Ultrastructural Pathology of Atherosclerosis, Calcific Aortic Valve Disease, and Bioprosthetic Heart Valve Degeneration: Commonalities and Differences

 ,
,  and
and
Abstract
:1. Introduction
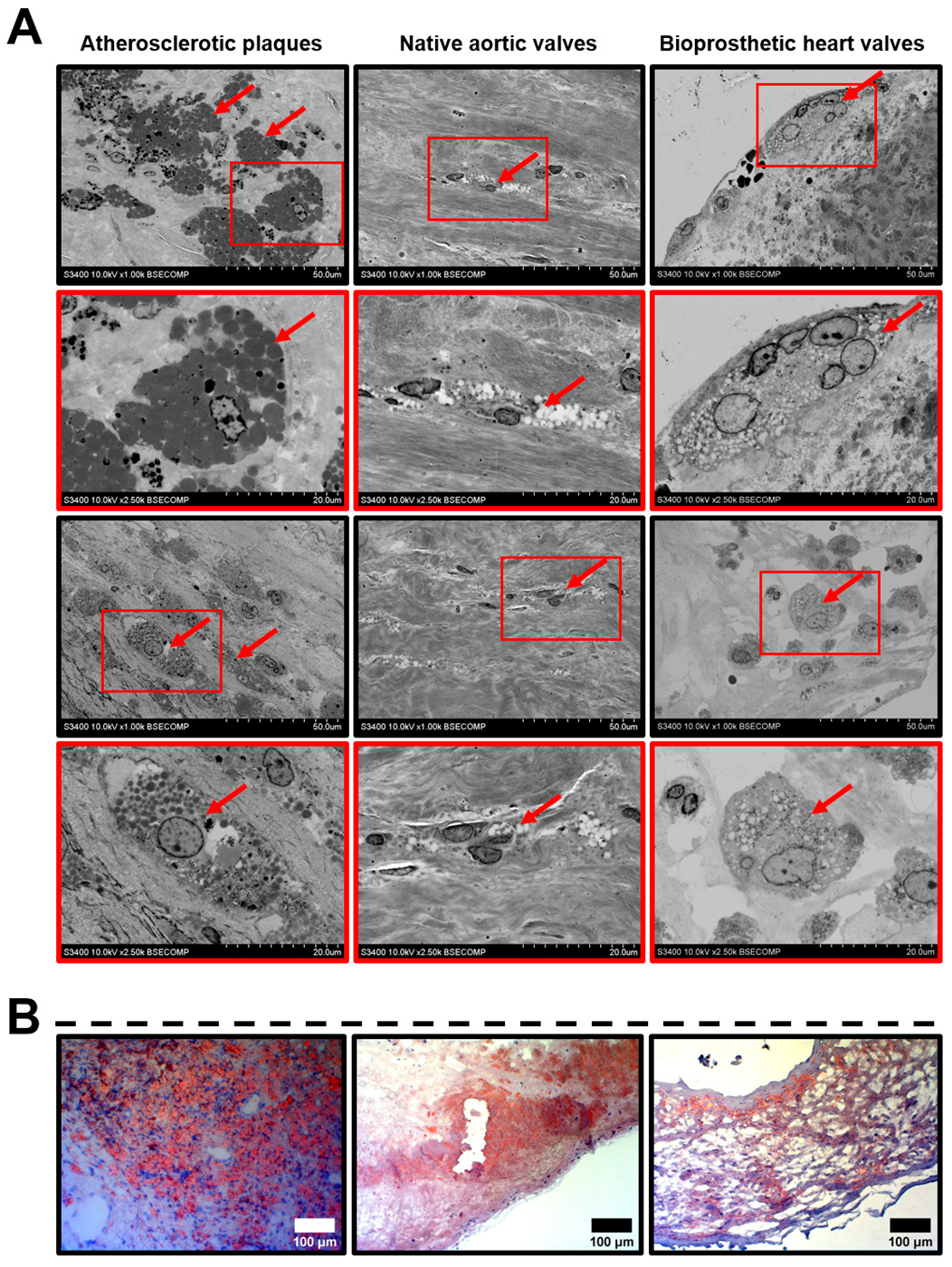
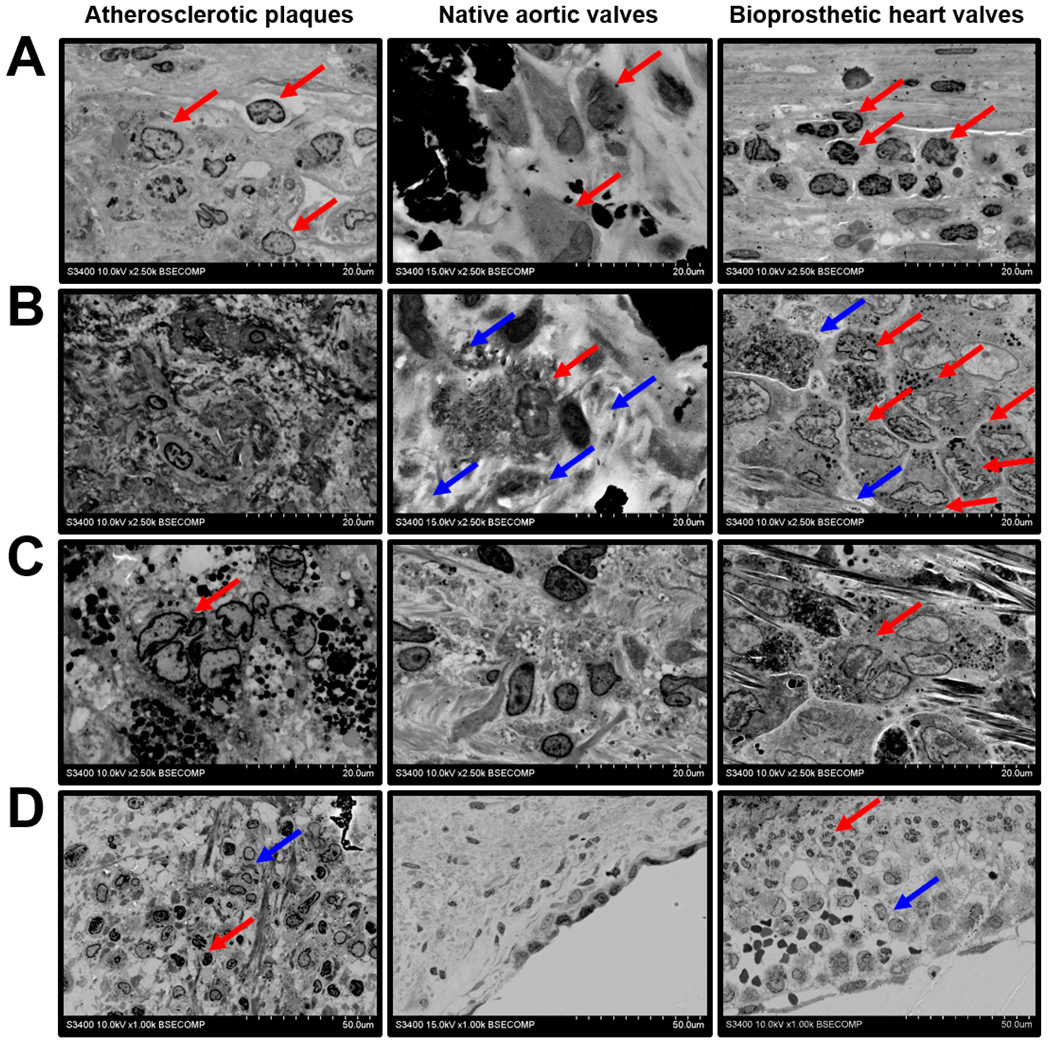
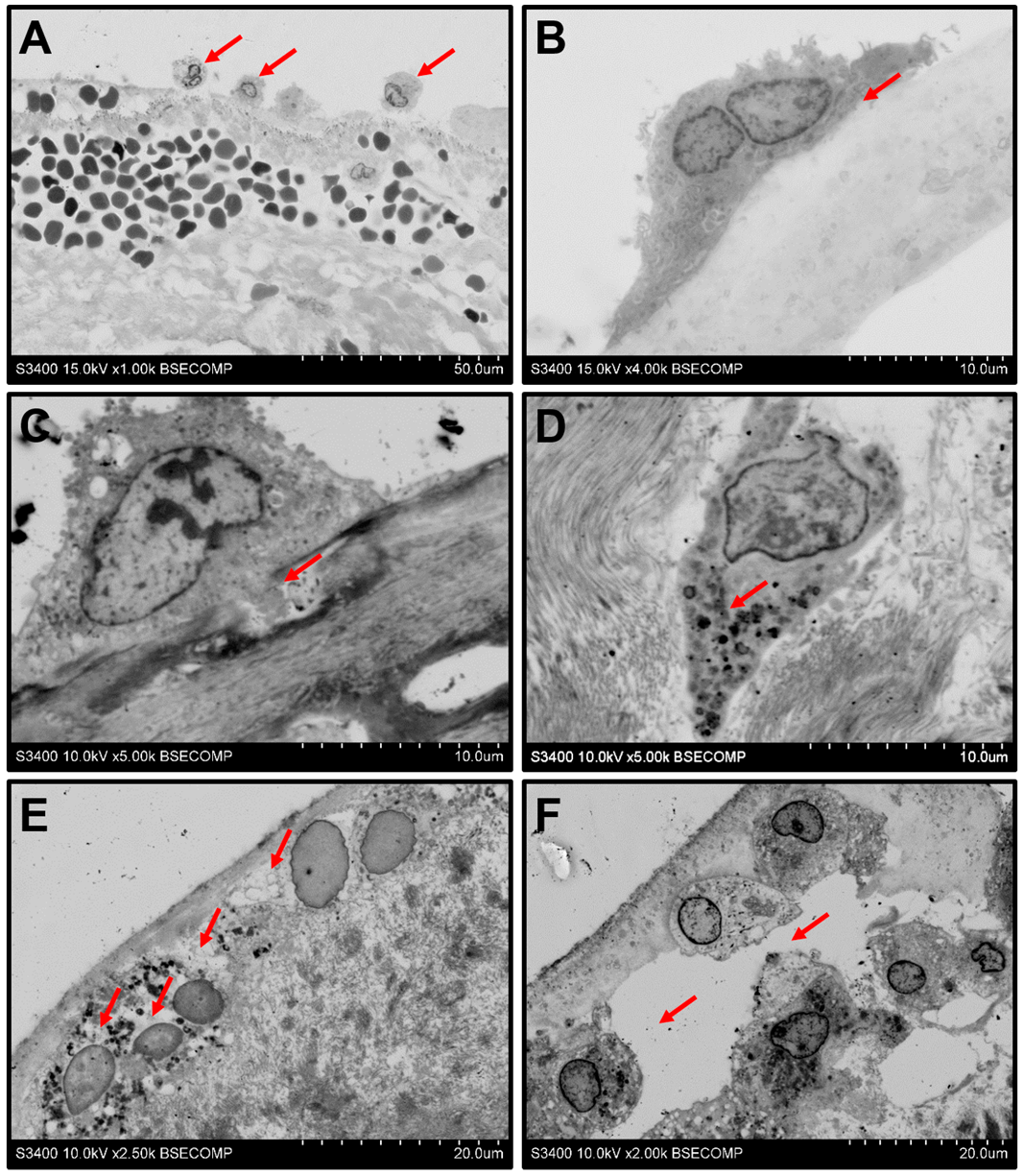
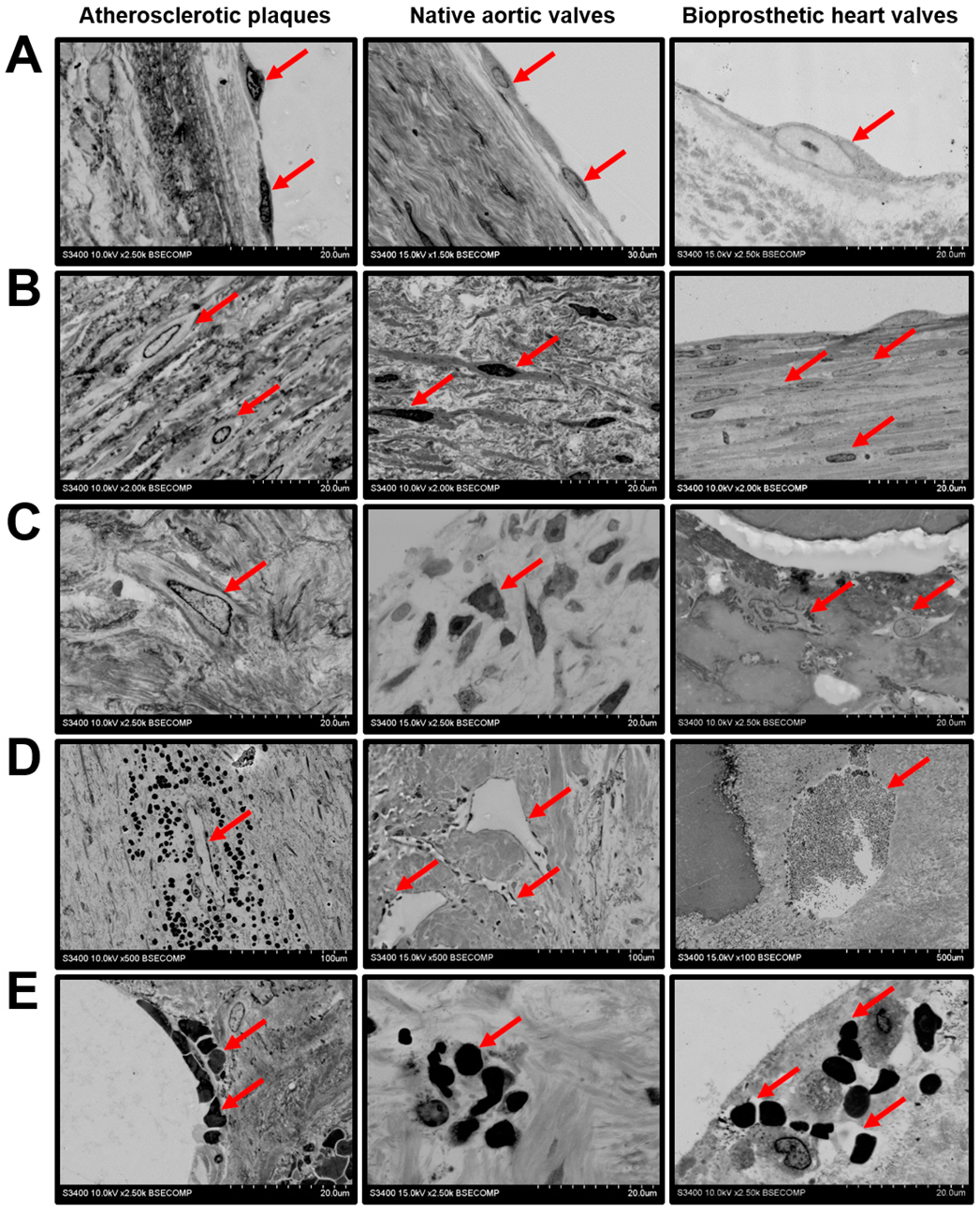
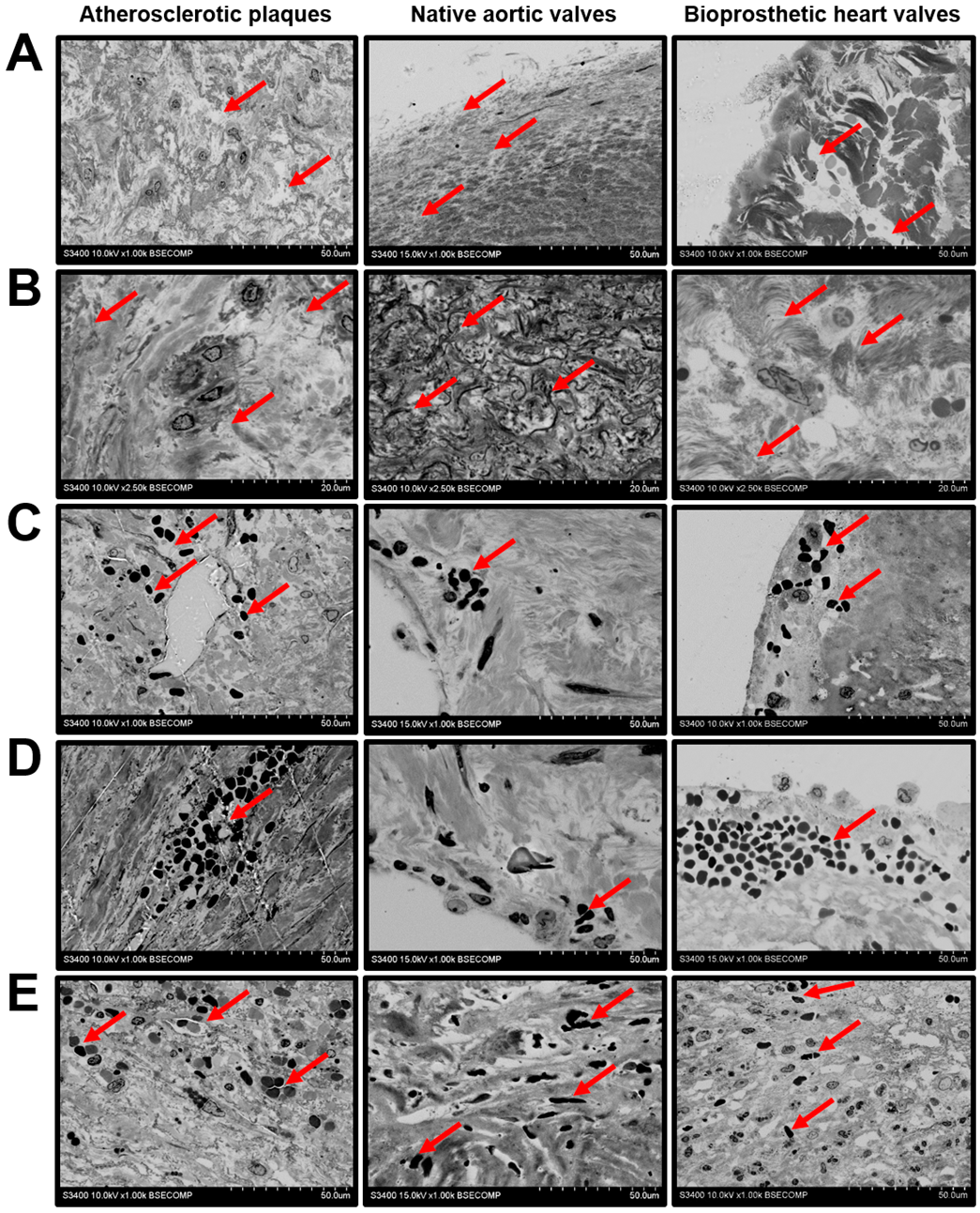
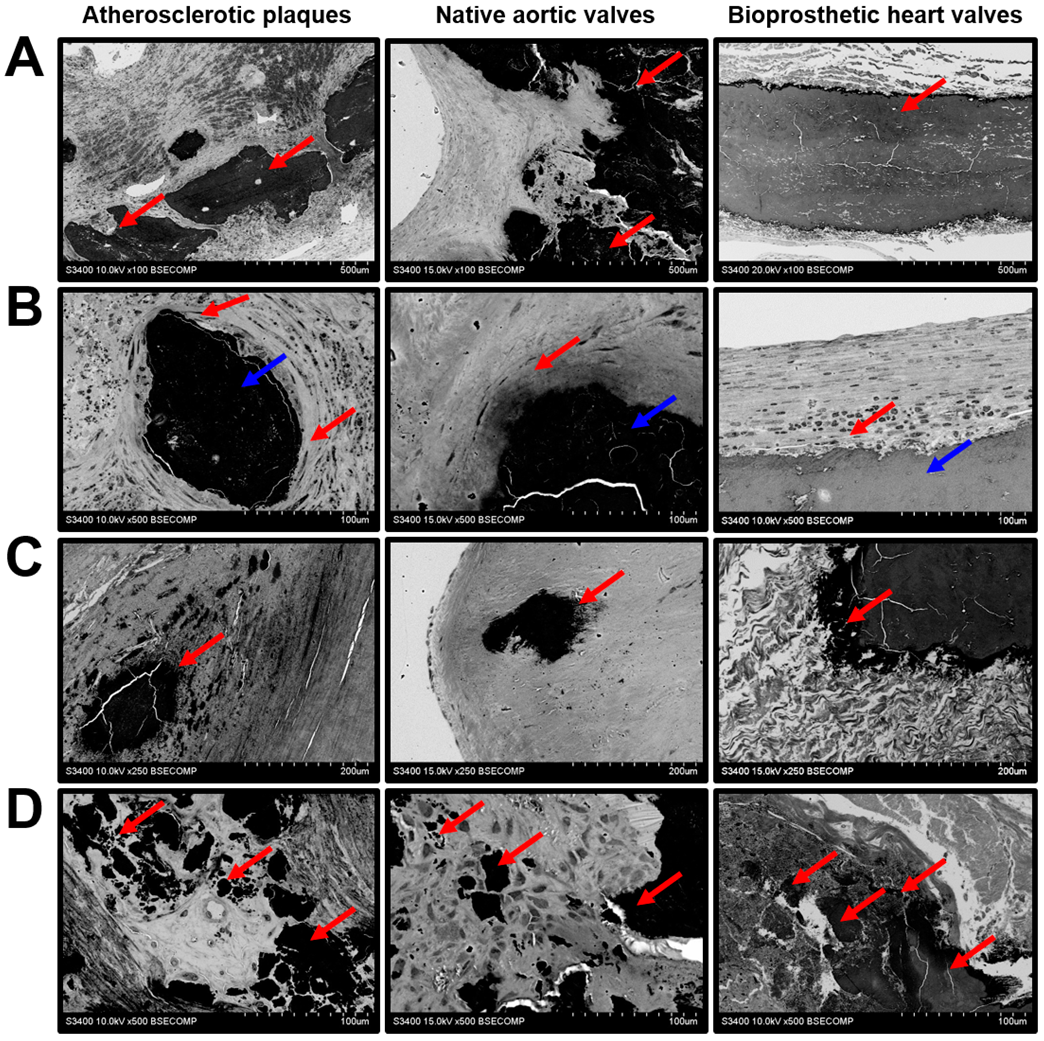
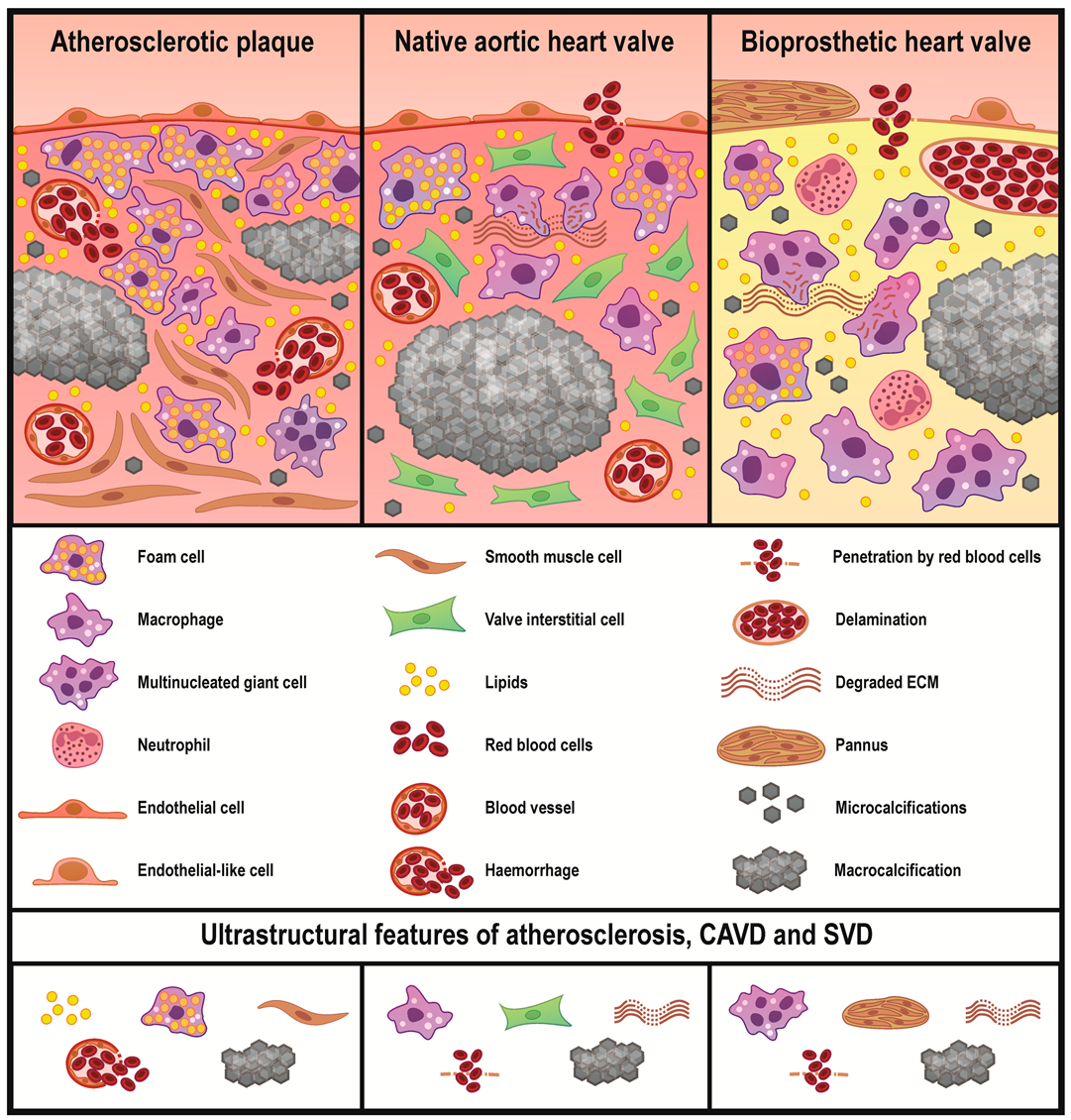
2. Results
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAVD | Calcific aortic valve disease |
| ECM | Extracellular matrix |
| VSMCs | Vascular smooth muscle cells |
| VICs | Valvular interstitial cells |
| BHVs | Bioprosthetic heart valves |
| SVD | Structural valve deterioration |
| AVs | Aortic valves |
| RBCs | Red blood cells |
| COPD | Chronic obstructive pulmonary disease |
References
- Kostyunin, A.E.; Yuzhalin, A.E.; Ovcharenko, E.A.; Kutikhin, A.G. Development of calcific aortic valve disease: Do we know enough for new clinical trials? J. Mol. Cell Cardiol. 2019, 132, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.X.; Mallat, Z. Targeting the immune system in atherosclerosis: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019, 73, 1691–1706. [Google Scholar] [CrossRef]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Capodanno, D.; Petronio, A.S.; Prendergast, B.; Eltchaninoff, H.; Vahanian, A.; Modine, T.; Lancellotti, P.; Sondergaard, L.; Ludman, P.F.; Tamburino, C.; et al. Standardized definitions of structural deterioration and valve failure in assessing long-term durability of transcatheter and surgical aortic bioprosthetic valves: A consensus statement from the European Association of Percutaneous Cardiovascular Interventions (EAPCI) endorsed by the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2017, 38, 3382–3390. [Google Scholar] [CrossRef] [Green Version]
- Dvir, D.; Bourguignon, T.; Otto, C.M.; Hahn, R.T.; Rosenhek, R.; Webb, J.G.; Treede, H.; Sarano, M.E.; Feldman, T.; Wijeysundera, H.C.; et al. VIVID (Valve in Valve International Data) Investigators. Standardized definition of structural valve degeneration for surgical and transcatheter bioprosthetic aortic valves. Circulation 2018, 137, 388–399. [Google Scholar] [CrossRef]
