Positron Emission Tomography (PET) and Neuroimaging in the Personalized Approach to Neurodegenerative Causes of Dementia

 ,
,  ,
,  and
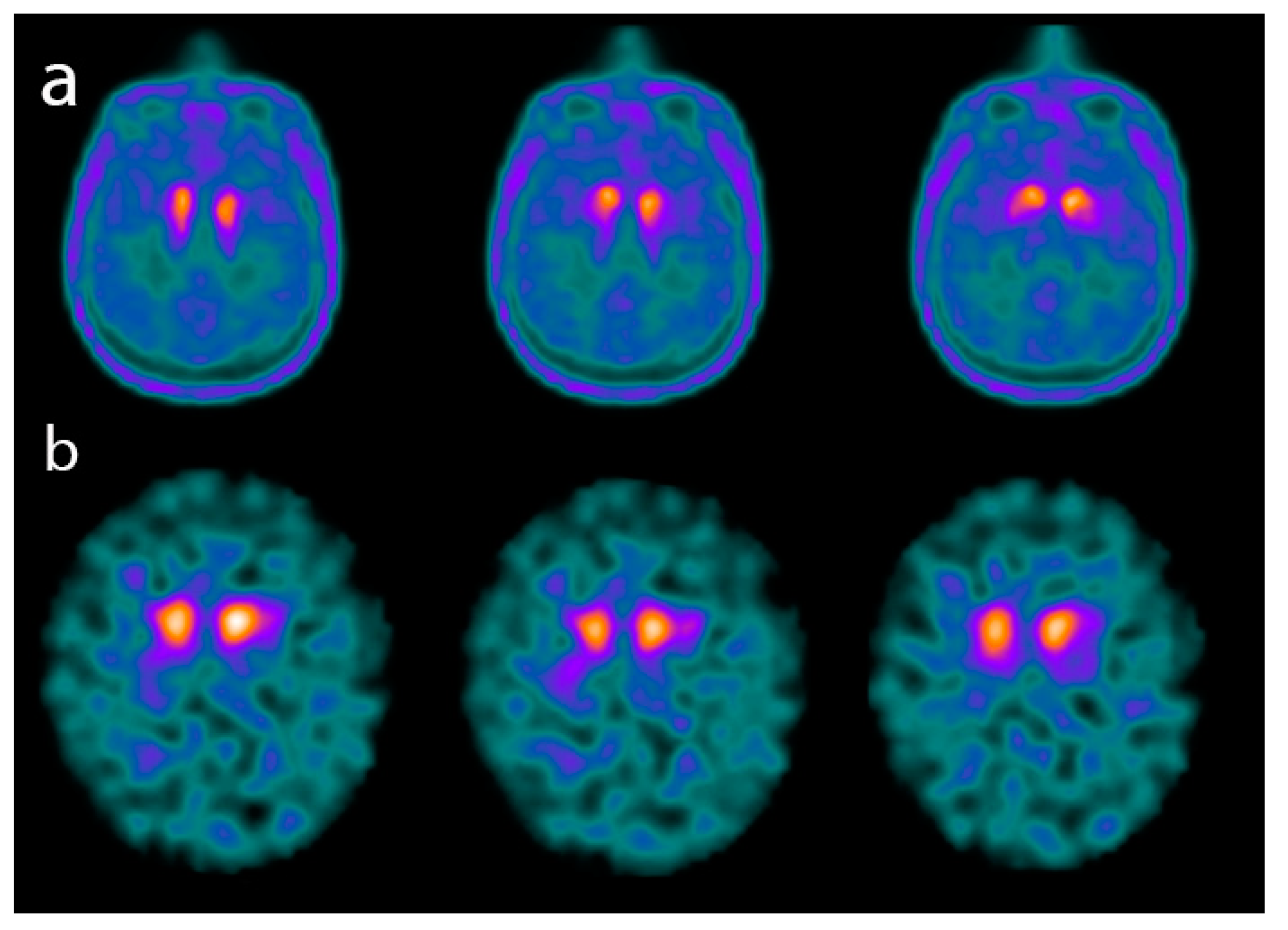
and 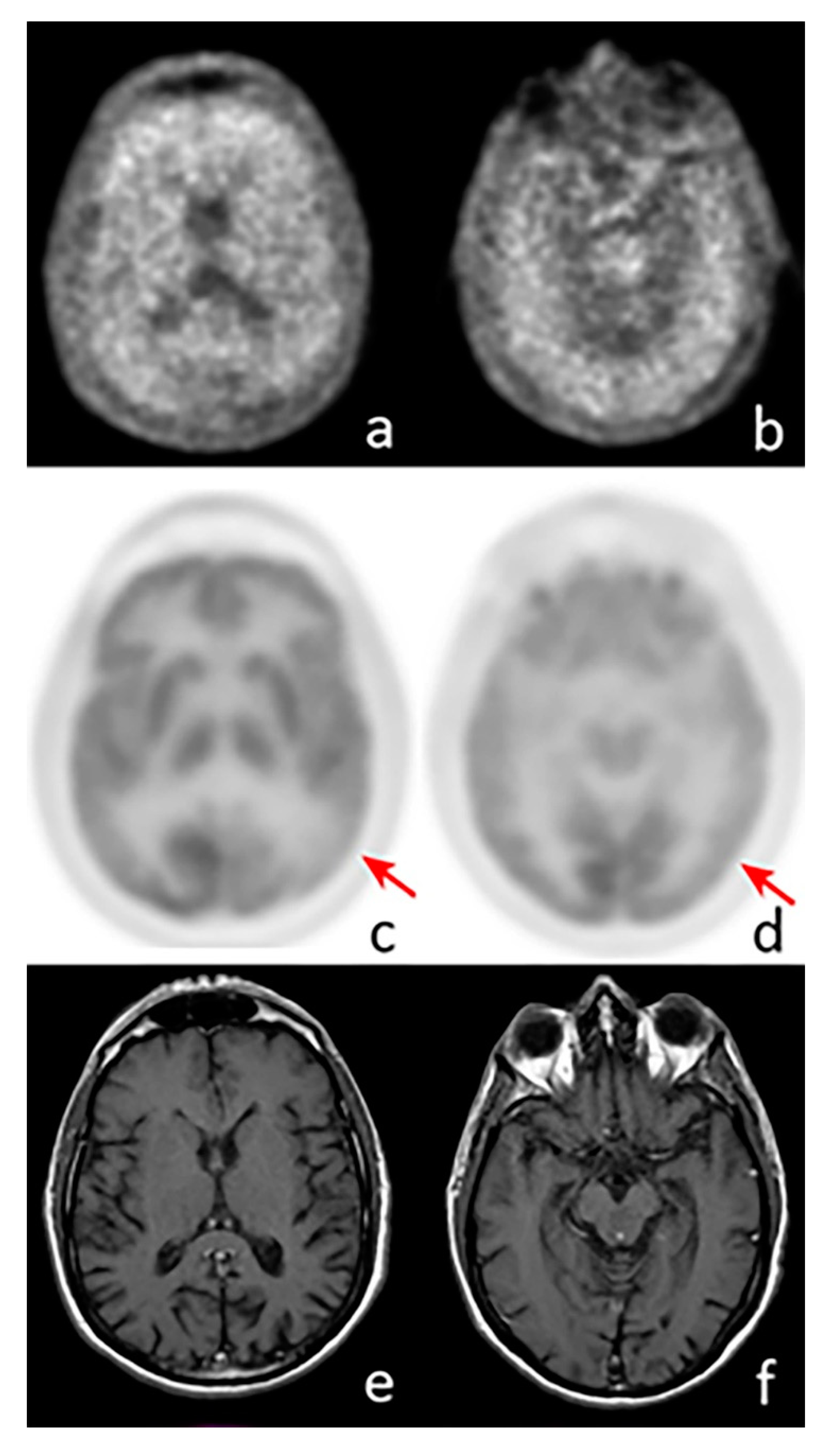{kind=link}
{kind=link}
{kind=link}
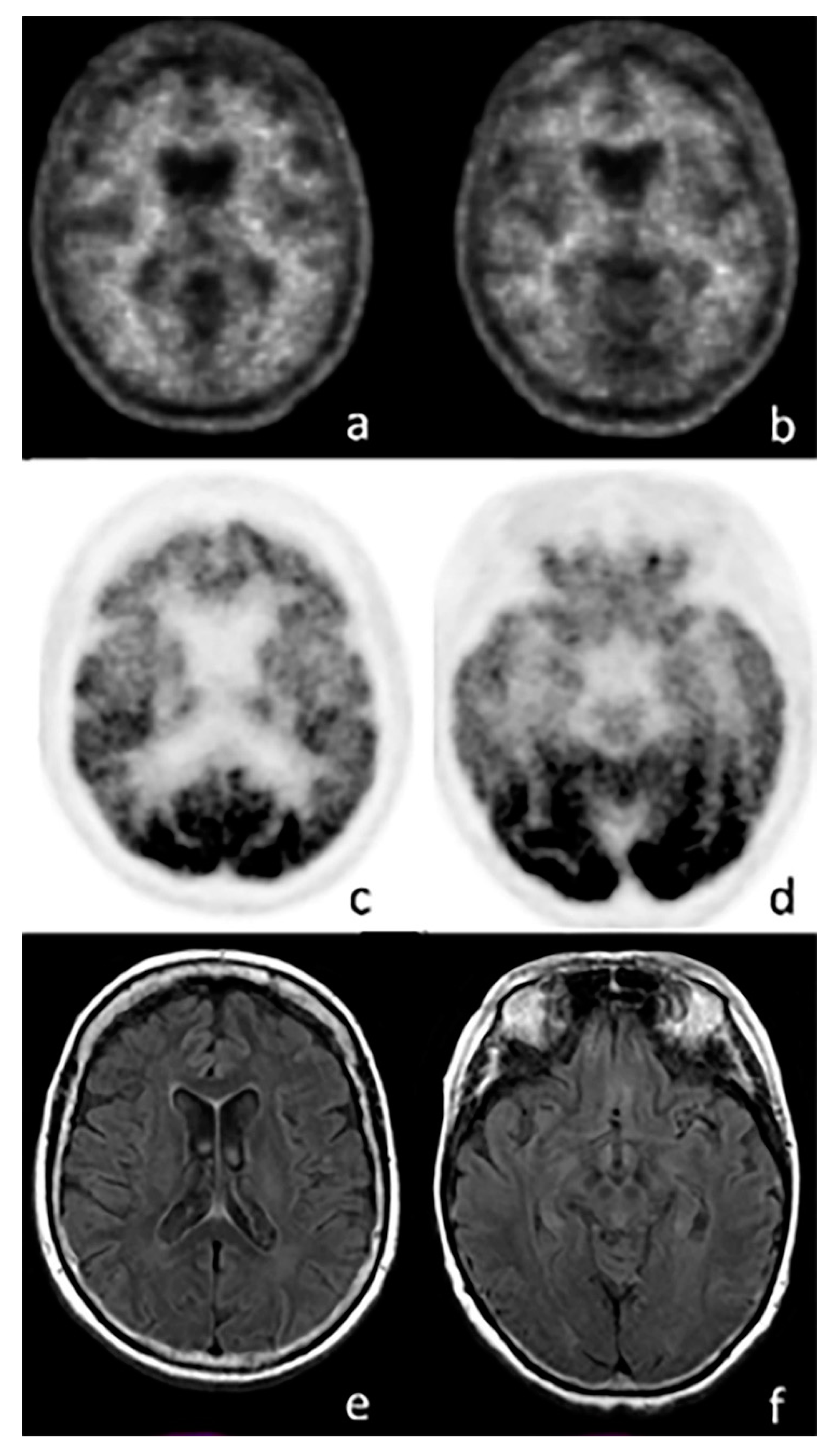
{kind=link}
Abstract
:1. Introduction
2. Alzheimer’s Disease (AD)
2.1. FDG Imaging in AD
2.2. Amyloid Imaging in AD
2.3. Tau Imaging in AD
3. Frontotemporal Dementia (FTD)
3.1. FDG-PET in FTD
3.2. Amyloid Imaging in FTD
3.3. Tau Imaging in FTD
4. The Alpha-Synucleinopathies (SNCApathies)
4.1. Dopamine Transporter (DAT) Imaging in SNCApathies
4.2. Cardiac Sympathetic Innervation Imaging in SNCApathies
4.3. FDG-PET Imaging in SNCApathies
4.4. Amyloid Imaging in SNCApathies
4.5. Tau Imaging in SNCApathies
4.6. Alpha-Synuclein Imaging in SNCApathies
5. Other Atypical Parkinsonian Syndromes
5.1. FDG-PET Imaging in Atypical Parkinsonian Syndromes
5.2. DAT Imaging in Other Atypical Parkinsonian Syndromes
5.3. Tau Imaging in Other Atypical Parkinsonian Syndromes
6. Summary of Techniques Validated for Clinical Practice
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| FTD | Frontotemporal dementia |
| SNCApathies | The alpha-synucleinopathies |
References
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013, 56, 963–975.e2. [Google Scholar] [CrossRef] [PubMed]
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 7, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Fox, N.C.; Jack, C.R., Jr.; Scheltens, P.; Thompson, P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Rathore, S.; Habes, M.; Iftikhar, M.A.; Shacklett, A.; Davatzikos, C. A review on neuroimaging-based classification studies and associated feature extraction methods for Alzheimer’s disease and its prodromal stages. Neuroimage 2017, 15, 530–548. [Google Scholar] [CrossRef]
- Mosconi, L.; Berti, V.; Glodzik, L.; Pupi, A.; De Santi, S.; de Leon, M.J. Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without amyloid imaging. J. Alzheimers Dis. 2010, 20, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Popuri, K.; Balachandar, R.; Alpert, K.; Lu, D. Development and validation of a novel dementia of Alzheimer’s type (DAT) score based on metabolism FDG-PET imaging. Neuroimage Clin. 2018, 10, 802–813. [Google Scholar] [CrossRef]
- Pagani, M.; De Carli, F.; Morbelli, S.; Öberg, J.; Chincarini, F.; Frisoni, G.B.; Galluzzi, S.; Perneczky, R.; Drzezga, A.; van Berckel, B.N.M.; et al. Volume of interest-based 18Ffluorodeoxyglucose PET discriminates MCI converting to Alzheimer’s disease from healthy controls. A European Alzheimer’s Disease Consortium (EADC) study. Neuroimage Clin. 2014, 7, 34–42. