procedure for efficient removal of albumin from serum. Electrophoresis 2005, 26, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Benabdelkamel, H.; Masood, A.; Jammah, A.A.; Ekhzaimy, A.A. Differences in the plasma proteome of patients with hypothyroidism before and after thyroid hormone replacement: A proteomic analysis. Int. J. Mol. Sci. 2018, 19, 88. [Google Scholar] [CrossRef] [Green Version]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Benabdelkamel, H.; Masood, A.; Moustafa, A.; Sallam, R.; Bassas, A.; Duncan, M. Proteomic analysis of mature adipocytes from obese patients in relation to aging. Exp. Gerontol. 2013, 48, 1196–1203. [Google Scholar] [CrossRef]
- Benabdelkamel, H.; Masood, A.; Almidani, G.M.; Alsadhan, A.A.; Bassas, A.F.; Duncan, M.W.; Alfadda, A.A. Mature adipocyte proteome reveals differentially altered protein abundances between lean, overweight and morbidly obese human subjects. Mol. Cell. Endocrinol. 2015, 401, 142–154. [Google Scholar] [CrossRef]
- Benabdelkamel, H.; Masood, A.; Okla, M.; Al-Naami, M.Y.; Alfadda, A.A. A Proteomics-Based Approach Reveals Differential Regulation of Urine Proteins between Metabolically Healthy and Unhealthy Obese Patients. Int. J. Mol. Sci. 2019, 20, 4905. [Google Scholar] [CrossRef] [Green Version]
- Malkawi, A.K.; Masood, A.; Shinwari, Z.; Jacob, M.; Benabdelkamel, H.; Matic, G.; Almuhanna, F.; Dasouki, M.; Alaiya, A.A.; Rahman, A.M.A. Proteomic Analysis of Morphologically Changed Tissues after Prolonged Dexamethasone Treatment. Int. J. Mol. Sci. 2019, 20, 3122. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Age (years) | Gender | CFTR Mutation * | CFTR Protein Mutation Class | Sweat Chloride (meq/L) | FEV1 L (%) | Pancreatic Status |
|---|---|---|---|---|---|---|---|
| CF1 | 23 | M | exon 12: c.1647 T>G; p.S549R | class III | NA | 3.03(70%) | PI |
| CF2 | 22 | F | exon 11: c.1418delG; p.G473EfsX54 | class III | NA | 0.47(15%) | PI |
| CF3 | 19 | F | exon 15: c.2619 G>C); p.E873D | class III | 105 | 1.17 (46%) | PI |
| CF4 | 20 | M | exon 14: c.1911 delG; p.Q637HfsX26 (het) | class III | 110 | 1.68(35%) | PI |
| CF5 | 22 | F | exon 22: c.3700 A>G; p.I1234V | class V | 100 | 0.92(31%) | PI |
| CF6 | 17 | F | exon11:c.1416 delG; p.M472fs | class III | NA | 1.23(41%) | PI |
| CF7 | 22 | M | exon 11: c.1418 delG; p.G473EfsX54 | class III | 79 | 1.12(31%) | PI |
| CF9 | 18 | F | exon 11: c.1418 delG; p.G473EfsX54 | class III | NA | 2.08(67%) | PI |
| CF10 | 28 | M | exon 4: c.416 A>T; p.H139L | class IV | NA | 0.6(16%) | PI |
| CF11 | 26 | M | exon 22: c.3700 A>G; p.I1234V | class V | NA | NA | PI |
| CF12 | 34 | F | exon 11: c.1418del G; p.G473EfsX54/Exon 12: c.1736 A>G; p.D579G | class III | 58 | 1.23(45%) | PI |
| CF13 | 20 | M | exon 22: c.3700 A>G; p.I1234V | class V | 108 | NA | PI |
| CF14 | 17 | M | exon 12: c.1647 T>G; p.S549R (het) | class III | 124 | 3.07(84%) | PI |