- Manji, R.A.; Lee, W.; Cooper, D.K.C. Xenograft bioprosthetic heart valves: Past, present and future. Int. J. Surg. 2015, 23, 280–284. [Google Scholar] [CrossRef]
- Cote, N.; Pibarot, P.; Clavel, M.A. Incidence, risk factors, clinical impact, and management of bioprosthesis structural valve degeneration. Curr. Opin. Cardiol. 2017, 32, 123–129. [Google Scholar] [CrossRef]
- Bourguignon, T.; Espitalier, F.; Pantaleon, C.; Vermes, E.; El-Arid, J.M.; Loardi, C.; Karam, E.; Candolfi, P.; Ivanes, F.; Aupart, M. Bioprosthetic mitral valve replacement in patients aged 65 years or younger: Long-term outcomes with the Carpentier-Edwards PERIMOUNT pericardial valve. Eur. J. Cardiothorac. Surg. 2018, 54, 302–309. [Google Scholar] [CrossRef]
- Foroutan, F.; Guyatt, G.H.; O’Brien, K.; Bain, E.; Stein, M.; Bhagra, S.; Sit, D.; Kamran, R.; Chang, Y.; Devji, T.; et al. Prognosis after surgical replacement with a bioprosthetic aortic valve in patients with severe symptomatic aortic stenosis: Systematic review of observational studies. BMJ 2016, 354, i5065. [Google Scholar] [CrossRef] [Green Version]
- Pibarot, P.; Dumesnil, J.G. Prosthetic heart valves: Selection of the optimal prosthesis and long-term management. Circulation 2009, 119, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Briand, M.; Pibarot, P.; Després, J.P.; Voisine, P.; Dumesnil, J.G.; Dagenais, F.; Mathieu, P. Metabolic syndrome is associated with faster degeneration of bioprosthetic valves. Circulation 2006, 114, I512–I517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farivar, R.S.; Cohn, L.H. Hypercholesterolemia is a risk factor for bioprosthetic valve calcification and explantation. J. Thorac. Cardiovasc. Surg. 2003, 126, 969–975. [Google Scholar] [CrossRef] [Green Version]
- Lorusso, R.; Gelsomino, S.; Lucà, F.; De Cicco, G.; Billè, G.; Carella, R.; Villa, E.; Troise, G.; Viganò, M.; Banfi, C.; et al. Type 2 diabetes mellitus is associated with faster degeneration of bioprosthetic valve: Results from a propensity score-matched Italian multicenter study. Circulation 2012, 125, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Nollert, G.; Miksch, J.; Kreuzer, E.; Reichart, B. Risk factors for atherosclerosis and the degeneration of pericardial valves after aortic valve replacement. J. Thorac. Cardiovasc. Surg. 2003, 126, 965–968. [Google Scholar] [CrossRef] [Green Version]
