Diabetes Mellitus-Related Dysfunction of the Motor System


Abstract
:1. Introduction
2. Alteration of the Neuromuscular Pathway
2.1. Cell Bodies of MNs
2.2. Motor Axon
2.3. NMJ and Muscle Fibers

2.4. Differences in Vulnerability to Diabetes between MNs
2.5. γ-MNs
3. Alteration of Corticomotoneuronal Pathway
3.1. Primary Motor Cortex and CST
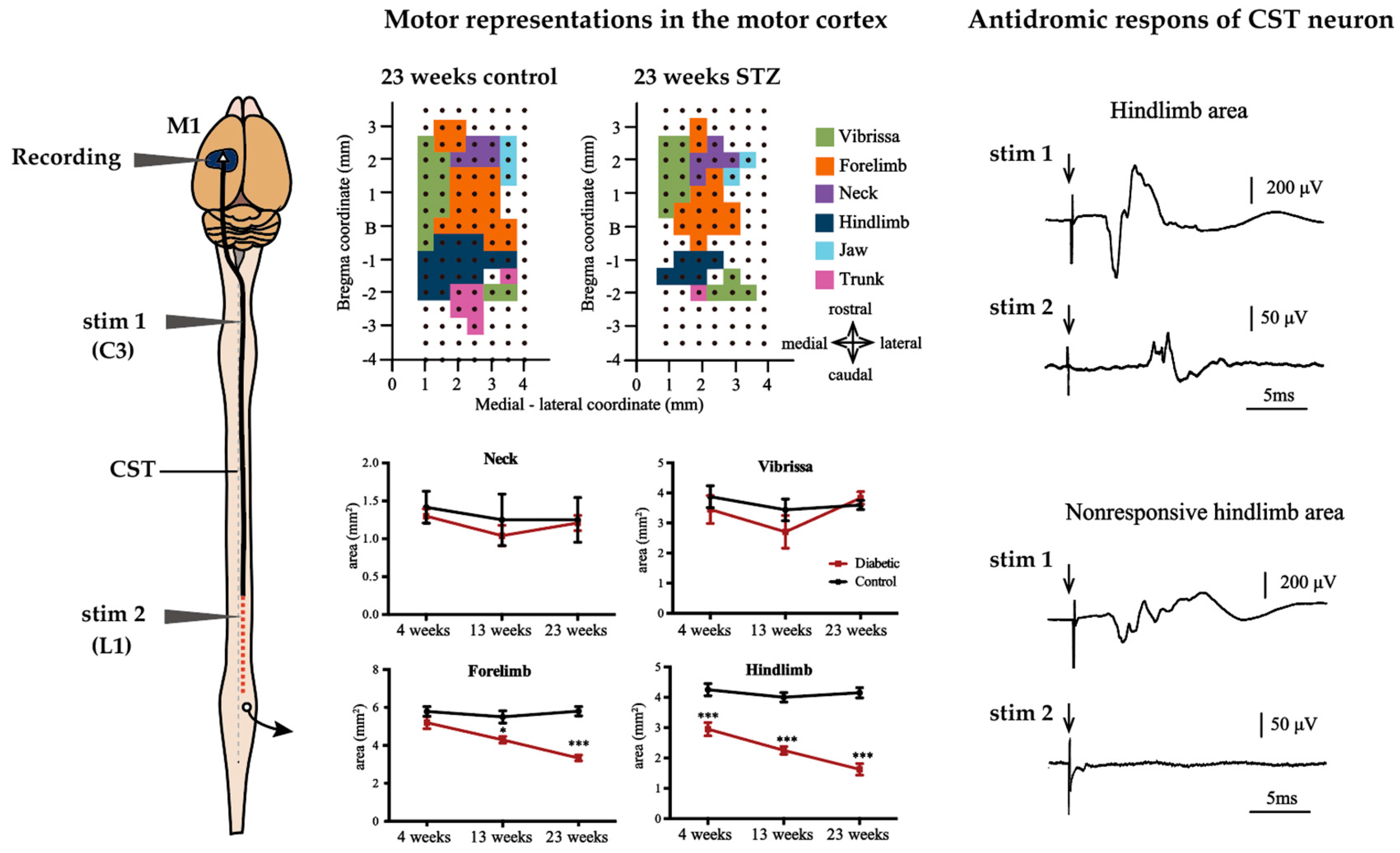
3.2. Alterations of M1
3.3. Alterations of CST
3.4. Possible Movement Disorders Caused by Diabetes-Induced Lesions of M1 and CST
3.5. Possible Therapies on Corticomotoneuronal Pathway Lesion in Diabetes
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DN | diabetic neuropathy |
| PNS | peripheral nervous system |
| DRG | dorsal root ganglion |
| BBB | blood-brain barrier |
| STZ | streptozotocin |
| BB | Bio-Breeding |
| MNCV | motor nerve conduction velocity |
| NMJ | neuromuscular junction |
| M1 | primary motor cortex |
| CST | corticospinal tract |
| MN | motoneuron |
| ACh | acetylcholine |
| ROS | reactive oxygen species |
| AGEs | advanced glycation end-products |
| RAGE | receptor for AGEs |
References
- Said, G. Diabetic neuropathy—A review. Nat. Rev. Neurol. 2007, 3, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, X.; Zhang, Q.; Zou, R. Diabetes mellitus and risk of falls in older adults: A systematic review and meta-analysis. Age Ageing 2016, 45, 761–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrofsky, J.; Lee, S.; Cuneo, M.L. Gait characteristics in patients with type 2 diabetes; improvement after administration of rosiglitazone. Med. Sci. Monit. 2005, 11, 43–51. [Google Scholar]
- Uccioli, L.; Giacomini, P.G.; Monticone, G.; Magrini, A.; Durola, L.; Bruno, E.; Parisi, L.; Di Girolamo, S.; Menzinger, G. Body sway in diabetic neuropathy. Diabetes Care 1995, 18, 339–344. [Google Scholar] [CrossRef]
- Volpato, S.; Leveille, S.G.; Blaum, C.; Fried, L.P.; Guralnik, J.M. Risk factors for falls in older disabled women with diabetes: The women’s health and aging study. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1539–1545. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.; Backholer, K.; Gearon, E.; Harding, J.; Freak-Poli, R.; Stevenson, C.; Peeters, A. Diabetes and risk of physical disability in adults: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2013, 1, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Zochodne, D.W.; Ramji, N.; Toth, C. Neuronal Targeting in Diabetes Mellitus: A Story of Sensory Neurons and Motor Neurons. Neuroscientist 2008, 14, 311–318. [Google Scholar] [CrossRef]
- Dyck, P.J.; Kratz, K.M.; Karnes, J.L.; Litchy, W.J.; Klein, R.; Pach, J.M.; Wilson, D.M.; O’Brien, P.C.; Melton, L.J. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: The Rochester Diabetic Neuropathy Study. Neurology 1993, 43, 817. [Google Scholar] [CrossRef]
- Zochodne, D.W.; Verge, V.M.K.; Cheng, C.; Sun, H.; Johnston, J. Does diabetes target ganglion neurones? Brain 2001, 124, 2319–2334. [Google Scholar] [CrossRef] [Green Version]
- Ramji, N.; Toth, C.; Kennedy, J.; Zochodne, D.W. Does diabetes mellitus target motor neurons? Neurobiol. Dis. 2007, 26, 301–311. [Google Scholar] [CrossRef]
- Feldman, E.L.; Nave, K.-A.; Jensen, T.S.; Bennett, D.L.H. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweetnam, D.; Holmes, A.; Tennant, K.A.; Zamani, A.; Walle, M.; Jones, P.; Wong, C.; Brown, C.E. Diabetes impairs cortical plasticity and functional recovery following ischemic stroke. J. Neurosci. 2012, 32, 5132–5143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.D. Stroke and diabetes mellitus. Handb. Clin. Neurol. 2014, 126, 167–174. [Google Scholar] [PubMed]
- Huynh, W.; Kwai, N.; Arnold, R.; Krishnan, A.V.; Lin, C.S.Y.; Vucic, S.; Kiernan, M.C. The Effect of Diabetes on Cortical Function in Stroke: Implications for Poststroke Plasticity. Diabetes 2017, 66, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, H.; Poulsen, P.L.; Mogensen, C.E.; Jakobsen, J. Isokinetic Muscle Strength in Long-Term IDDM Patients in Relation to Diabetic Complications. Diabetes 1996, 45, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.; Nielsen, S.; Mogensen, C.E.; Jakobsen, J. Muscle Strength in Type 2 Diabetes. Diabetes 2004, 53, 1543–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, T.N.; Tuttle, L.J.; Bohnert, K.L.; Mueller, M.J.; Sinacore, D.R. Excessive Adipose Tissue Infiltration in Skeletal Muscle in Individuals with Obesity, Diabetes Mellitus, and Peripheral Neuropathy: Association with Performance and Function. Phys. Ther. 2008, 88, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.; Sommers, M.S.; Lucki, I. Effects of diabetes on hippocampal neurogenesis: Links to cognition and depression. Neurosci. Biobehav. Rev. 2013, 37, 1346–1362. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the Brain: Its Pathophysiological Implications for States Related with Central Insulin Resistance, Type 2 Diabetes and Alzheimer’s Disease. Front. Endocrinol. 2014, 5, 102. [Google Scholar] [CrossRef] [Green Version]