| CF16 | 18 | F | exon12:c.1647T>G; p.S549R | class III | NA | 2.56(85%) | PI |
| CF22 | 14 | F | exon 4: c.416 A>T; p.H139L | class IV | NA | 0.68(24.9%) | PI |
| CF24 | 17 | F | exon 13: c.2043 delG | class III | NA | 2.32(84%) | PI |
| CF25 | 20 | F | exon 11: c.1418 delG; p.G473EfsX54 | class III | NA | 1.97(70%) | PI |
| CF26 | 26 | M | exon 22: c.3700 A>G; p.I1234V | class V | NA | 0.99 (27%) | PI |
| CF27 | 14 | M | exon11:c.1416 delG; p.M472fs (het) | class III | NA | 2.47(64%) | PI |
| CF29 | 26 | F | exon 22: c.3700 A>G; p.I1234V | class V | 60 | 2.71(88%, post TX) | PI |
| CF30 | 12 | M | exon 22: c.3700 A>G; p.I1234V | class V | 88 | 2.58(81%) | PI |
| CF31 | 24 | F | exon 22: c.3700 A>G; p.I1234V | class V | NA | 1.68(61%) | PI |
| CF32 | 20 | F | exon 22: c.3700 A>G; p.I1234V | class V | NA | 1.3(47%) | PI |
| CF33 | 22 | F | exon 22: c.3700 A>G; p.I1234V | Class V | NA | NA | NA |
| CF34 | 18 | M | exon 11: c.1416 delG; p.M472fs (het) | class III | 81 | 0.95(24%) | PI |
| CF40 | 18 | F | exon4:c.416A>T; p.H139L | class IV | NA | 0.63(16%) | PI |
| CF41 | 14 | M | exon 4: c.416 A>T; p.H139L | class IV | NA | 1.57(48%) | PI |
| CF42 | 18 | M | exon 19-21 del | class IV | NA | 3.08(77%) | PI |
| Spot No | Accession No | Protein Name | p-Value (ANOVA) | CF/C Ratio | EXP |
|---|---|---|---|---|---|
| 804 | P32019 | Type II inositol 1,4,5-trisphosphate | 7.28 × 10−14 | −13.06 | Down |
| 998 | Q9UL68 | Myelin transcription factor 1-like | 1.57 × 10−7 | −9.94 | Down |
| 811 | P32019 | Type II inositol 1,4,5-trisphosphate | 2.54 × 10−9 | −9.45 | Down |
| 853 | Q8TDY2 | RB1-inducible coiled-coil protein 1 | 5.48 × 10−11 | −9.22 | Down |
| 796 | Q8IZP2 | Putative protein FAM10A4 | 7.08 × 10−11 | −8.3 | Down |
| 987 | Q86WV5 | CST complex subunit TEN1 | 2.75 × 10−7 | −7.35 | Down |
| 792 | O95932 | Protein-glutamine gamma-glutamyltransferase 6 | 5.04 × 10−12 | −7.23 | Down |
| 932 | Q8TDE3 | Ribonuclease 8 | 1.94 × 10−9 | −6.95 | Down |
| 848 | Q96LK0 | Centrosomal protein of 19 kDa | 4.05 × 10−8 | −6.77 | Down |
| 787 | O75923 | Dysferlin | 9.09 × 10−10 | −6.4 | Down |
| 808 | Q2M2D7 | TBC1 domain family member 28 | 1.94 × 10−9 | −6.21 | Down |
| 700 | P01877 | Immunoglobulin heavy constant alpha 2 | 7.09 × 10−4 | −5.6 | Down |
| 967 | Q9UL68 | Myelin transcription factor 1-like protein | 8.76 × 10−6 | −4.2 | Down |
| 823 | P06858 | Lipoprotein lipase | 1.15 × 10−9 | −4.2 | Down |
| 863 | Q6UWF9 | Protein FAM180A | 1.87 × 10−6 | −3.9 | Down |
| 907 | P02647 | Apolipoprotein A-I | 2.95 × 10−6 | −3.36 | Down |
| 782 | P00738 | Haptoglobin | 6.59 × 10−5 | −3.15 | Down |
| 834 | O60869 | Endothelial differentiation-related factor 1 | 4.43 × 10−7 | −3.1 | Down |
| 714 | Q96QP1 | Alpha-protein kinase 1 | 2.02 × 10−5 | −2.98 | Down |
| 730 | Q6JEL2 | Kelch-like protein 10 | 1.15 × 10−5 | −2.97 | Down |
| 966 | P00738 | Haptoglobin | 0.005 | −2.94 | Down |