- Mukhamadiyarov, R.A.; Sevostyanova, V.V.; Shishkova, D.K.; Nokhrin, A.V.; Sidorova, O.D.; Kutikhin, A.G. Grinding and polishing instead of sectioning for the tissue samples with a graft: Implications for light and electron microscopy. Micron 2016, 85, 1–7. [Google Scholar] [CrossRef]
- Mukhamadiyarov, R.A.; Bogdanov, L.A.; Mishinov, S.V.; Kutikhin, A.G. A novel technique for preparation, staining, and visualization of tissue with metal implants and extraskeletal calcification areas. Sovrem. Tehnol. V. Med. 2020, 12, 13–22. [Google Scholar] [CrossRef]
- Mukhamadiyarov, R.A.; Kutikhin, A.G. Backscattered Scanning Electron Microscopy Approach for Assessment of Microvessels under Conditions of Normal Microanatomy and Pathological Neovascularization. Bull. Exp. Biol. Med. 2020, 169, 525–530. [Google Scholar] [CrossRef]
- Bloebaum, R.D.; Skedros, J.G.; Vajda, E.G.; Bachus, K.N.; Constantz, B.R. Determining mineral content variations in bone using backscattered electron imaging. Bone 1997, 20, 485–490. [Google Scholar] [CrossRef]
- Fratzl-Zelman, N.; Roschger, P.; Gourrier, A.; Weber, M.; Misof, B.M.; Loveridge, N.; Reeve, J.; Klaushofer, K.; Fratzl, P. Combination of nanoindentation and quantitative backscattered electron imaging revealed altered bone material properties associated with femoral neck fragility. Calcif. Tissue Int. 2009, 85, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthis, A.H.C.; Tsolaki, E.; Didierlaurent, L.; Staubli, S.; Zboray, R.; Neels, A.; Dietrich, D.; Manser, P.; Desbiolles, L.M.; Leschka, S.; et al. Nano-analytical characterization of endogenous minerals in healthy placental tissue: Mineral distribution, composition and ultrastructure. Analyst 2019, 144, 6850–6857. [Google Scholar] [CrossRef] [PubMed]
- Shah, F.A.; Ruscsák, K.; Palmquist, A. 50 years of scanning electron microscopy of bone-a comprehensive overview of the important discoveries made and insights gained into bone material properties in health, disease, and taphonomy. Bone Res. 2019, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Beach, J.M.; Mihaljevic, T.; Svensson, L.G.; Rajeswaran, J.; Marwick, T.; Griffin, B.; Johnston, D.R.; Sabik, J.F., 3rd; Blackstone, E.H. Coronary artery disease and outcomes of aortic valve replacement for severe aortic stenosis. J. Am. Coll. Cardiol. 2013, 61, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yutzey, K.E.; Demer, L.L.; Body, S.C.; Huggins, G.S.; Towler, D.A.; Giachelli, C.M.; Hofmann-Bowman, M.A.; Mortlock, D.P.; Rogers, M.B.; Sadeghi, M.M.; et al. Calcific aortic valve disease: A consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arter. Thromb. Vasc. Biol. 2014, 34, 2387–2393. [Google Scholar] [CrossRef] [Green Version]