- Malone, J.I. Diabetic Central Neuropathy: CNS Damage Related to Hyperglycemia. Diabetes 2016, 65, 355–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Maciejczyk, M.; Żebrowska, E.; Chabowski, A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Fang, P.; An, J.; Lin, H.; Liang, Y.; Shen, W.; Leng, X.; Zhang, C.; Zheng, Y.; Qiu, S. Micro-structural white matter abnormalities in type 2 diabetic patients: A DTI study using TBSS analysis. Neuroradiology 2016, 58, 1209–1216. [Google Scholar] [CrossRef]
- Eaton, S.E.; Harris, N.D.; Rajbhandari, S.M.; Greenwood, P.; Wilkinson, I.D.; Ward, J.D.; Griffiths, P.D.; Tesfaye, S. Spinal-cord involvement in diabetic peripheral neuropathy. Lancet 2001, 358, 35–36. [Google Scholar] [CrossRef]
- Xiong, Y.; Sui, Y.; Xu, Z.; Zhang, Q.; Karaman, M.M.; Cai, K.; Anderson, T.M.; Zhu, W.; Wang, J.; Zhou, X.J. A Diffusion Tensor Imaging Study on White Matter Abnormalities in Patients with Type 2 Diabetes Using Tract-Based Spatial Statistics. Am. J. Neuroradiol. 2016, 37, 1462–1469. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, J.; Liu, X.; Wang, X.; Xu, X.; Li, H.; Cao, B.; Yang, Y.; Lu, J.; Chen, Z. Abnormal subcortical nuclei shapes in patients with type 2 diabetes mellitus. Eur. Radiol. 2017, 27, 4247–4256. [Google Scholar] [CrossRef]
- Emerick, A.J.; Richards, M.P.; Kartje, G.L.; Neafsey, E.J.; Stubbs, E.B. Experimental diabetes attenuates cerebral cortical-evoked forelimb motor responses. Diabetes 2005, 54, 2764–2771. [Google Scholar] [CrossRef]
- Muramatsu, K.; Ikutomo, M.; Tamaki, T.; Shimo, S.; Niwa, M. Effect of streptozotocin-induced diabetes on motor representations in the motor cortex and corticospinal tract in rats. Brain Res. 2018, 1680, 115–126. [Google Scholar] [CrossRef]
- Svoboda, K.; Li, N. Neural mechanisms of movement planning: Motor cortex and beyond. Curr. Opin. Neurol. 2018, 49, 33–41. [Google Scholar] [CrossRef]
- Lemon, R.N. Neural control of dexterity: What has been achieved? Exp. Brain Res. 1999, 128, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.K.; Inglis, J.T.; Madden, K.M.; Boyd, L.A. Brain and Body: A Review of Central Nervous System Contributions to Movement Impairments in Diabetes. Diabetes 2020, 69, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Isa, T.; Ohki, Y.; Alstermark, B.; Pettersson, L.-G.; Sasaki, S. Direct and Indirect Cortico-Motoneuronal Pathways and Control of Hand/Arm Movements. Physiology 2007, 22, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Manuel, M.; Zytnicki, D. Alpha, beta and gamma motoneurons: Functional diversity in the motor system’s final pathway. J. Integr. Neurosci. 2012, 10, 243–276. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.F. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [Green Version]
- Calderón, J.C.; Bolaños, P.; Caputo, C. The excitation–contraction coupling mechanism in skeletal muscle. Biophys. Rev. 2014, 6, 133–160. [Google Scholar] [CrossRef] [Green Version]
- Sherrington, C.S. The Integrative Action of the Nervous System; Archibald Constable and Co., Ltd.: London, UK, 1906; pp. 36–69. [Google Scholar]
- Burke, R.E.; Levine, D.N.; Tsairis, P.; Zajac, F.E. Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J. Physiol. 1973, 234, 723–748. [Google Scholar] [CrossRef]
- Eldred, E.; Granit, R.; Merton, P.A. Supraspinal control of the muscle spindles and its significance. J. Physiol. 1953, 122, 498–523. [Google Scholar] [CrossRef]
- Yagihashi, S.; Mizukami, H.; Sugimoto, K. Mechanism of diabetic neuropathy: Where are we now and where to go? J. Diabetes. Investig. 2011, 2, 18–32. [Google Scholar] [CrossRef] [Green Version]
- Pop-Busui, R.; Boulton, A.J.M.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic Neuropathy: A Position Statement by the American Diabetes Association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Reske-Nielsen, E.; Lundbaek, K. Pathological changes in the central and peripheral nervous system of young long-term diabetics. II. The spinal cord and peripheral nerves. Diabetologia 1968, 4, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonell, C.W.; Chopek, J.W.; Gardiner, K.R.; Gardiner, P.F. α-Motoneurons maintain biophysical heterogeneity in obesity and diabetes in Zucker rats. J. Neurophysiol. 2017, 118, 2318–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidenius, P.; Jakobsen, J. Reduced Perikaryal Volume of Lower Motor and Primary Sensory Neurons in Early Experimental Diabetes. Diabetes 1980, 29, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, V.B.; Vega, M.C.; Coirini, H. Reduction of the spinal nucleus of the bulbocavernosous volume by experimental diabetes. Brain Res. 2004, 1019, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.J.; Martinez, J.A.; Liu, W.Q.; Zochodne, D.W.; Hanson, L.R.; Frey, W.H.; Toth, C. Motor end plate innervation loss in diabetes and the role of insulin. J. Neuropathol. Exp. Neurol. 