| 591 | O00487 | 26S proteasome non-ATPase regulatory subunit 14 | 6.53 × 10−5 | −2.82 | Down |
| 857 | Q9UL68 | Myelin transcription factor 1-like protein | 6.76 × 10−7 | −2.82 | Down |
| 763 | P62906 | 60S ribosomal protein L10a | 7.13 × 10−5 | −2.77 | Down |
| 785 | P02774 | Vitamin D-binding protein, DBP | 2.61 × 10-5 | −2.7 | Down |
| 856 | Q49AM3 | Tetratricopeptide repeat protein 31 | 3.24 × 10−8 | −2.64 | Down |
| 789 | Q99570 | Phosphoinositide 3-kinase regulatory subunit 4 | 1.09 × 10−6 | −2.55 | Down |
| 570 | P02768 | Serum albumin | 8.38 × 10−6 | −2.47 | Down |
| 893 | P02647 | Apolipoprotein A-I | 1.27 × 10−6 | −2.45 | Down |
| 829 | P24310 | Cytochrome c oxidase subunit 7A1 | 1.52 × 10−4 | −2.27 | Down |
| 919 | P02753 | Retinol-binding protein 4 | 7.97 × 10−5 | −2.25 | Down |
| 900 | P02647 | Apolipoprotein A-I | 8.06 × 10−4 | −2.16 | Down |
| 589 | P02768 | Serum albumin | 9.00 × 10−5 | −2.16 | Down |
| 1012 | Q6ZN57 | Zinc finger protein 2 homolog, Zfp-2 | 0.002 | −2.13 | Down |
| 982 | Q8TAA9 | Vang-like protein 1 | 0.013 | −2.09 | Down |
| 840 | O95922 | Probable tubulin polyglutamylase TTLL1 | 0.004 | −2.06 | Down |
| 820 | P02647 | Apolipoprotein A-I | 8.46 × 10−4 | −2.02 | Down |
| 567 | Q96PX9 | Pleckstrin | 3.10 × 10−6 | −2.02 | Down |
| 862 | P24310 | Cytochrome c oxidase subunit 7A1 | 0.044 | −1.97 | Down |
| 759 | P00738 | Haptoglobin | 0.005 | −1.95 | Down |
| 801 | Q96SI1 | BTB/POZ domain-containing protein KCTD15 | 9.35 × 10−5 | −1.88 | Down |
| 839 | P31751 | RAC-beta serine/threonine-protein kinase | 0.003 | −1.86 | Down |
| 709 | Q8TDE3 | Ribonuclease 8 | 1.61 × 10−4 | −1.86 | Down |
| 710 | P01008 | Antithrombin-III | 0.011 | −1.8 | Down |
| 896 | P02647 | Apolipoprotein A-I | 0.002 | −1.66 | Down |
| 668 | Q9BQ50 | Three prime repair exonuclease 2 | 2.32 × 10−4 | −1.62 | Down |
| 604 | P02790 | Hemopexin | 0.017 | −1.5 | Down |
| 473 | P24666 | Low molecular weight phosphotyrosine protein phosphatase | 0.002 | −1.5 | Down |
| 773 | P05156 | Complement factor I | 7.98 × 10−4 | −1.5 | Down |
| 895 | P02647 | Apolipoprotein A-I, | 0.026 | −1.35 | Down |
| 598 | P02790 | Hemopexin | 0.028 | −1.26 | Down |
| 566 | P04217 | Alpha-1B-glycoprotein | 0.02 | −1.24 | Down |
| 972 | Q9NR11 | Zinc finger protein 302 | 3.53 × 10−4 | 4.94 | Up |
| 689 | P01009 | Alpha-1-antitrypsin | 1.97 × 10−6 | 4.77 | Up |
| 908 | Q9NT22 | EMILIN-3 | 0.001 | 4.49 | Up |
| 261 | P01023 | Alpha-2-macroglobulin | 2.35 × 10−8 | 4.45 | Up |
| 916 | P01042 | Kininogen-1 | 0.004 | 4.3 | Up |
| 264 | P01023 | Alpha-2-macroglobulin | 3.13 × 10−8 | 3.786 | Up |
| 688 | P01009 | Alpha-1-antitrypsin | 5.99 × 10−6 | 3.78 | Up |
| 256 | P01023 | Alpha-2-macroglobulin | 7.45 × 10−7 | 3.68 | Up |
| 596 | P00450 | Ceruloplasmin | 2.96 × 10−5 | 3.67 | Up |
| 695 | P01009 | Alpha-1-antitrypsin | 2.16 × 10−6 | 3.6 | Up |
| 184 | P01023 | Alpha-2-macroglobulin | 1.06 × 10−9 | 3.43 | Up |
