Discovery of Novel Fetal Hemoglobin Inducers through Small Chemical Library Screening

 ,
, 

Abstract
:1. Introduction
2. Results
2.1. Screening of the Chemical Library Using the K562.GR Cellular Biosensor
2.2. Analysis of HbF Induction in a Human Erythroleukemic K562 Cell Line
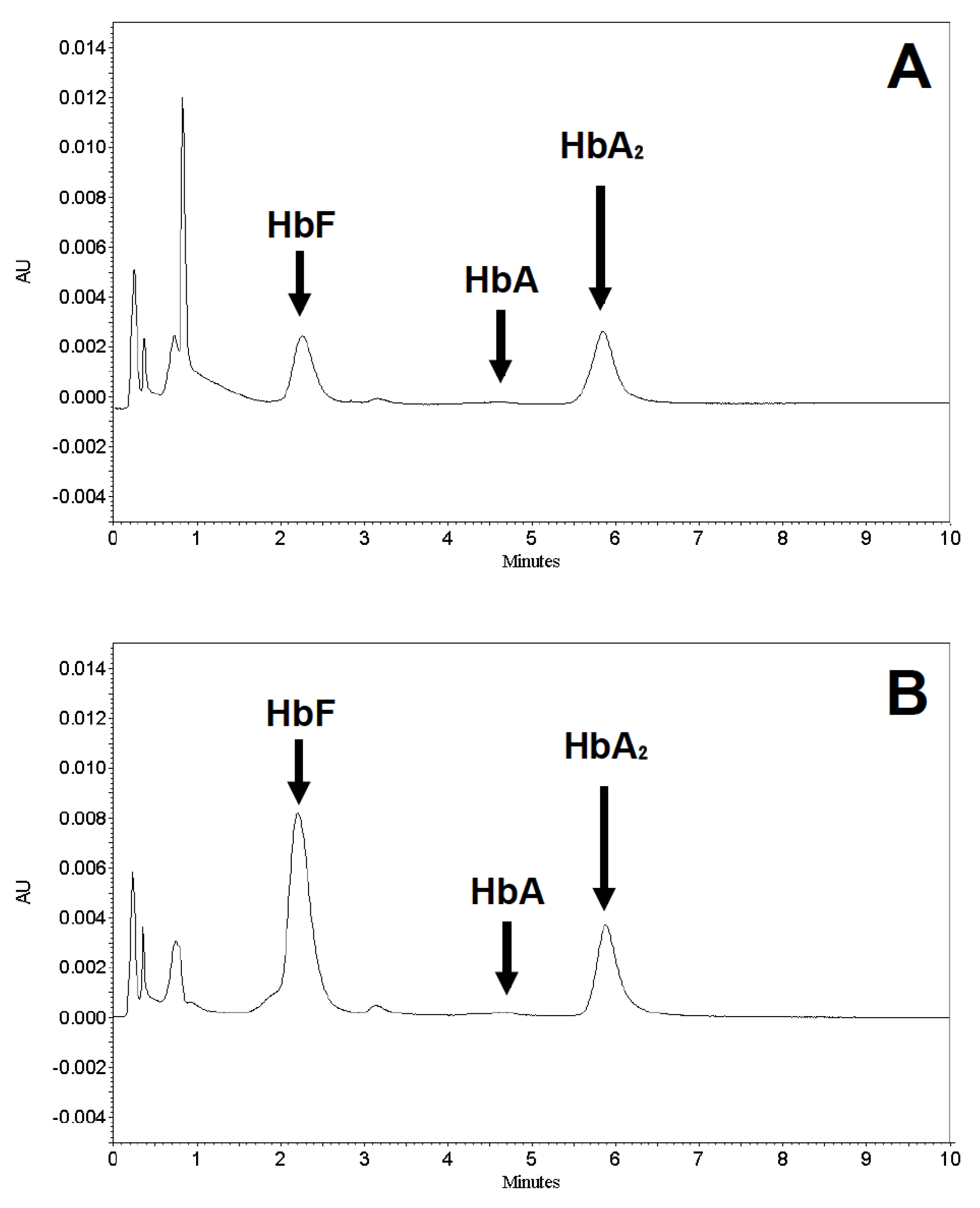
2.3. HbF Induction of Erythroid Precursors from β-Thalassemia Patients
3. Discussion
4. Materials and Methods
4.1. Chemical Library
4.2. Cell Lines and Culture Conditions
4.3. FACS Analysis
4.4. Erythroid Precursor Cultures Obtained from Blood of Thalassemia Patients
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, S.H.; Parveen, S.; Siddiqui, S.; Perveen, K.; Ahmed, G.; Kaleem, B.; Ahmed, S.; Zohaib, M.; Farzana, T.; Shamsi, T. Managing thalassemia in the developing world: An evidence-based approach for prevention, transfusion independency, and curative treatment with hematopoietic stem cell transplantation. Blood Adv. 2018, 2, 42–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T. An overview of current treatment strategies for β-thalassemia. Expert Opin. Orphan Drugs 2014, 7, 665–679. [Google Scholar] [CrossRef]
- Asadov, C.; Alimirzoeva, Z.; Mammadova, T.; Aliyeva, G.; Gafarova, S.; Mammadov, J. β-Thalassemia intermedia: A comprehensive overview and novel approaches. Int. J. Hematol. 2018, 108, 5–21. [Google Scholar] [CrossRef]
- Poggiali, E.; Cassinerio, E.; Zanaboni, L.; Cappellini, M.D. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411–422. [Google Scholar]
- King, A.; Shenoy, S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood 2014, 123, 3089–3094. [Google Scholar] [CrossRef] [Green Version]
- Ikawa, Y.; Miccio, A.; Magrin, E.; Kwiatkowski, J.L.; Rivella, S.; Cavazzana, M. Gene therapy of hemoglobinopathies: Progress and future challenges. Hum. Mol Genet. 2019, 28, R24–R30. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef]
- Piga, A.; Perrotta, S.; Gamberini, M.R.; Voskaridou, E.; Melpignano, A.; Filosa, A.; Caruso, V.; Pietrangelo, A.; Longo, F.; Tartaglione, I.; et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with β-thalassemia. Blood 2019, 133, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers. 2018, 4, 18010. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, M.H. Genetic etiologies for phenotypic diversity in sickle cell anemia. Sci. World J. 2009, 9, 46–67. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Porter, J.B.; Viprakasit, V.; Taher, A.T. A paradigm shift on beta-thalassaemia treatment: How will we manage this old disease with new therapies? Blood Rev. 2018, 32, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Testa, U. Fetal hemoglobin chemical inducers for treatment of hemoglobinopathies. Ann. Hematol. 2009, 88, 505–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucharoen, S.; Weatherall, D.J. Progress Toward the Control and Management of the Thalassemias. Hematol. Oncol. Clin. North. Am. 2016, 30, 359–371. [Google Scholar] [CrossRef]
- Anagnou, N.P.; Papayannopoulou, T.; Nienhuis, A.W.; Stamatoyannopoulos, G. Molecular characterization of a novel form of (A gamma delta beta)zero-thalassemia deletion with a 3′ breakpoint close to those of HPFH-3 and HPFH-4: Insights for a common regulatory mechanism. Nucleic Acids Res. 1988, 16, 6057–6066. [Google Scholar] [CrossRef] [Green Version]
- Murray, N.; Serjeant, B.E.; Serjeant, G.R. Sickle cell-hereditary persistence of fetal haemoglobin and its differentiation from other sickle cell syndromes. Br. J. Haematol. 1988, 69, 89–92. [Google Scholar] [CrossRef]
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Xie, F.; Muench, M.O.; Kan, Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc. Natl. Acad. Sci. USA 2016, 113, 10661–10665. [Google Scholar] [CrossRef] [Green Version]
- Finotti, A.; Gambari, R. Recent trends for novel options in experimental biological therapy of β-thalassemia. Expert Opin. Biol. Ther. 2014, 14, 1443–1454. [Google Scholar] [CrossRef]
- Suzuki, M.; Yamamoto, M.; Engel, J.D. Fetal globin gene repressors as drug targets for molecular therapies to treat the β-globinopathies. Mol. Cell. Biol. 2014, 34, 3560–3569. [Google Scholar] [CrossRef] [Green Version]
- Pace, B.S.; Liu, L.; Li, B.; Makala, L.H. Cell signaling pathways involved in drug-mediated fetal hemoglobin induction: Strategies to treat sickle cell disease. Exp. Biol. Med. 2015, 240, 1050–1064. [Google Scholar] [CrossRef] [Green Version]
- Nathan, D.G. Thalassemia: A look to the future. Ann. N. Y. Acad. Sci. 2016, 1368, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Martyn, G.E.; Funnell, A.P.W.; Quinlan, K.G.R.; Crossley, M. Wake-up Sleepy Gene: Reactivating Fetal Globin for β-Hemoglobinopathies. Trends Genet. 2018, 34, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Chui, D.H.; Dover, G.J.; Sebastiani, P.; Alsultan, A. Fetal hemoglobin in sickle cell anemia: A glass half full? Blood 2014, 123, 481–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foong, W.C.; Ho, J.J.; Loh, C.K.; Viprakasit, V. Hydroxyurea for reducing blood transfusion in non-transfusion dependent beta thalassaemias. Cochrane Database of Syst. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bayanzay, K.; Khan, R. Meta-analysis on effectiveness of hydroxyurea to treat transfusion-dependent β-thalassemia. Hematology 2015, 20, 469–476. [Google Scholar] [CrossRef]