- Alushi, B.; Curini, L.; Christopher, M.R.; Grubitzch, H.; Landmesser, U.; Amedei, A.; Lauten, A. Calcific Aortic Valve Disease-Natural History and Future Therapeutic Strategies. Front. Pharm. 2020, 11, 685. [Google Scholar] [CrossRef]
- Fukumoto, R.; Kawai, M.; Minai, K.; Ogawa, K.; Yoshida, J.; Inoue, Y.; Morimoto, S.; Tanaka, T.; Nagoshi, T.; Ogawa, T.; et al. Conflicting relationship between age-dependent disorders, valvular heart disease and coronary artery disease by covariance structure analysis: Possible contribution of natriuretic peptide. PLoS ONE 2017, 12, e0181206. [Google Scholar] [CrossRef]
- Makeeva, O.A.; Sleptsov, A.A.; Kulish, E.V.; Barbarash, O.L.; Mazur, A.M.; Prokhorchuk, E.B.; Chekanov, N.N.; Stepanov, V.A.; Puzyrev, V.P. Genomic Study of Cardiovascular Continuum Comorbidity. Acta Nat. 2015, 7, 89–99. [Google Scholar] [CrossRef]
- Smith, J.G.; Newton-Cheh, C. Genome-wide association studies of late-onset cardiovascular disease. J. Mol. Cell Cardiol. 2015, 83, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.C.; Mason, A.M.; Bäck, M.; Klarin, D.; Damrauer, S.M.; Million Veteran Program; Michaëlsson, K.; Burgess, S. Genetic predisposition to smoking in relation to 14 cardiovascular diseases. Eur. Heart J. 2020, ehaa193. [Google Scholar] [CrossRef]
- Dotsenko, O.; Chackathayil, J.; Patel, J.V.; Gill, P.S.; Lip, G.Y. Candidate circulating biomarkers for the cardiovascular disease continuum. Curr. Pharm. Des. 2008, 14, 2445–2461. [Google Scholar] [CrossRef] [PubMed]
- Battistoni, A.; Rubattu, S.; Volpe, M. Circulating biomarkers with preventive, diagnostic and prognostic implications in cardiovascular diseases. Int. J. Cardiol. 2012, 157, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Clemente, A.; Traghella, I.; Mazzone, A.; Sbrana, S.; Vassalle, C. Vascular and valvular calcification biomarkers. Adv. Clin. Chem. 2020, 95, 73–103. [Google Scholar] [CrossRef]