2011, 70, 323–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolopoulos-Stournaras, S.; Iles, J.F. Motor neuron columns in the lumbar spinal cord of the rat. J. Comp. Neurol. 1983, 217, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, K.; Niwa, M.; Nagai, M.; Kamimura, T.; Sasaki, S.-I.; Ishiguro, T. The size of motoneurons of the gastrocnemius muscle in rats with diabetes. Neurosci. Lett. 2012, 531, 109–113. [Google Scholar] [CrossRef]
- Muramatsu, K.; Niwa, M.; Tamaki, T.; Ikutomo, M.; Masu, Y.; Hasegawa, T.; Shimo, S.; Sasaki, S.-I. Effect of streptozotocin-induced diabetes on motoneurons and muscle spindles in rats. Neurosci. Res. 2017, 115, 21–28. [Google Scholar] [CrossRef]
- Tamaki, T.; Muramatsu, K.; Ikutomo, M.; Oshiro, N.; Hayashi, H.; Niwa, M. Effects of streptozotocin-induced diabetes on leg muscle contractile properties and motor neuron morphology in rats. Anat. Sci. Int. 2018, 93, 502–513. [Google Scholar] [CrossRef]
- Wilson, N.M.; Wright, D.E. Experimental motor neuropathy in diabetes. Handb. Clin. Neurol. 2014, 126, 461–467. [Google Scholar]
- Sima, A.A.F.; Zhang, W.-X.; Greene, D.A. Diabetic and hypoglycemic neuropathy—A comparison in the BB rat. Diabetes Res. Clin. Pract. 1989, 6, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, S. Hypoglycaemic neuropathy in diabetic BB/Wor rats treated with insulin implants affects ventral root axons but not dorsal root axons. Acta Neuropathol. 2000, 100, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.G.; Locke, S. Motor Nerve Conduction Velocity in Diabetes. Arch. Neurol. 1961, 5, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, G. Variations in motor conduction velocity produced by acute changes of the metabolic state in diabetic patients. Diabetologia 1968, 4, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrington, A.L.; Shaw, J.E.; Van Schie, C.H.M.; Abbott, C.A.; Vileikyte, L.; Boulton, A.J.M. Can Motor Nerve Conduction Velocity Predict Foot Problems in Diabetic Subjects Over a 6-Year Outcome Period? Diabetes Care 2002, 25, 2010–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, J.; Yamada, T.; Nelson, P.S. Distal slowing of motor nerve conduction velocity in diabetic polyneuropathy. J. Neurol. Sci. 1979, 42, 291–302. [Google Scholar] [CrossRef]
- Coppey, L.J.; Davidson, E.P.; Dunlap, J.A.; Lund, D.D.; Yorek, M.A. Slowing of motor nerve conduction velocity in streptozotocin-induced diabetic rats is preceded by impaired vasodilation in arterioles that overlie the sciatic nerve. Int. J. Exp. Diabetes Res. 2000, 1, 131–143. [Google Scholar] [CrossRef]
- Brismar, T. Nodal function of pathological nerve fibers. Cell. Mol. Life Sci. 1983, 39, 946–953. [Google Scholar] [CrossRef]
- Sima, A.A.F.; Brismar, T. Reversible diabetic nerve dysfunction: Structural correlates to electrophysiological abnormalities. Ann. Neurol. 1985, 18, 21–29. [Google Scholar] [CrossRef]
- VargheseCherian, P.; Kamijo, M.; Angelides, K.J.; Sima, A.A.F. Nodal Na+-channel displacement is associated with nerve-conduction slowing in the chronically diabetic BB/W rat: Prevention by aldose reductase inhibition. J. Diabetes. Complicat. 1996, 10, 192–200. [Google Scholar]
- Sima, A.A.; Nathaniel, V.; Bril, V.; McEwen, T.A.; Greene, D.A. Histopathological heterogeneity of neuropathy in insulin-dependent and non-insulin-dependent diabetes, and demonstration of axo-glial dysjunction in human diabetic neuropathy. J. Clin. Investig. 1988, 81, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Sima, A.A.; Lattimer, S.A.; Yagihashi, S.; Greene, D.A. Axo-glial dysjunction. A novel structural lesion that accounts for poorly reversible slowing of nerve conduction in the spontaneously diabetic bio-breeding rat. J. Clin. Investig. 1986, 77, 474–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sima, A.A.; Zhang, W.; Xu, G.; Sugimoto, K.; Guberski, D.; Yorek, M.A. A comparison of diabetic polyneuropathy in type II diabetic BBZDR/Wor rats and in type I diabetic BB/Wor rats. Diabetologia 2000, 43, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Cheng, C.; Sun, H.; Zochodne, D.W. Near nerve local insulin prevents conduction slowing in experimental diabetes. Brain Res. 1997, 763, 209–214. [Google Scholar] [CrossRef]
- Sima, A.A.F.; Zhang, W.; Li, Z.-G.; Murakawa, Y.; Pierson, C.R. Molecular alterations underlie nodal and paranodal degeneration in type 1 diabetic neuropathy and are prevented by C-peptide. Diabetes 2004, 53, 1556–1563. [Google Scholar] [CrossRef] [Green Version]