| 270 | P08603 | Complement factor H | 5.71 × 10−6 | 3.41 | Up |
| 163 | P01023 | Alpha-2-macroglobulin | 3.48 × 10−9 | 3.35 | Up |
| 243 | P01023 | Alpha-2-macroglobulin | 5.45 × 10−9 | 3.23 | Up |
| 234 | P01023 | Alpha-2-macroglobulin | 1.06 × 10−6 | 2.97 | Up |
| 771 | P01024 | Complement C3 | 8.69 × 10−6 | 2.9 | Up |
| 260 | P01023 | Alpha-2-macroglobulin | 2.21 × 10−7 | 2.85 | Up |
| 614 | P01011 | Alpha-1-antichymotrypsin | 2.30 × 10−8 | 2.85 | Up |
| 225 | P01023 | Alpha-2-macroglobulin | 6.36 × 10−7 | 2.8 | Up |
| 273 | P01023 | Alpha-2-macroglobulin | 3.16 × 10−7 | 2.8 | Up |
| 247 | P01023 | Alpha-2-macroglobulin | 5.45 × 10−9 | 2.74 | Up |
| 291 | P01023 | Alpha-2-macroglobulin | 2.00 × 10−6 | 2.7 | Up |
| 693 | P01009 | Alpha-1-antitrypsin | 0.001 | 2.68 | Up |
| 277 | P01023 | Alpha-2-macroglobulin | 1.31 × 10−6 | 2.67 | Up |
| 294 | P01023 | Alpha-2-macroglobulin | 1.37 × 10−6 | 2.66 | Up |
| 592 | P15622 | Zinc finger protein 250 | 3.30 × 10−4 | 2.65 | Up |
| 766 | Q6UXP9 | Putative uncharacterized protein | 0.004 | 2.6 | Up |
| 283 | P01023 | Alpha-2-macroglobulin | 3.64 × 10−7 | 2.58 | Up |
| 150 | Q9H2F9 | Coiled-coil domain-containing protein 68 | 1.19 × 10−6 | 2.54 | Up |
| 259 | P01023 | Alpha-2-macroglobulin | 9.20 × 10−7 | 2.53 | Up |
| 276 | P01023 | Alpha-2-macroglobulin | 3.98 × 10−7 | 2.51 | Up |
| 281 | P01023 | Alpha-2-macroglobulin | 2.08 × 10−6 | 2.5 | Up |
| 601 | P00450 | Ceruloplasmin | 0.003 | 2.43 | Up |
| 285 | P01023 | Alpha-2-macroglobulin | 1.97 × 10−6 | 2.4 | Up |
| 266 | P01023 | Alpha-2-macroglobulin | 6.86 × 10−5 | 2.38 | Up |
| 87 | P01023 | Alpha-2-macroglobulin | 2.58 × 10−6 | 2.34 | Up |
| 635 | P01042 | Kininogen-1 | 4.46 × 10−9 | 2.33 | Up |
| 289 | P01023 | Alpha-2-macroglobulin | 1.67 × 10−6 | 2.3 | Up |
| 149 | P01023 | Alpha-2-macroglobulin | 9.97 × 10−7 | 2.3 | Up |
| 334 | P07911 | Uromodulin | 3.79 × 10−6 | 2.25 | Up |
| 610 | P01011 | Alpha-1-antichymotrypsin | 1.65 × 10−6 | 2.23 | Up |
| 367 | P00450 | Ceruloplasmin | 9.27 × 10−5 | 2.18 | Up |
| 255 | P01023 | Alpha-2-macroglobulin | 8.08 × 10−6 | 2.15 | Up |
| 253 | P01023 | Alpha-2-macroglobulin | 3.64 × 10−6 | 2.12 | Up |
| 331 | P00450 | Ceruloplasmin | 7.60 × 10−6 | 2.09 | Up |
| 338 | P00450 | Ceruloplasmin | 1.83 × 10−5 | 2.07 | Up |
| 611 | Q9H0J9 | Poly [ADP-ribose] polymerase 12 | 0.003 | 2.06 | Up |
| 326 | P00450 | Ceruloplasmin | 5.77 × 10−6 | 2.05 | Up |
| 362 | P01023 | Alpha-2-macroglobulin | 2.62 × 10−5 | 2.03 | Up |
| 374 | P01023 | Alpha-2-macroglobulin | 0.002 | 2.01 | Up |
| 388 | Q14833 | Metabotropic glutamate receptor 4, mGluR4 | 0.002 | 1.96 | Up |
| 377 | P01023 | Alpha-2-macroglobulin | 0.005 | 1.93 | Up |
| 257 | P01023 | Alpha-2-macroglobulin | 4.37 × 10−5 | 1.93 | Up |
| 376 | P00450 | Ceruloplasmin | 0.004 | 1.9 | Up |
| 638 | P01042 | Kininogen-1 | 7.75 × 10−6 | 1.9 | Up |
| 405 | P00450 | Ceruloplasmin | 4.61 × 10−4 | 1.89 | Up |