- Hinton, R.B., Jr.; Lincoln, J.; Deutsch, G.H.; Osinska, H.; Manning, P.B.; Benson, D.W.; Yutzey, K.E. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 2006, 98, 1431–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fondard, O.; Detaint, D.; Iung, B.; Choqueux, C.; Adle-Biassette, H.; Jarraya, M.; Hvass, U.; Couetil, J.P.; Henin, D.; Michel, J.B.; et al. Extracellular matrix remodelling in human aortic valve disease: The role of matrix metalloproteinases and their tissue inhibitors. Eur. Heart J. 2005, 26, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N. Vascular extracellular matrix in atherosclerosis. Cardiol. Rev. 2013, 21, 270–288. [Google Scholar] [CrossRef]
- Skjøt-Arkil, H.; Barascuk, N.; Register, T.; Karsdal, M.A. Macrophage-mediated proteolytic remodeling of the extracellular matrix in atherosclerosis results in neoepitopes: A potential new class of biochemical markers. Assay Drug Dev. Technol 2010, 8, 542–552. [Google Scholar] [CrossRef]
- Xu, H.; Jiang, J.; Chen, W.; Li, W.; Chen, Z. Vascular Macrophages in Atherosclerosis. J. Immunol. Res. 2019, 4354786. [Google Scholar] [CrossRef] [Green Version]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arter. Thromb. Vasc. Biol 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostyunin, A.E.; Yuzhalin, A.E.; Rezvova, M.A.; Ovcharenko, E.A.; Glushkova, T.V.; Kutikhin, A.G. Degeneration of Bioprosthetic Heart Valves: Update 2020. J. Am. Heart Assoc. 2020, e018506. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.W.; Kim, K.W.; Jang, M.Y.; Seok Jang, H.; Yun, T.J.; Lee, S.H.; et al. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Invest. 2018, 128, 1106–1124. [Google Scholar] [CrossRef]
- Baidžajevas, K.; Hadadi, É.; Lee, B.; Lum, J.; Shihui, F.; Sudbery, I.; Kiss-Tóth, E.; Wong, S.C.; Wilson, H.L. Macrophage polarisation associated with atherosclerosis differentially affects their capacity to handle lipids. Atherosclerosis 2020, 305, 10–18. [Google Scholar] [CrossRef]
- Perota, A.; Lagutina, I.; Duchi, R.; Zanfrini, E.; Lazzari, G.; Judor, J.P.; Conchon, S.; Bach, J.M.; Bottio, T.; Gerosa, G.; et al. Generation of cattle knockout for galactose-α1,3-galactose and N-glycolylneuraminic acid antigens. Xenotransplantation 2019, 26, e12524. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.R.; Quax, P.H. Plaque angiogenesis and its relation to inflammation and atherosclerotic plaque destabilization. Curr. Opin. Lipidol. 2016, 27, 499–506. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Contribution of neovascularization and intraplaque haemorrhage to atherosclerotic plaque progression and instability. Acta Physiol. (Oxf.) 2015, 213, 539–553. [Google Scholar] [CrossRef]