- Kosaryan, M.; Zafari, M.; Alipur, A.; Hedayatizadeh-Omran, A. The effect and side effect of hydroxyurea therapy on patients with β-thalassemia: A systematic review to December 2012. Hemoglobin 2014, 38, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Cohan, N.; Mousavizadeh, K.; Falahi, M.J.; Haghpanah, S. Adverse effects of hydroxyurea in beta-thalassemia intermedia patients: 10 years’ experience. Pediatr. Hematol. Oncol. 2010, 27, 205–211. [Google Scholar] [CrossRef]
- Ma, Q.; Wyszynski, D.F.; Farrell, J.J.; Kutlar, A.; Farrer, L.A.; Baldwin, C.T.; Steinberg, M.H. Fetal hemoglobin in sickle cell anemia: Genetic determinants of response to hydroxyurea. Pharm. J. 2007, 7, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Breveglieri, G.; Salvatori, F.; Finotti, A.; Cosenza, L.C.; Zuccato, C.; Bianchi, N.; Breda, L.; Rivella, S.; Bresciani, A.; Bisbocci, M.; et al. Development and characterization of cellular biosensors for HTS of erythroid differentiation inducers targeting the transcriptional activity of γ-globin and β-globin gene promoters. Anal. Bioanal. Chem. 2019, 411, 7669–7680. [Google Scholar] [CrossRef]
- Breveglieri, G.; Salvatori, F.; Finotti, A.; Bertuzzi, I.; Destro, F.; Falzoni, S.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Feriotto, G.; et al. Cellular biosensors for the identification of fetal hemoglobin inducers. Minerva Biotecnol. 2007, 19, 123–132. [Google Scholar]
- Gambari, R.; Fibach, E. Medicinal chemistry of fetal hemoglobin inducers for treatment of beta-thalassemia. Curr. Med. Chem. 2007, 14, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Paikari, A.; Sumazin, P.; Ginter Summarell, C.C.; Crosby, J.R.; Boerwinkle, E.; Weiss, M.J.; Sheehan, V.A. Metformin induces FOXO3-dependent fetal hemoglobin production in human primary erythroid cells. Blood 2018, 132, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fibach, E. Erythropoiesis In Vitro-A Research and Therapeutic Tool in Thalassemia. J. Clin Med. 2019, 8, 2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampronti, I.; Bianchi, N.; Zuccato, C.; Dall’Acqua, F.; Vedaldi, D.; Viola, G.; Potenza, R.; Chiavilli, F.; Breveglieri, G.; Borgatti, M.; et al. Increase in gamma-globin mRNA content in human erythroid cells treated with angelicin analogs. Int. J. Hematol. 