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaravalloti, A.; Koch, G.; Toniolo, S.; Belli, L.; Di Lorenzo, F.; Gaudenzi, S.; Schillaci, O.; Bozzali, M.; Sancesario, G.; Martorana, A. Comparison between Early-Onset and Late-Onset Alzheimer’s Disease Patients with Amnestic Presentation: CSF and (18)F-FDG PET Study. Dement. Geriatr. Cogn. Dis. Extra 2016, 6, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Nobili, F.; Arbizu, J.; Bouwman, F.; Drzezga, A.; Agosta, F.; Nestor, P.; Walker, Z.; Boccardi, M. European Association of Nuclear Medicine and European Academy of Neurology recommendations for the use of brain 18 F-fluorodeoxyglucose positron emission tomography in neurodegenerative cognitive impairment and dementia: Delphi consensus. Eur. J. Neurol. 2018, 25, 1201–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, M.; Chiaravalloti, A.; Martorana, A.; Koch, G.; De Lucia, V.; Barbagallo, G.; Schillaci, O. The role of epsilon phenotype in brain glucose consumption in Alzheimer’s disease. Ann. Nucl. Med. 2020, 34, 254–262. [Google Scholar] [CrossRef]
- Frantellizzi, V.; Pani, A.; Ricci, M.; Locuratolo, N.; Fattapposta, F.; De Vincentis, G. Neuroimaging in Vascular Cognitive Impairment and Dementia: A Systematic Review. J. Alzheimers Dis. 2020, 73, 1279–1294. [Google Scholar] [CrossRef]
- Shimada, A.; Hashimoto, H.; Kawabe, J.; Higashiyama, S.; Kai, T.; Kataoka, K.; Tagawa, R.; Kawarada, Y.; Nakanishi, A.; Inoue, K.; et al. Evaluation of therapeutic response to donepezil by positron emission tomography. Osaka City Med. J. 2011, 57, 11–19. [Google Scholar]
- Mega, M.S.; Dinov, I.D.; Porter, V.; Chow, G.; Reback, E.; Davoodi, P.; O’Connor, S.M.; Carter, M.F.; Amezcua, H.; Cummings, J.L. Metabolic patterns associated with the clinical response to galantamine therapy: A fludeoxyglucose f 18 positron emission tomographic study. Arch. Neurol. 2005, 62, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Huang, Q.; Reiman, E.M.; Chen, K.; Li, X.; Li, G.; Lin, Z.; Li, C.; Xiao, S. Effects of memantine on clinical ratings, fluorodeoxyglucose positron emission tomography measurements, and cerebrospinal fluid assays in patients with moderate to severe Alzheimer dementia: A 24-week, randomized, clinical trial. J. Clin. Psychopharmacol. 2013, 33, 636–642. [Google Scholar] [CrossRef] [Green Version]
- Sultzer, D.L.; Melrose, R.J.; Harwood, D.G.; Campa, O.; Mandelkern, M.A. Effect of memantine treatment on regional cortical metabolism in Alzheimer’s disease. Am. J. Geriatr. Psychiatr. 2010, 18, 606–614. [Google Scholar] [CrossRef]
- Rice, L.; Bisdas, S. The diagnostic value of FDG and amyloid PET in Alzheimer’s disease-A systematic review. Eur. J. Radiol. 2017, 94, 16–24. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–419. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.C.; Villemagne, V.L. Brain amyloid imaging. J. Nucl. Med. 2011, 52, 1733–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, C.C.; Ackerman, U.; Browne, W.; Mulligan, R.; Pike, K.L.; O’Keefe, G.; Tochon-Danguy, H.; Chan, G.; Berlangieri, S.U.; Jones, G.; et al. Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: Proof of mechanism. Lancet Neurol. 2008, 7, 129–135. [Google Scholar] [CrossRef]