- Kitagawa, T.; Yamamoto, H.; Sentani, K.; Takahashi, S.; Tsushima, H.; Senoo, A.; Yasui, W.; Sueda, T.; Kihara, Y. The relationship between inflammation and neoangiogenesis of epicardial adipose tissue and coronary atherosclerosis based on computed tomography analysis. Atherosclerosis 2015, 243, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Katsi, V.; Magkas, N.; Antonopoulos, A.; Trantalis, G.; Toutouzas, K.; Tousoulis, D. Aortic valve: Anatomy and structure and the role of vasculature in the degenerative process. Acta Cardiol. 2020, 1–14. [Google Scholar] [CrossRef]
- Javid, F.; Shahmansouri, N.; Angeles, J.; Mongrain, R. Fatigue exhaustion of the mitral valve tissue. Biomech. Model. Mechanobiol. 2019, 18, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Akahori, H.; Tsujino, T.; Naito, Y.; Matsumoto, M.; Lee-Kawabata, M.; Ohyanagi, M.; Mitsuno, M.; Miyamoto, Y.; Daimon, T.; Hao, H.; et al. Intraleaflet haemorrhage is associated with rapid progression of degenerative aortic valve stenosis. Eur. Heart J. 2011, 32, 888–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stam, O.C.G.; Daemen, M.J.A.P.; van Rijswijk, J.W.; de Mol, B.A.J.M.; van der Wal, A.C. Intraleaflet hemorrhages are a common finding in symptomatic aortic and mitral valves. Cardiovasc. Pathol. 2017, 30, 12–18. [Google Scholar] [CrossRef]
- Morvan, M.; Arangalage, D.; Franck, G.; Perez, F.; Cattan-Levy, L.; Codogno, I.; Jacob-Lenet, M.P.; Deschildre, C.; Choqueux, C.; Even, G.; et al. Relationship of Iron Deposition to Calcium Deposition in Human Aortic Valve Leaflets. J. Am. Coll. Cardiol. 2019, 73, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Tziakas, D.N.; Chalikias, G.; Pavlaki, M.; Kareli, D.; Gogiraju, R.; Hubert, A.; Böhm, E.; Stamoulis, P.; Drosos, I.; Kikas, P.; et al. Lysed Erythrocyte Membranes Promote Vascular Calcification. Circulation 2019, 139, 2032–2048. [Google Scholar] [CrossRef]
- Rogers, M.A.; Aikawa, E. Cardiovascular calcification: Artificial intelligence and big data accelerate mechanistic discovery. Nat. Rev. Cardiol. 2019, 16, 261–274. [Google Scholar] [CrossRef]