- Wada, R.; Yagihashi, S. Role of Advanced Glycation End Products and Their Receptors in Development of Diabetic Neuropathy. Ann. N. Y. Acad. Sci. 2005, 1043, 598–604. [Google Scholar] [CrossRef]
- Schlaepfer, W.W.; Gerritsen, G.C.; Dulin, W.E. Segmental demyelination in the distal peripheral nerves of chronically diabetic chinese hamsters. Diabetologia 1974, 10, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Almeida, S.; Riddell, M.C.; Cafarelli, E. Slower conduction velocity and motor unit discharge frequency are associated with muscle fatigue during isometric exercise in type 1 diabetes mellitus. Muscle Nerve 2008, 37, 231–240. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Cragg, B.G. What is the signal for chromatolysis? Brain Res. 1970, 23, 1–21. [Google Scholar] [CrossRef]
- Fink, D.J.; Purkiss, D.; Mata, M. Alterations in retrograde axonal transport in streptozocin-induced diabetic rats. Diabetes 1987, 36, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Kou, Z.; Li, C.; Hu, J.; Zhang, D.L.; Wu, Z.Y.; Ding, T.; Qu, J.; Li, H.; Li, Y.Q. Alterations in the neural circuits from peripheral afferents to the spinal cord: Possible implications for diabetic polyneuropathy in streptozotocin-induced type 1 diabetic rats. Front. Neural Circuits 2014, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.G.; Hohman, T.C.; Cai, F.; Regalia, J.; Helke, C.J. Streptozotocin-Induced Diabetes Causes Metabolic Changes and Alterations in Neurotrophin Content and Retrograde Transport in the Cervical Vagus Nerve. Exp. Neurol. 2001, 170, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Kablar, B.; Belliveau, A.C. Presence of neurotrophic factors in skeletal muscle correlates with survival of spinal cord motor neurons. Dev. Dyn. 2005, 234, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.; Sidenius, P. Decreased axonal flux of retrogradely transported glycoproteins in early experimental diabetes. J. Neurochem. 1979, 33, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Meiri, K.F.; McLean, W.G. Axonal transport of protein in motor fibres of experimentally diabetic rats—Fast anterograde transport. Brain Res. 1982, 238, 77–88. [Google Scholar] [CrossRef]
- Schmidt, R.E.; Modert, C.W.; Yip, H.K.; Johnson, E.M. Retrograde axonal transport of intravenously administered 125I-nerve growth factor in rats with streptozotocin-induced diabetes. Diabetes 1983, 32, 654–663. [Google Scholar] [CrossRef]
- Jakobsen, J.; Sidenius, P. Decreased axonal transport of structural proteins in streptozotocin diabetic rats. J. Clin. Investig. 1980, 66, 292–297. [Google Scholar] [CrossRef]
- Mayer, J.H.; Tomlinson, D.R.; McLean, W.G. Slow Orthograde Axonal Transport of Radiolabelled Protein in Sciatic Motoneurones of Rats with Short-Term Experimental Diabetes: Effects of Treatment with an Aldose Reductase Inhibitor or myo-Inositol. J. Neurochem. 1984, 43, 1265–1270. [Google Scholar] [CrossRef]
- Medori, R.; Autilio-Gambetti, L.; Jenich, H.; Gambetti, P. Changes in axon size and slow axonal transport are related in experimental diabetic neuropathy. Neurology 1988, 38, 597. [Google Scholar] [CrossRef]
- Tomlinson, D.R.; Filliatreau, G.; Figliomeni, B.; Hassig, R.; Di Giamberardino, L.; Willars, G.B. Proteins of slow axonal transport in sciatic motoneurones of rats with streptozotocin-induced diabetes or galactosaemia. Diabetes Res. Clin. Pract. 1990, 9, 15–21. [Google Scholar] [CrossRef]
- Medori, R.; Autilio-Gambetti, L.; Monaco, S.; Gambetti, P. Experimental diabetic neuropathy: Impairment of slow transport with changes in axon cross-sectional area. Proc. Natl. Acad. Sci. USA 1985, 82, 7716–7720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sango, K.; Mizukami, H.; Horie, H.; Yagihashi, S. Impaired Axonal Regeneration in Diabetes. Perspective on the Underlying Mechanism from In Vivo and In Vitro Experimental Studies. Front. Endocrinol. 2017, 8, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier-Larsen, E.; Holzbaur, E.L.F. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1094–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mclean, W.G. The Role of the Axonal Cytoskeleton in Diabetic Neuropathy. Neurochem. Res. 1997, 22, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Buras, E.; Terashima, T.; Serrano, F.; Massaad, C.A.; Hu, L.; Bitner, B.; Inoue, T.; Chan, L.; Pautler, R.G. Hyperglycemia Induces Oxidative Stress and Impairs Axonal Transport Rates in Mice. PLoS ONE 2010, 5, e13463. [Google Scholar] [CrossRef] [Green Version]
- Juranek, J.K.; Geddis, M.S.; Rosario, R.; Schmidt, A.M. Impaired slow axonal transport in diabetic peripheral nerve is independent of RAGE. Eur. J. Neurosci. 2013, 38, 3159–3168. [Google Scholar] [CrossRef]