| 389 | P01023 | Alpha-2-macroglobulin | 6.16 × 10−4 | 1.88 | Up |
| 332 | Q14833 | Metabotropic glutamate receptor 4 | 1.69 × 10−4 | 1.85 | Up |
| 263 | P01023 | Alpha-2-macroglobulin | 7.51 × 10−5 | 1.82 | Up |
| 408 | P43652 | Afamin | 2.83 × 10−4 | 1.76 | Up |
| 460 | P00450 | Ceruloplasmin | 0.011 | 1.75 | Up |
| 949 | Q8TDE3 | Ribonuclease 8 | 0.017 | 1.74 | UP |
| 651 | P01009 | Alpha-1-antitrypsin | 1.62 × 10−5 | 1.7 | Up |
| 645 | P01009 | Alpha-1-antitrypsin | 1.72 × 10−4 | 1.69 | Up |
| 641 | P01009 | Alpha-1-antitrypsin | 0.003 | 1.68 | Up |
| 343 | P00450 | Ceruloplasmin | 8.46 × 10−4 | 1.68 | Up |
| 659 | P01009 | Alpha-1-antitrypsin | 1.26 × 10−5 | 1.68 | Up |
| 345 | P00450 | Ceruloplasmin | 4.95 × 10−4 | 1.67 | Up |
| 656 | P01009 | Alpha-1-antitrypsin | 1.08 × 10−5 | 1.67 | Up |
| 339 | P00450 | Ceruloplasmin | 9.42 × 10−5 | 1.66 | Up |
| 649 | P01009 | Alpha-1-antitrypsin | 3.34 × 10−5 | 1.65 | Up |
| 650 | P01009 | Alpha-1-antitrypsin | 4.28 × 10−4 | 1.61 | Up |
| 642 | P01009 | Alpha-1-antitrypsin | 0.007 | 1.59 | Up |
| 351 | Q8NDZ2 | SUMO-interacting motif-containing protein 1 | 0.007 | 1.55 | Up |
| 463 | Q8TCP9 | Protein FAM200A | 0.007 | 1.51 | Up |
| 502 | P04217 | Alpha-1B-glycoprotein | 0.002 | 1.51 | Up |
| 560 | P04217 | Alpha-1B-glycoprotein | 0.008 | 1.5 | Up |
| 558 | Q9NXU5 | ADP-ribosylation factor-like protein 15 | 0.003 | 1.5 | Up |
| 404 | P00450 | Ceruloplasmin | 0.022 | 1.47 | Up |
| 465 | P04217 | Alpha-1B-glycoprotein | 0.02 | 1.37 | Up |
| 559 | P00450 | Ceruloplasmin | 0.01 | 1.36 | Up |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benabdelkamel, H.; Alamri, H.; Okla, M.; Masood, A.; Abdel Jabar, M.; Alanazi, I.O.; Alfadda, A.A.; Nizami, I.; Dasouki, M.; Abdel Rahman, A.M. Serum-Based Proteomics Profiling in Adult Patients with Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 7415. https://doi.org/10.3390/ijms21197415
Benabdelkamel H, Alamri H, Okla M, Masood A, Abdel Jabar M, Alanazi IO, Alfadda AA, Nizami I, Dasouki M, Abdel Rahman AM. Serum-Based Proteomics Profiling in Adult Patients with Cystic Fibrosis. International Journal of Molecular Sciences. 2020; 21(19):7415. https://doi.org/10.3390/ijms21197415
Chicago/Turabian StyleBenabdelkamel, Hicham, Hanadi Alamri, Meshail Okla, Afshan Masood, Mai Abdel Jabar, Ibrahim O. Alanazi, Assim A. Alfadda, Imran Nizami, Majed Dasouki, and Anas M. Abdel Rahman. 2020. "Serum-Based Proteomics Profiling in Adult Patients with Cystic Fibrosis" International Journal of Molecular Sciences 21, no. 19: 7415. https://doi.org/10.3390/ijms21197415
APA StyleBenabdelkamel, H., Alamri, H., Okla, M., Masood, A., Abdel Jabar, M., Alanazi, I. O., Alfadda, A. A., Nizami, I., Dasouki, M., & Abdel Rahman, A. M. (2020). Serum-Based Proteomics Profiling in Adult Patients with Cystic Fibrosis. International Journal of Molecular Sciences, 21(19), 7415. https://doi.org/10.3390/ijms21197415