2009, 90, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.A.; Turner, A.R.; Mannoni, P.; Locksley, E.M.; Turc, J.M. Differentiation of K562 leukemia cells along erythroid, macrophage, and megakaryocyte lineages. J. Biol. Response Mod. 1985, 5, 250–262. [Google Scholar]
- Guerrasio, A.; Vainchenker, W.; Breton-Gorius, J.; Testa, U.; Rosa, R.; Thomopoulos, P.; Titeux, M.; Guichard, J.; Beuzard, Y. Embryonic and fetal hemoglobin synthesis in K562 cell line. Blood Cells 1981, 7, 165–176. [Google Scholar]
- Guerrini, A.; Lampronti, I.; Bianchi, N.; Zuccato, C.; Breveglieri, G.; Salvatori, F.; Mancini, I.; Rossi, D.; Potenza, R.; Chiavilli, F.; et al. Bergamot (Citrus bergamia Risso) fruit extracts as γ-globin gene expression inducers: Phytochemical and functional perspectives. J. Agric. Food. Chem. 2009, 57, 4103–4111. [Google Scholar] [CrossRef]
- Kohne, E. Hemoglobinopathies: Clinical Manifestations, Diagnosis, and Treatment. Dtsch. Arztebl. Int. 2011, 108, 532–540. [Google Scholar]
- Zhu, X.; Hu, T.; Ho, M.H.; Wang, Y.; Yu, M.; Patel, N.; Pi, W.; Choi, J.H.; Xu, H.; Ganapathy, V.; et al. Hydroxyurea differentially modulates activator and repressors of γ-globin gene in erythroblasts of responsive and non-responsive patients with sickle cell disease in correlation with Index of Hydroxyurea Responsiveness. Haematologica 2017, 102, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Sankaran, V.G.; Menne, T.F.; Šćepanović, D.; Vergilio, J.A.; Ji, P.; Kim, J.; Thiru, P.; Orkin, S.H.; Lander, E.S.; Lodish, H.F. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc. Natl. Acad. Sci. USA 2011, 108, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Angelis, N.; Thein, S.L. MYB—A regulatory factor in hematopoiesis. Gene 2018, 665, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, Y.; Shi, L. BCL11A Down-Regulation Induces γ-Globin in Human β-Thalassemia Major Erythroid Cells. Hemoglobin 2018, 42, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Norton, L.J.; Funnell, A.P.W.; Burdach, J.; Wienert, B.; Kurita, R.; Nakamura, Y.; Philipsen, S.; Pearson, R.C.M.; Quinlan, K.G.R.; Crossley, M. KLF1 directly activates expression of the novel fetal globin repressor ZBTB7A/LRF in erythroid cells. Blood Adv. 2017, 1, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Métais, J.Y.; Doerfler, P.A.; Mayuranathan, T.; Bauer, D.E.; Fowler, S.C.; Hsieh, M.M.; Katta, V.; Keriwala, S.; Lazzarotto, C.R.; Luk, K.; et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019, 3, 3379–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi, M.A.; Abbasalipour, M.; Concordet, J.P.; Berg, J.V.; Zeinali, S.; Arashkia, A.; Azadmanesh, K.; Buch, T.; Karimipoor, M. Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: A promising approach for gene therapy of beta thalassemia disease. Eur. J. Pharmacol. 2019, 854, 398–405. [Google Scholar] [CrossRef]
- Demirci, S.; Leonard, A.; Tisdale, J.F. Genome editing strategies for fetal hemoglobin induction in beta-hemoglobinopathies. Hum. Mol. Genet. 2020, 29, R100–R106. [Google Scholar] [CrossRef]
- Lozzio, C.B.; Lozzio, B.B. Human chronic myelogenous leukemia cell-line with positive Philadelphia-chromosome. Blood 1975, 45, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.; Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Effects of rapamycin on accumulation of α-, β- and γ-globin mRNAs in erythroid precursor cells from β-thalassaemia patients. Eur. J. Haematol. 2006, 77, 437–441. [Google Scholar]



{kind=link}
{kind=link}
| Compound | Structure | 10 nM | 100 nM | 1 µM | 10 µM |
|---|---|---|---|---|---|
| 6 |  | 1.15 | 1.05 | 1.00 | 1.57 |
| 17 |  | 0.90 | 0.94 | 0.99 | 1.48 |
| 22 |  | 0.94 | 0.92 | 1.02 | 1.53 |
| 56 | ND * | 1.02 | 1.05 | 1.59 | - |
| 57 | ND * | 1.10 | 1.00 | 1.07 | 1.36 |
| 58 |  | 0.99 | 1.13 | 1.55 | 1.62 |
| 59 |  | 1.03 | 1.20 | 2.00 | 2.10 |
| 62 |  | 1.01 | 0.96 | 0.98 | 1.35 |
| 63 |  | 1.02 | 1.01 | 1.04 | 1.78 |
| Compd | 10 nM | 100 nM | 1 µM | 10 µM |
|---|---|---|---|---|
| 6 | 1.09 ± 0.08 | 1.11 ± 0.07 | 1.14 ± 0.06 | 1.47 ± 0.27 |
| 17 | 1.13 ± 0.07 | 1.17 ± 0.05 | 1.18 ± 0.04 | 1.42 ± 0.13 |
| 22 | 1.11 ± 0.08 | 1.19 ± 0.06 | 1.29 ± 0.05 | 1.89 ± 0.27 |
| 56 | 1.11 ± 0.05 | 1.19 ± 0.12 | 1.47 ± 0.20 | - |
| 57 | 1.13 ± 0.12 | 1.13 ± 0.09 | 1.21 ± 0.10 | 1.60 ± 0.30 |
| 58 | 1.13 ± 0.07 | 1.20 ± 0.11 | 1.53 ± 0.31 | 1.54 ± 0.20 |
| 59 | 1.14 ± 0.06 | 1.38 ± 0.24 | 1.77 ± 0.15 | - |
| 62 | 1.15 ± 0.11 | 1.18 ± 0.05 | 1.24 ± 0.09 | 1.61 ± 0.28 |
| 63 | 1.14 ± 0.09 | 1.17 ± 0.07 | 1.20 ± 0.08 | 1.63 ± 0.28 |
| Compound | 10 nM | 100 nM | 1 µM | 10 µM |
|---|---|---|---|---|
| 6 | 0.76 ± 0.17 | 0.67 ± 0.15 | 0.72 ± 0.17 | 0.57 ± 0.02 |
| 17 | 0.70 ± 0.19 | 0.62 ± 0.16 | 0.58 ± 0.12 | 0.50 ± 0.03 |
| 22 | 0.70 ± 0.23 | 0.62 ± 0.15 | 0.61 ± 0.13 | 0.49 ± 0.04 |
| 56 | 0.75 ± 0.15 | 0.69 ± 0.14 | 0.72 ± 0.24 | - |
| 57 | 0.75 ± 0.25 | 0.63 ± 0.22 | 0.65 ± 0.21 | 0.63 ± 0.22 |