- Tomlinson, D.R.; Moriarty, R.J.; Mayer, J.H. Prevention and Reversal of Defective Axonal Transport and Motor Nerve Conduction Velocity in Rats with Experimental Diabetes by Treatment with the Aldose Reductase Inhibitor Sorbinil. Diabetes 1984, 33, 470–476. [Google Scholar] [CrossRef]
- Picconi, F.; Mataluni, G.; Ziccardi, L.; Parravano, M.; Di Renzo, A.; Ylli, D.; Pasqualetti, P.; Studer, V.; Chioma, L.; Marfia, G.A.; et al. Association between Early Neuroretinal Dysfunction and Peripheral Motor Unit Loss in Patients with Type 1 Diabetes Mellitus. J. Diabetes Res. 2018, 2018, 9763507. [Google Scholar] [CrossRef] [Green Version]
- Reske-Nielsen, E.; Gregersen, G.; Harmsen, A.; Lundbaek, K. Morphological abnormalities of the terminal neuromuscular apparatus in recent juvenile diabetes. Diabetologia 1970, 6, 104–109. [Google Scholar] [CrossRef] [Green Version]
- Fahim, M.A.; El-Sabban, F.; Davidson, N. Muscle contractility decrement and correlated morphology during the pathogenesis of streptozotocin-diabetic mice. Anat. Rec. 1998, 251, 240–244. [Google Scholar] [CrossRef]
- Fahim, M.A.; Hasan, M.Y.; Alshuaib, W.B. Early morphological remodeling of neuromuscular junction in a murine model of diabetes. J. Appl. Physiol. 2000, 89, 2235–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, I.; Okazaki, M.; Kimura, M. Streptozocin-Diabetes Modifies Acetylcholine Release from Mouse Phrenic Nerve Terminal and Presynaptic Sensitivity to Succinylcholine. Jpn. J. Pharmacol. 1993, 62, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, T.; Kawae, T.; Kataoka, H.; Ikeda, Y. Loss of lower extremity muscle strength based on diabetic polyneuropathy in older patients with type 2 diabetes: Multicenter Survey of the Isometric Lower Extremity Strength in Type 2 Diabetes: Phase 2 study. J. Diabetes. Investig. 2020, 2019, 170. [Google Scholar] [CrossRef]
- Ijzerman, T.H.; Schaper, N.C.; Melai, T.; Meijer, K.; Willems, P.J.B.; Savelberg, H.H.C.M. Lower extremity muscle strength is reduced in people with type 2 diabetes, with and without polyneuropathy, and is associated with impaired mobility and reduced quality of life. Diabetes Res. Clin. Pract. 2012, 95, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.D.; Kimpinski, K.; Doherty, T.J.; Rice, C.L. Length dependent loss of motor axons and altered motor unit properties in human diabetic polyneuropathy. Clin. Neurophysiol. 2014, 125, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Choi, I.H.; Kimpinski, K.; Doherty, T.J.; Rice, C.L. Motor unit loss and weakness in association with diabetic neuropathy in humans. Muscle Nerve 2013, 48, 298–300. [Google Scholar] [CrossRef]
- Andersen, H.; Stålberg, E.; Gjerstad, M.D.; Jakobsen, J. Association of muscle strength and electrophysiological measures of reinnervation in diabetic neuropathy. Muscle Nerve 1998, 21, 1647–1654. [Google Scholar] [CrossRef]
- Andersen, H.; Gadeberg, P.C.; Brock, B.; Jakobsen, J. Muscular atrophy in diabetic neuropathy: A stereological magnetic resonance imaging study. Diabetologia 1997, 40, 1062–1069. [Google Scholar] [CrossRef]
- Andersen, H.; Gjerstad, M.D.; Jakobsen, J. Atrophy of foot muscles: A measure of diabetic neuropathy. Diabetes Care 2004, 27, 2382–2385. [Google Scholar] [CrossRef] [Green Version]
- Thorne, R.G.; Pronk, G.J.; Padmanabhan, V.; Frey, W.H., II. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 2004, 127, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Murakawa, Y.; Sima, A.A.F. Expression and localization of insulin receptor in rat dorsal root ganglion and spinal cord. J. Peripher. Nerv. Syst. 2002, 7, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Grote, C.W.; Wright, D.E. A Role for Insulin in Diabetic Neuropathy. Front. Neurosci. 2016, 10, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, H. Motor function in diabetic neuropathy. Acta Neurol. Scand. 1999, 100, 211–220. [Google Scholar] [CrossRef]
- Andersen, H. Motor dysfunction in diabetes. Diabetes Metab. Res. Rev. 2012, 28, 89–92. [Google Scholar] [CrossRef]
- Andersen, H. Motor neuropathy. Handb. Clin. Neurol. 2014, 126, 81–95. [Google Scholar]
- Perry, B.D.; Caldow, M.K.; Brennan-Speranza, T.C.; Sbaraglia, M.; Jerums, G.; Garnham, A.; Wong, C.; Levinger, P.; Asrar Ul Haq, M.; Hare, D.L.; et al. Muscle atrophy in patients with Type 2 Diabetes Mellitus: Roles of inflammatory pathways, physical activity and exercise. Exerc. Immunol. Rev. 2016, 22, 94–109. [Google Scholar]
- Monaco, C.M.F.; Gingrich, M.A.; Hawke, T.J. Considering Type 1 Diabetes as a Form of Accelerated Muscle Aging. Exerc. Sport Sci. Rev. 2019, 47, 98–107. [Google Scholar] [CrossRef]
- Mesinovic, J.; Zengin, A.; De Courten, B.; Ebeling, P.R.; Scott, D. Sarcopenia and type 2 diabetes mellitus: A bidirectional relationship. Diabetes Metab. Syndr. Obes. 2019, 12, 1057–1072. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, E.M.; Helber, M.D.; Dealva, D.; Ashton-Miller, J.A.; Richardson, J.K. Mild diabetic neuropathy affects ankle motor function. Clin. Biomech. 2001, 16, 522–528. [Google Scholar] [CrossRef]