- Syed, Y.Y.; Deeks, E. [(18)F]Florbetaben: A review in β-amyloid PET imaging in cognitive impairment. CNS Drugs 2015, 29, 605–613. [Google Scholar] [CrossRef]
- Sabri, O.; Seibyl, J.; Rowe, C.; Barthel, H. Beta-amyloid imaging with florbetaben. Clin. Transl. Imaging 2015, 3, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Oya, S.; Kung, M.; Hou, C.; Maier, D.L.; Kung, H.F. F-18 Polyethyleneglycol stilbenes as PET imaging agents targeting Abeta aggregates in the brain. Nucl. Med. Biol. 2005, 32, 799–809. [Google Scholar] [CrossRef]
- Sabri, O.; Sabbagh, M.N.; Seibyl, J.; Barthel, H.; Akatsu, H.; Ouchi, Y.; Senda, K.; Murayama, S.; Ishii, K.; Takao, M.; et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: Phase 3 study. 1. Alzheimers Dement. 2015, 11, 964–974. [Google Scholar] [CrossRef] [Green Version]
- Ikonomovic, M.D.; Klunk, W.E.; Abrahamson, E.E.; Mathis, C.A.; Price, J.C.; Tsopelas, N.D.; Lopresti, B.J.; Ziolko, S.; Bi, W.; Paljung, W.R.; et al. Postmortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 2008, 131 Pt 6, 1630–1645. [Google Scholar] [CrossRef] [Green Version]
- Ong, K.T.; Villemagne, V.L.; Bahar-Fuchs, A.; Lamb, F.; Langdon, N.; Catafau, A.M.; Stephens, A.W.; Seibyl, J.; Dinkelborg, L.M.; Reininger, C.B.; et al. Aβ imaging with 18F-florbetaben in prodromal Alzheimer’s disease: A prospective outcome study. J. Neurol. Neurosurg. Psychiatry 2015, 86, 431–436. [Google Scholar] [CrossRef]
- Koivunen, J.; Scheinin, N.; Virta, J.R.; Aalto, S.; Vahlberg, T.; Nagren, K.; Helin, S.; Parkkola, R.; Viitanen, M.; Rinne, J.O. Amyloid PET imaging in patients with mild cognitive impairment: A 2-year follow-up study. Neurology 2011, 76, 1085–1090. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Honigberg, L.A.; Cho, W.; Ward, M.; Friesenhahn, M.; Brunstein, F.; Quartino, A.; Clayton, D.; Mortensen, D.; Bittner, T.; et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res. Ther. 2018, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017, 9, 95. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Mormino, E.C.; Betensky, R.A.; Hedden, T.; Schultz, A.P.; Amariglio, R.E.; Rentz, D.M.; Johnson, K.A.; Sperling, R.A. Synergistic effect of β-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol. 2014, 71, 1379–1485. [Google Scholar] [CrossRef]
- Tiepolt, S.; Hesse, S.; Patt, M.; Luthardt, J.; Schroeter, M.L.; Hoffmann, K.; Weise, D.; Gertz, H.J.; Sabri, O.; Barthel, H.; et al. Early [(18)F]florbetaben and [(11)C]PiB PET images are a surrogate biomarker of neuronal injury in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1700–1709. [Google Scholar] [CrossRef]