| 58 | 0.73 ± 0.18 | 0.67 ± 0.18 | 1.07 ± 0.43 | 0.79 ± 0.24 |
| 59 | 0.76 ± 0.20 | 0.66 ± 0.18 | 0.83 ± 0.43 | - |
| 62 | 0.69 ± 0.25 | 0.60 ± 0.24 | 0.61 ± 0.22 | 0.60 ± 0.23 |
| 63 | 0.74 ± 0.27 | 0.63 ± 0.21 | 0.61 ± 0.23 | 0.58 ± 0.19 |
| Compound | IC50 (µM) | Benzidine Assay | |
|---|---|---|---|
| Concentration | Benzidine-Positive Cells (%) | ||
| 6 | 5.00 | 1 µM | 4 |
| 5 µM | 9 | ||
| 10 µM | 15 | ||
| 17 | 3.60 | 1 µM | 6 |
| 5 µM | 10 | ||
| 10 µM | 11 | ||
| 22 | 1.20 | 1 µM | 4 |
| 5 µM | 7 | ||
| 10 µM | 18 | ||
| 56 | 0.85 | 0.5 µM | 1 |
| 1 µM | 2 | ||
| 5 µM | 1 | ||
| 57 | >10.00 | 1 µM | 5 |
| 5 µM | 6 | ||
| 10 µM | 8 | ||
| 58 | 0.69 | 0.5 µM | 6 |
| 1 µM | 5 | ||
| 5 µM | 3 | ||
| 59 | 0.10 | 0.5 µM | 2 |
| 1 µM | 3 | ||
| 5 µM | 2 | ||
| 62 | >10.00 | 1 µM | 3 |
| 5 µM | 4 | ||
| 10 µM | 8 | ||
| 63 | >10.00 | 1 µM | 2 |
| 5 µM | 3 | ||
| 10 µM | 6 | ||

 not treated;
not treated;  not analyzable;
not analyzable;  fold induction <50% HU;
fold induction <50% HU;  50% HU <fold induction <HU;
50% HU <fold induction <HU;  fold induction >HU.
fold induction >HU.© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breveglieri, G.; Pacifico, S.; Zuccato, C.; Cosenza, L.C.; Sultan, S.; D’Aversa, E.; Gambari, R.; Preti, D.; Trapella, C.; Guerrini, R.; et al. Discovery of Novel Fetal Hemoglobin Inducers through Small Chemical Library Screening. Int. J. Mol. Sci. 2020, 21, 7426. https://doi.org/10.3390/ijms21197426
Breveglieri G, Pacifico S, Zuccato C, Cosenza LC, Sultan S, D’Aversa E, Gambari R, Preti D, Trapella C, Guerrini R, et al. Discovery of Novel Fetal Hemoglobin Inducers through Small Chemical Library Screening. International Journal of Molecular Sciences. 2020; 21(19):7426. https://doi.org/10.3390/ijms21197426
Chicago/Turabian StyleBreveglieri, Giulia, Salvatore Pacifico, Cristina Zuccato, Lucia Carmela Cosenza, Shaiq Sultan, Elisabetta D’Aversa, Roberto Gambari, Delia Preti, Claudio Trapella, Remo Guerrini, and et al. 2020. "Discovery of Novel Fetal Hemoglobin Inducers through Small Chemical Library Screening" International Journal of Molecular Sciences 21, no. 19: 7426. https://doi.org/10.3390/ijms21197426
APA StyleBreveglieri, G., Pacifico, S., Zuccato, C., Cosenza, L. C., Sultan, S., D’Aversa, E., Gambari, R., Preti, D., Trapella, C., Guerrini, R., & Borgatti, M. (2020). Discovery of Novel Fetal Hemoglobin Inducers through Small Chemical Library Screening. International Journal of Molecular Sciences, 21(19), 7426. https://doi.org/10.3390/ijms21197426