- Sacchetti, M.; Scotto Sacchetti, M.; Balducci, S.; Bazzucchi, I.; Carlucci, F.; Scotto di Palumbo, A.; Di Palumbo, A.S.; Haxhi, J.; Conti, F.; Di Biase, N.; et al. Neuromuscular dysfunction in diabetes: Role of nerve impairment and training status. Med. Sci. Sports Exerc. 2013, 45, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Goodpaster, B.H.; Strotmeyer, E.S.; Kuller, L.H.; Broudeau, R.; Kammerer, C.; de Rekeneire, N.; Harris, T.B.; Schwartz, A.V.; Tylavsky, F.A.; et al. Accelerated Loss of Skeletal Muscle Strength in Older Adults with Type 2 Diabetes. Diabetes Care 2007, 30, 1507–1512. [Google Scholar] [CrossRef] [Green Version]
- Lesniewski, L.A.; Miller, T.A.; Armstrong, R.B. Mechanisms of force loss in diabetic mouse skeletal muscle. Muscle Nerve 2003, 28, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Eshima, H.; Tamura, Y.; Kakehi, S.; Nakamura, K.; Kurebayashi, N.; Murayama, T.; Kakigi, R.; Sakurai, T.; Kawamori, R.; Watada, H. Dysfunction of muscle contraction with impaired intracellular Ca2+ handling in skeletal muscle and the effect of exercise training in male db/dbmice. J. Appl. Physiol. 2019, 126, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.B.; Gollnick, P.D.; Ianuzzo, C.D. Histochemical properties of skeletal muscle fibers in streptozotocin-diabetic rats. Cell Tissue Res. 1975, 162, 387–393. [Google Scholar] [CrossRef] [PubMed]
- MÅrin, P.; Andersson, B.; Krotkiewski, M.; Björntorp, P. Muscle Fiber Composition and Capillary Density in Women and Men with NIDDM. Diabetes Care 1994, 17, 382–386. [Google Scholar] [CrossRef]
- Greene, D.A.; Lewis, R.A.; Lattimer, S.A.; Brown, M.J. Selective effects of myo-inositol administration on sciatic and tibial motor nerve conduction parameters in the streptozocin-diabetic rat. Diabetes 1982, 31, 573–578. [Google Scholar] [CrossRef]
- Cameron, N.E.; Cotter, M.A.; Harrison, J. Effect of diabetes on motor conduction velocity in different branches of the rat sciatic nerve. Exp. Neurol. 1986, 92, 757–761. [Google Scholar] [CrossRef]
- Fritzsch, B. Fast axonal diffusion of 3000 molecular weight dextran amines. J. Neurosci. Methods 1993, 50, 95–103. [Google Scholar] [CrossRef]
- Ishihara, A.; Naitoh, H.; Araki, H.; Nishihira, Y. Soma size and oxidative enzyme activity of motoneurones supplying the fast twitch and slow twitch muscles in the rat. Brain Res. 1988, 446, 195–198. [Google Scholar] [CrossRef]
- Cameron, N.E.; Cotter, M.A.; Robertson, S. Changes in skeletal muscle contractile properties in streptozocin-induced diabetic rats and role of polyol pathway and hypoinsulinemia. Diabetes 1990, 39, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Major, B.; Kimpinski, K.; Doherty, T.J.; Rice, C.L. Skeletal muscle morphology and contractile function in relation to muscle denervation in diabetic neuropathy. J. Appl. Physiol. 2014, 116, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshitake, Y.; Shinohara, M.; Kouzaki, M.; Fukunaga, T. Fluctuations in plantar flexion force are reduced after prolonged tendon vibration. J. Appl. Physiol. 2004, 97, 2090–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Deursen, R.W.; Sanchez, M.M.; Ulbrecht, J.S.; Cavanagh, P.R. The role of muscle spindles in ankle movement perception in human subjects with diabetic neuropathy. Exp. Brain Res. 1998, 120, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Swash, M.; Fox, K.P. The pathology of the human muscle spindle: Effect of denervation. J. Neurol. Sci. 1974, 22, 1–24. [Google Scholar] [CrossRef]
- Weis, J.; Schr der, J.M.; Dimpfel, W. Nerve conduction changes and fine structural alterations of extra- and intrafusal muscle and nerve fibers in streptozotocin diabetic rats. Muscle Nerve 1995, 18, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.F.; Andersen, H.; Sinkjaer, T. Decreased stiffness at the ankle joint in patients with long-term Type 1 diabetes. Diabet. Med. 2004, 21, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.A.; Ryals, J.M.; Feldman, E.L.; Wright, D.E. Abnormal muscle spindle innervation and large-fiber neuropathy in diabetic mice. Diabetes 2008, 57, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Friese, A.; Friese, A.; Kaltschmidt, J.A.; Kaltschmidt, J.A.; Ladle, D.R.; Ladle, D.R.; Sigrist, M.; Sigrist, M.; Jessell, T.M.; Jessell, T.M.; et al. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc. Natl. Acad. Sci. USA 2009, 106, 13588–13593. [Google Scholar] [CrossRef] [Green Version]
- Kucera, J. Splitting of the nuclear bag fiber in the course of muscle spindle denervation and reinnervation. J. Histochem. Cytochem. 1977, 25, 1102–1104. [Google Scholar] [CrossRef] [Green Version]