- Filippi, L.; Chiaravalloti, A.; Bagni, O.; Schillaci, O. 18 F-labeled radiopharmaceuticals for the molecular neuroimaging of amyloid plaques in Alzheimer’s disease. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 268–281. [Google Scholar]
- Florek, L.; Tiepolt, S.; Schroeter, M.L.; Berrouschot, J.; Saur, D.; Hesse, S.; Jochimsen, T.; Luthardt, J.; Sattler, B.; Patt, M.; et al. Dual Time-Point 18FFlorbetaben PET Delivers Dual Biomarker Information in Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimers Dis. 2018, 66, 1105–1116. [Google Scholar] [CrossRef]
- Kolb, H.C.; Andrés, J.I. Tau Positron Emission Tomography Imaging. Cold Spring Harb. Perspect Biol. 2017, 9, a023721. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Dickerson, B.C. Multimodal PET Imaging of Amyloid and Tau Pathology in Alzheimer Disease and Non-Alzheimer Disease Dementias. PET Clin. 2017, 12, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K.; et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholl, M.; Lockhart, S.M.; Schonhaut, D.R.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.D.; et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 2016, 89, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Blennow, K.; Johnson, K.; Keeley, M.; Bateman, R.J.; Molinuevo, J.L.; Touchon, J.; Aisen, P.; Vellas, B. Anti-Tau Trials for Alzheimer’s Disease: A Report from the EU/US/CTAD Task Force. J. Prev. Alz. Dis. 2019, 6, 157–163. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Fodero-Tavoletti, M.T.; Masters, C.L.; Rowe, C.C. Tau imaging: Early progress and future directions. Lancet Neurol. 2015, 14, 114–124. [Google Scholar] [CrossRef]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal dementia: Latest evidence and clinical implications. Ther. Adv. Psychopharmacol. 2018, 8, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [Green Version]
- Morgan, S.; Kemp, P.; Booji, J.; Costa, D.C.; Padayachee, S.; Lee, L.; Barber, C.; Carter, J.; Walker, Z. Differentiation of frontotemporal dementia from dementia with Lewy bodies using FP-CIT SPECT. J. Neurol. Neurosurg. Psychiatry 2012, 83, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Claassen, D.O.; Parisi, J.E.; Giannini, C.; Boeve, B.F.; Dickson, D.W.; Josephs, K.A. Frontotemporal dementia mimicking dementia with Lewy bodies. Cogn. Behav. Neurol. 2008, 21, 157–163. [Google Scholar] [CrossRef]