- Ruiz, J.L.; Hutcheson, J.D.; Aikawa, E. Cardiovascular calcification: Current controversies and novel concepts. Cardiovasc. Pathol. 2015, 24, 207–212. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Goettsch, C.; Rogers, M.A.; Aikawa, E. Revisiting cardiovascular calcification: A multifaceted disease requiring a multidisciplinary approach. Semin. Cell Dev. Biol. 2015, 46, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef]
- Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Aikawa, E.; Cardoso, L.; Weinbaum, S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. USA 2013, 110, 10741–10746. [Google Scholar] [CrossRef] [Green Version]
- Vengrenyuk, Y.; Carlier, S.; Xanthos, S.; Cardoso, L.; Ganatos, P.; Virmani, R.; Einav, S.; Gilchrist, L.; Weinbaum, S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc. Natl. Acad. Sci. USA 2006, 103, 14678–14683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Zhang, Y.; Hou, J.; Lin, G.; Yu, B. Relation Between Superficial Calcifications and Plaque Rupture: An Optical Coherence Tomography Study. Can. J. Cardiol. 2017, 33, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Akers, E.J.; Nicholls, S.J.; Di Bartolo, B.A. Plaque Calcification: Do Lipoproteins Have a Role? Arter. Thromb. Vasc. Biol. 2019, 39, 1902–1910. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature/Sample | Patients with Atherosclerosis (n = 36) | Patient with CAVD (n = 12) | Patients with BHV Failure (SVD) (n = 12) | p Value |
|---|---|---|---|---|
| Male gender, n (%) | 25 (69.4) | 8 (66.7) | 6 (50.0) | 0.47 |
| Age, years, Me (25th; 75th) | 64 (58; 69) | 63.5 (55; 66) | 64.5 (61; 71.25) | 0.92 |
| Arterial hypertension, n (%) | 34 (94.4) | 10 (83.3) | 10 (83.3) | 0.37 |
| Chronic heart failure, n (%) | 34 (94.4) | 12 (100.0) | 12 (100.0) | 0.50 |
| COPD or asthma, n (%) | 9 (25.0) | 3 (25.0) | 0 (0.0) | 0.15 |
| Chronic kidney disease, n (%) | 8 (22.2) | 4 (33.3) | 7 (58.3) | 0.07 |
| Diabetes mellitus, n (%) | 6 (16.7) | 0 (0.0) | 0 (0.0) | 0.11 |
| Overweight or obesity, n (%) | 17 (47.2) | 6 (50.0) | 8 (66.7) | 0.50 |
| Feature/Sample | Atherosclerotic Plaques (n = 36) | Calcified Native AVs (n = 12) | Failed BHVs (n = 12) | p Value |
|---|---|---|---|---|
| Foam cells, n (%) | 36 (100.0) | 3 (25.0) | 4 (33.3) | 0.0001 |