- Sveen, K.A.; Karimé, B.; Jørum, E.; Mellgren, S.I.; Fagerland, M.W.; Monnier, V.M.; Dahl-Jørgensen, K.; Hanseen, K.F. Small- and Large-Fiber Neuropathy After 40 Years of Type 1 Diabetes: Associations with glycemic control and advanced protein glycation: The Oslo Study. Diabetes Care 2013, 36, 3712–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemon, R.N. Descending pathways in motor control. Annu. Rev. Neurosci. 2008, 31, 195–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddle, C.N.; Baker, S.N. Convergence of Pyramidal and Medial Brain Stem Descending Pathways onto Macaque Cervical Spinal Interneurons. J. Neurophysiol. 2010, 103, 2821–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nudo, R.J. Adaptive plasticity in motor cortex: Implications for rehabilitation after brain injury. J. Rehabil. Med. 2003, 41, 7–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Lewallen, K.A.; Pfaff, S.L. Spatial organization of cortical and spinal neurons controlling motor behavior. Curr. Opin. Neurol. 2012, 22, 812–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welniarz, Q.; Dusart, I.; Roze, E. The corticospinal tract: Evolution, development, and human disorders. Dev. Neurobiol. 2017, 77, 810–829. [Google Scholar] [CrossRef]
- Seki, K.; Perlmutter, S.I.; Fetz, E.E. Sensory input to primate spinal cord is presynaptically inhibited during voluntary movement. Nat. Neurosci. 2003, 6, 1309–1316. [Google Scholar] [CrossRef]
- Ueno, M.; Nakamura, Y.; Li, J.; Gu, Z.; Niehaus, J.; Maezawa, M.; Crone, S.A.; Goulding, M.; Baccei, M.L.; Yoshida, Y. Corticospinal Circuits from the Sensory and Motor Cortices Differentially Regulate Skilled Movements through Distinct Spinal Interneurons. Cell Rep. 2018, 23, 1286–1300.e7. [Google Scholar] [CrossRef]
- Lawrence, D.G.; Kuypers, H.G.J.M. The functional organization of the motor system in the monkey. Brain 1968, 91, 1–14. [Google Scholar] [CrossRef]
- Sasaki, S.; Isa, T.; Pettersson, L.-G.; Alstermark, B.; Naito, K.; Yoshimura, K.; Seki, K.; Ohki, Y. Dexterous Finger Movements in Primate Without Monosynaptic Corticomotoneuronal Excitation. J. Neurophysiol. 2004, 92, 3142–3147. [Google Scholar] [CrossRef] [Green Version]
- Brands, A.M.A.; Biessels, G.-J.; de Haan, E.H.F.; Kappelle, L.J.; Roy, P.C. Kessels The Effects of Type 1 Diabetes on Cognitive Performance: A meta-analysis. Diabetes Care 2005, 28, 726–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, A.; Biessels, G.-J.; Gispen, W.H.; Ramakers, G.M.J. Synaptic transmission changes in the pyramidal cells of the hippocampus in streptozotocin-induced diabetes mellitus in rats. Brain Res. 2006, 1073–1074, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Gold, S.M.; Dziobek, I.; Sweat, V.; Tirsi, A.; Rogers, K.; Bruehl, H.; Tsui, W.; Richardson, S.; Javier, E.; Convit, A. Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia 2007, 50, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008, 11, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reijmer, Y.D.; van den Berg, E.; de Bresser, J.; Kessels, R.P.C.; Kappelle, L.J.; Algra, A.; Biessels, G.-J. Accelerated cognitive decline in patients with type 2 diabetes: MRI correlates and risk factors. Diabetes. Metab. Res. Rev. 2011, 27, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.N.; Younan, S.M.; Youssef, M.F.; Rashed, L.A.; Mohamady, I. A histological and functional study on hippocampal formation of normal and diabetic rats. F1000Res. 2013, 2, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Xu, L.; He, D.; Ling, S. Endoplasmic reticulum stress-mediated hippocampal neuron apoptosis involved in diabetic cognitive impairment. Biomed. Res. Int. 2013, 2013, 924327. [Google Scholar] [CrossRef]
- Marks, J.L.; Porte, D., Jr.; Stahl, W.L.; Baskin, D.G. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 1990, 127, 3234–3236. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Parihar, M.S.; Chaudhary, M.; Shetty, R.; Hemnani, T. Susceptibility of hippocampus and cerebral cortex to oxidative damage in streptozotocin treated mice: Prevention by extracts of Withania somnifera and Aloe vera. J. Clin. Neurosci. 2004, 11, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Srivastava, S.; Kakkar, P. Bacopa monnieri modulates antioxidant responses in brain and kidney of diabetic rats. Environ. Toxicol. Pharmacol. 2009, 27, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Restivo, F.; Vercellinatto, I.; Danni, O.; Brignardello, E.; Aragno, M.; Boccuzzi, G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J. Endocrinol. 2005, 187, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Aragno, M.; Brignardello, E.; Tamagno, E.; Gatto, V.; Danni, O.; Boccuzzi, G. Dehydroepiandrosterone administration prevents the oxidative damage induced by acute hyperglycemia in rats. J. Endocrinol. 1997, 155, 233–240. [Google Scholar] [CrossRef]