- Chow, T.; Graff-Guerrero, A.; Verhoeff, N.P.L.G.; Binns, M.A.; Tang-Wai, D.F.; Freedman, M.; Masellis, M.; Black, S.E.; Wilson, A.A.; Houle, S.; et al. Open-label study of the short-term effects of memantine on FDG-PET in frontotemporal dementia. Neuropsychiatr. Dis. Treat. 2011, 7, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Villemagne, V.L.; Ong, K.; Mulligan, R.S.; Holl, G.; Pejoska, S.; Jones, G.; O’Keefe, G.; Ackerman, U.; Tochon-Danguy, H.; Gordon Chan, J.; et al. Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J. Nucl. Med. 2011, 52, 1210–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, H.; Frizell Santillo, A.; Wang, S.X.; Lindau, M.; Savitcheva, I.; Nordberg, A.; Lannfelt, L.; Langstrom, B.; Kilander, L. In vivo amyloid imaging with PET in frontotemporal dementia. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Minoshima, S.; Bohnen, N.I.; Donohoe, K.J.; Foster, N.L.; Herscovitch, P.; Karlawish, J.H.; Rowe, C.C.; Carrillo, M.C.; Hartley, D.M.; et al. Appropriate use criteria for amyloid PET: A report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement 2013, 9, e-1-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, J.A.; Burns, J.M.; Hou, C.E.; McKeel, D.W., Jr.; Storandt, M.; Morris, J.C. Progressive posterior cortical dysfunction: A clinicopathologic series. Neurology 2004, 63, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Merusalam, M.; Wicklund, A.; Johnson, N.; Rogalski, E.; Leger, G.C.; Rademaker, A.; Weintraub, S.; Bigio, E.H. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann. Neurol. 2008, 63, 709–719. [Google Scholar] [CrossRef]
- Sander, K.; Lashley, T.; Gami, P.; Gendron, T.; Lythgoe, M.F.; Rohrer, J.D. Characterization of tau positron emission tomography tracer [18 F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimers Dement 2016, 12, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousaf, T.; Dervenoulas, G.; Valkimadi, P.; Politis, M. Neuroimaging in Lewy body dementia. J. Neurol. 2019, 266, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Arbizu, J.; Luquin, M.R.; Abella, J.; de la Fuente-Fernández, R.; Fernandez-Torrón, R.; García-Solís, D.; Garrastachu, P.; Jimenez-Hoyuela, J.M.; Llaneza, M.; Lomena, F.; et al. Functional neuroimaging in the diagnosis of patients with Parkinsonism: Update and recommendations for clinical use. Rev. Esp. Med. Nucl. Imagen Mol. 2014, 33, 215–226. [Google Scholar] [CrossRef]
- Palma, J.; Norcliffe-Kaufmann, L.; Kaufmann, H. Diagnosis of multiple system atrophy. Auton. Neurosci. 2018, 211, 15–25. [Google Scholar] [CrossRef] [Green Version]
- McCleery, J.; Morgan, S.; Bradley, K.M.; Noel-Storr, A.H.; Ansorge, O.; Hyde, C. Dopamine transporter imaging for the diagnosis of dementia with Lewy bodies. Cochrane Database Syst. Rev. 2015, 1, CD010633. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.; O’Brien, J.; Walker, Z.; Tatsch, K.; Booij, J.; Darcourt, J.; Padovani, A.; Giubbini, R.; Bonuccelli, U.; Volterrani, D.; et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: A phase III, multicentre study. Lancet Neurol. 2007, 6, 305–313. [Google Scholar] [CrossRef]