| Canonical macrophages, n (%) | 36 (100.0) | 12 (100.0) | 12 (100.0) | 0.99 |
| Multinucleated giant cells, n (%) | 8 (22.2) | 0 (0.00) | 9 (75.0) | 0.0001 |
| Neutrophil infiltration, n (%) | 13 (36.1) | 0 (0.00) | 7 (58.3) | 0.009 |
| (Pseudo)endothelialisation, n (%) | 26 (72.2) | 12 (100.0) | 6 (50.0) | 0.02 |
| Neovascularisation, n (%) | 35 (97.2) | 5 (41.7) | 0 (0.0) | 0.0001 |
| Haemorrhages, n (%) | 17 (47.2) | 2 (16.7) | 4 (33.3) | 0.16 |
| Leaky microvessels, n (%) | 21 (58.3) | 0 (0.00) | 0 (0.0) | 0.0001 |
| Penetration by RBCs, n (%) | 5 (13.9) | 5 (41.7) | 11 (91.7) | 0.0001 |
| Delamination, n (%) | 4 (11.1) | 2 (16.7) | 9 (75.0) | 0.0001 |
| Microcalcification, n (%) | 36 (100.0) | 12 (100.0) | 10 (83.3) | 0.02 |
| Macrocalcification, n (%) | 36 (100.0) | 12 (100.0) | 10 (83.3) | 0.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostyunin, A.; Mukhamadiyarov, R.; Glushkova, T.; Bogdanov, L.; Shishkova, D.; Osyaev, N.; Ovcharenko, E.; Kutikhin, A. Ultrastructural Pathology of Atherosclerosis, Calcific Aortic Valve Disease, and Bioprosthetic Heart Valve Degeneration: Commonalities and Differences. Int. J. Mol. Sci. 2020, 21, 7434. https://doi.org/10.3390/ijms21207434
Kostyunin A, Mukhamadiyarov R, Glushkova T, Bogdanov L, Shishkova D, Osyaev N, Ovcharenko E, Kutikhin A. Ultrastructural Pathology of Atherosclerosis, Calcific Aortic Valve Disease, and Bioprosthetic Heart Valve Degeneration: Commonalities and Differences. International Journal of Molecular Sciences. 2020; 21(20):7434. https://doi.org/10.3390/ijms21207434
Chicago/Turabian StyleKostyunin, Alexander, Rinat Mukhamadiyarov, Tatiana Glushkova, Leo Bogdanov, Daria Shishkova, Nikolay Osyaev, Evgeniy Ovcharenko, and Anton Kutikhin. 2020. "Ultrastructural Pathology of Atherosclerosis, Calcific Aortic Valve Disease, and Bioprosthetic Heart Valve Degeneration: Commonalities and Differences" International Journal of Molecular Sciences 21, no. 20: 7434. https://doi.org/10.3390/ijms21207434
APA StyleKostyunin, A., Mukhamadiyarov, R., Glushkova, T., Bogdanov, L., Shishkova, D., Osyaev, N., Ovcharenko, E., & Kutikhin, A. (2020). Ultrastructural Pathology of Atherosclerosis, Calcific Aortic Valve Disease, and Bioprosthetic Heart Valve Degeneration: Commonalities and Differences. International Journal of Molecular Sciences, 21(20), 7434. https://doi.org/10.3390/ijms21207434