- Gebus, O.; Montaut, S.; Monga, B.; Wirth, T.; Cheraud, C.; Alves Do Rego, C.; Zinchenko, I.; Carrè, G.; Hamdaoui, M.; Hautecloque, G.; et al. Deciphering the causes of sporadic late-onset cerebellar ataxias: A prospective study with implications for diagnostic work. J. Neurol. 2017, 264, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J.; Seppi, K.; Neuroimaging Working Group on MSA. Proposed neuroimaging criteria for the diagnosis of multiple system atrophy. Mov. Disord. 2009, 24, 949–964. [Google Scholar] [CrossRef]
- Goldstein, D.S. Dysautonomia in Parkinson disease. Compr. Physiol. 2014, 4, 805–826. [Google Scholar] [CrossRef] [Green Version]
- Nagayama, H.; Ueda, M.; Yamazaki, M.; Nishiyama, Y.; Hamamoto, M.; Katayama, Y. Abnormal cardiac [(123)I]-meta-iodobenzylguanidine uptake in multiple system atrophy. Mov. Disord. 2010, 25, 1744–1747. [Google Scholar] [CrossRef]
- Noordzij, W.; Glaudemans, A.W.J.M.; Juarez-Orozco, L.E.; Slart, R.H.J.A. Towards consensus in acquisition and image analysis of PET and SPECT in the assessment of cardiac sympathetic innervation: A mini-review. Clin. Transl. Imaging 2019, 7, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.C.; Poston, K.L.; Eckert, T.; Feigin, A.; Frucht, S.; Gudesblatt, M.; Dhawan, V.; Lesser, M.; Vonsattel, J.P.; Fahn, S.; et al. Differential diagnosis of Parkinsonism: A metabolic imaging study using pattern analysis. Lancet Neurol. 2010, 9, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Sarro, L.; Senjem, M.L.; Lundt, E.S.; Przybelski, S.A.; Lesnick, T.G.; Graff-Radford, J.; Boeve, B.F.; Lowe, V.L.; Ferman, T.J.; Knopman, D.S.; et al. Amyloid-β deposition and regional grey matter atrophy rates in dementia with Lewy bodies. Brain 2016, 139 Pt 10, 2740–2750. [Google Scholar] [CrossRef]
- Saeed, U.; Compagnone, J.; Aviv, R.I.; Strafella, A.P.; Black, S.E.; Lang, A.E.; Masellis, M. Imaging biomarkers in Parkinson’s disease and Parkinsonian syndromes: Current and emerging concepts. Transl. Neurodegener. 2017, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Catafau, A.M.; Bullich, S. Amyloid PET imaging: Applications beyond Alzheimer’s disease. Clin. Transl. Imaging 2015, 3, 39–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomperts, S.N.; Locascio, J.J.; Marquie, M.; Santarlasci, A.L.; Rentz, D.M.; Maye, J.; Johnson, K.A.; Growdon, J.H. Brain amyloid and cognition in Lewy body diseases. Mov. Disord. 2012, 27, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, S.N.; Locascio, J.J.; Makaretz, S.J.; Schultz, A.; Caso, C.; Vasdev, N.; Sperling, R.; Growdon, J.H.; Dickerson, B.C.; Johnson, K. Tau Positron Emission Tomographic Imaging in the Lewy Body Diseases. JAMA Neurol. 2016, 73, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, K.; Lowe, V.J.; Boeve, B.F.; Senjem, M.L.; Tosakulwong, N.; Lesnick, T.G.; Spychalla, A.J.; Gunter, J.L.; Fields, J.A.; Graff-Radford, J.; et al. AV-1451 tau and β-amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann. Neurol. 2017, 81, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Verdurand, M.; Levigoureux, E.; Lancelot, S.; Zeinyeh, W.; Billard, T.; Quadrio, I.; Perret-Liaudet, A.; Zimmer, L.; Chauveau, F. Amyloid-Beta Radiotracer [18 F]BF-227 Does Not Bind to Cytoplasmic Glial Inclusions of Postmortem Multiple System Atrophy Brain Tissue. Contrast Media Mol. Imaging 2018, 2018, 9165458. [Google Scholar] [CrossRef] [Green Version]
- Fodero-Tavoletti, M.T.; Mulligan, R.S.; Okamura, N.; Furumoto, S.; Rowe, C.C.; Kudo, Y.; Master, C.L.; Cappai, R.; Yanai, K.; Villemagne, V.L. In vitro characterisation of BF227 binding to alpha-synuclein/Lewy bodies. Eur. J. Pharmacol. 2009, 617, 54–58. [Google Scholar] [CrossRef]
- Chu, W.; Zhou, D.; Gaba, V.; Liu, J.; Li, S.; Peng, X.; Xu, J.; Dhavale, D.; Bagchi, D.P.; d’Avignon, A.; et al. Design, Synthesis, and Characterization of 3-(Benzylidene)indolin-2-one Derivatives as Ligands for α-Synuclein Fibrils. J. Med. Chem. 2015, 58, 6002–6017. [Google Scholar] [CrossRef] [Green Version]
- Dickson, D.W.; Bergeron, C.; Chin, S.S.; Duyckaerts, C.; Horoupian, D.; Ikeda, K.; Jellinger, K.; Lantos, P.L.; Lippa, C.F.; Mirra, S.S.; et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J. Neuropathol. Exp. Neurol. 2002, 61, 935–946. [Google Scholar] [CrossRef]
- Dąbrowska, M.; Schinwelski, M.; Sitek, E.J.; Muraszko-Klaudel, A.; Brockhuis, B.; Jamrozik, Z.; Slawek, J. The role of neuroimaging in the diagnosis of the atypical parkinsonian syndromes in clinical practice. Neurol. Neurochir. Pol. 2015, 49, 421–431. [Google Scholar] [CrossRef]
- Rolland, Y.; Vérin, M.; Payan, C.A.; Duchesne, S.; Kraft, E.; Hauser, T.K.; Jarosz, J.; Deasy, N.; Defevbre, L.; Delmaire, C.; et al. A new MRI rating scale for progressive supranuclear palsy and multiple system atrophy: Validity and reliability. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.S.; Chung, S.J.; Kim, S.; Oh, J.S.; Kim, J.S.; Ye, B.S.; Sohn, Y.H.; Lee, P.H. The role of 18F-FP-CIT PET in differentiation of progressive supranuclear palsy and frontotemporal dementia in the early stage. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Arai, K.; Hattori, T. Study of the rostral midbrain atrophy in progressive supranuclear palsy. J. Neurol. Sci. 2003, 210, 57–60. [Google Scholar] [CrossRef]
- Brooks, D.J. Molecular imaging of dopamine transporters. Ageing Res. Rev. 2016, 30, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Schöll, M.; Widner, H.; van Westen, D.; Svenningsson, P.; Hägerström, D.; Ohlsson, T.; Jogi, J.; Nilsson, C.; Hansson, O. In vivo retention of 18 F-AV-1451 in corticobasal syndrome. Neurology 2017, 89, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, A.; Okamura, N.; Hasegawa, T.; Harada, R.; Watanuki, S.; Funaki, Y.; Hiraoka, K.; Baba, T.; Sugeno, N.; Oshima, R.; et al. In vivo visualization of tau deposits in corticobasal syndrome by 18F-THK5351 PET. Neurology 2016, 87, 2309–2316. [Google Scholar] [CrossRef] [Green Version]
- Kepe, V.; Bordelon, Y.; Boxer, A.; Huang, S.; Liu, J.; Thiede, F.C.; Mazziotta, J.C.; Mendez, M.F.; Donoghue, N.; Small, G.W.; et al. PET imaging of neuropathology in tauopathies: Progressive supranuclear palsy. J. Alzheimers Dis. 2013, 36, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Lee, S.H.; Ryu, Y.H.; Lee, M.S.; Lyoo, C.H. Subcortical 18 F-AV-1451 binding patterns in progressive supranuclear palsy. Mov. Disord. 2017, 32, 134–140. [Google Scholar] [CrossRef]
- Ishiki, A.; Harada, R.; Okamura, N.; Tomita, N.; Rowe, C.C.; Villemagne, V.L.; Yanai, K.; Kudo, Y.; Arai, H.; Furumoto, S.; et al. Tau imaging with [18 F]THK-5351 in progressive supranuclear palsy. Eur. J. Neurol. 2017, 24, 130–136. [Google Scholar] [CrossRef]
- Minoshima, S.; Drzezga, A.E.; Barthel, H.; Bohnen, N.; Djekidel, M.; Lewis, D.H.; Mathis, C.A.; McConathy, J.; Nordberg, A.; Sabri, O.; et al. SNMMI Procedure Standard/EANM Practice Guideline for Amyloid PET Imaging of the Brain 1.0. J. Nucl. Med. 2016, 57, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Morbelli, S.; Esposito, G.; Arbizu, J.; Barthel, H.; Boellaard, R.; Bohnen, N.I.; Brooks, D.J.; Darcourt, J.; Dickson, J.C.; Douglas, D.; et al. EANM practice guideline/SNMMI procedure standard for dopaminergic imaging in Parkinsonian syndromes 1.0. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1885–1912. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, M.; Cimini, A.; Chiaravalloti, A.; Filippi, L.; Schillaci, O. Positron Emission Tomography (PET) and Neuroimaging in the Personalized Approach to Neurodegenerative Causes of Dementia. Int. J. Mol. Sci. 2020, 21, 7481. https://doi.org/10.3390/ijms21207481
Ricci M, Cimini A, Chiaravalloti A, Filippi L, Schillaci O. Positron Emission Tomography (PET) and Neuroimaging in the Personalized Approach to Neurodegenerative Causes of Dementia. International Journal of Molecular Sciences. 2020; 21(20):7481. https://doi.org/10.3390/ijms21207481
Chicago/Turabian StyleRicci, Maria, Andrea Cimini, Agostino Chiaravalloti, Luca Filippi, and Orazio Schillaci. 2020. "Positron Emission Tomography (PET) and Neuroimaging in the Personalized Approach to Neurodegenerative Causes of Dementia" International Journal of Molecular Sciences 21, no. 20: 7481. https://doi.org/10.3390/ijms21207481
APA StyleRicci, M., Cimini, A., Chiaravalloti, A., Filippi, L., & Schillaci, O. (2020). Positron Emission Tomography (PET) and Neuroimaging in the Personalized Approach to Neurodegenerative Causes of Dementia. International Journal of Molecular Sciences, 21(20), 7481. https://doi.org/10.3390/ijms21207481