- Andersen, H.; Nielsen, S.; Nielsen, J.F. Motor cortical excitability remains unaffected of short-term hyperglycemia in Type 1 diabetic patients. J. Diabetes Complicat. 2006, 20, 51–55. [Google Scholar] [CrossRef]
- Andersen, H.; Nielsen, J.F.; Poulsen, P.L.; Mogensen, C.E.; Jakobsen, J. Motor pathway function in normoalbuminuric IDDM patients. Diabetologia 1995, 38, 1191–1196. [Google Scholar] [CrossRef]
- Liu, D.; Duan, S.; Zhang, J.; Zhou, C.; Liang, M.; Yin, X.; Wei, P.; Wang, J. Aberrant Brain Regional Homogeneity and Functional Connectivity in Middle-Aged T2DM Patients: A Resting-State Functional MRI Study. Front. Hum. Neurosci. 2016, 10, 539. [Google Scholar] [CrossRef]
- Hughes, T.M.; Ryan, C.M.; Aizenstein, H.J.; Nunley, K.; Gianaros, P.J.; Miller, R.; Costacou, T.; Strotmeyer, E.S.; Orchard, T.J.; Rosano, C. Frontal gray matter atrophy in middle aged adults with type 1 diabetes is independent of cardiovascular risk factors and diabetes complications. J. Diabetes Complicat. 2013, 27, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Chen, Z.; Ma, L.; Dai, Y. Cerebral alterations of type 2 diabetes mellitus on MRI: A pilot study. Neurosci. Lett. 2015, 606, 100–105. [Google Scholar] [CrossRef]
- Chen, Z.; Li, L.; Sun, J.; Ma, L. Mapping the brain in type II diabetes: Voxel-based morphometry using DARTEL. Eur. J. Radiol. 2012, 81, 1870–1876. [Google Scholar] [CrossRef]
- Yoon, S.; Cho, H.; Kim, J.; Lee, D.-W.; Kim, G.H.; Hong, Y.S.; Moon, S.; Park, S.; Lee, S.; Lee, S.; et al. Brain changes in overweight/obese and normal-weight adults with type 2 diabetes mellitus. Diabetologia 2017, 60, 1207–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Duinkerken, E.; Schoonheim, M.M.; IJzerman, R.G.; Klein, M.; Ryan, C.M.; Moll, A.C.; Snoek, F.J.; Barkhof, F.; Diamant, M.; Pouwels, P.J.W. Diffusion tensor imaging in type 1 diabetes: Decreased white matter integrity relates to cognitive functions. Diabetologia 2012, 55, 1218–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Bloemendaal, L.; IJzerman, R.G.; Jennifer, S.; Barkhof, F.; Diamant, M.; Veltman, D.J.; van Duinkerken, E. Alterations in white matter volume and integrity in obesity and type 2 diabetes. Metab. Brain Dis. 2016, 31, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Fonseca, J.P.; Rincón, J. Structural and ultrastructural analysis of cerebral cortex, cerebellum, and hypothalamus from diabetic rats. Exp. Diabetes Res. 2009, 2009, 329632. [Google Scholar] [CrossRef]
- Martínez-Tellez, R.; Gómez-Villalobos, M.d.J.; Flores, G. Alteration in dendritic morphology of cortical neurons in rats with diabetes mellitus induced by streptozotocin. Brain Res. 2005, 1048, 108–115. [Google Scholar] [CrossRef]
- Mukai, N.; Hori, S.; Pomeroy, M. Cerebral lesions in rats with streptozotocin-induced diabetes. Acta Neuropathol. 1980, 51, 79–84. [Google Scholar] [CrossRef]
- Jakobsen, J.; Sidenius, P.; Gundersen, H.J.G.; Østerby, R. Quantitative Changes of Cerebral Neocortical Structure in Insulin-Treated Long-Term Streptozocin-lnduced Diabetes in Rats. Diabetes 1987, 36, 597–601. [Google Scholar] [CrossRef]
- Reske-Nielsen, E.; Lundbaek, K.; Rafaelsen, O.J. Pathological changes in the central and peripheral nervous system of young long-term diabetics: I. Diabetic encephalopathy. Diabetologia 1966, 1, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Salkovic-Petrisic, M.; Osmanovic-Barilar, J.; Brückner, M.K.; Hoyer, S.; Arendt, T.; Riederer, P. Cerebral amyloid angiopathy in streptozotocin rat model of sporadic Alzheimer’s disease: A long-term follow up study. J. Neural. Transm. 2011, 118, 765–772. [Google Scholar] [CrossRef]
- Larsson, M.; Lietzau, G.; Nathanson, D.; Östenson, C.-G.; Mallard, C.; Johansson, M.E.; Nyström, T.; Patrone, C.; Darsalia, V. Diabetes negatively affects cortical and striatal GABAergic neurons: An effect that is partially counteracted by exendin-4. Biosci. Rep. 2016, 36, e00421. [Google Scholar] [CrossRef]
- Aye, T.; Barnea-Goraly, N.; Ambler, C.; Hoang, S.; Schleifer, K.; Park, Y.; Drobny, J.; Wilson, D.M.; Reiss, A.L.; Buckingham, B.A. White Matter Structural Differences in Young Children with Type 1 Diabetes: A Diffusion Tensor Imaging Study. Diabetes Care 2012, 35, 2167–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, Y.; WANG, J.; Zhou, X.; Shu, N.; Wang, Y.; Zhang, Z. White Matter Integrity Disruptions Associated with Cognitive Impairments in Type 2 Diabetic Patients. Diabetes 2014, 63, 3596–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perantie, D.C.; Wu, J.; Koller, J.M.; Lim, A.; Warren, S.L.; Black, K.J.; Sadler, M.; White, N.H.; Hershey, T. Regional brain volume differences associated with hyperglycemia and severe hypoglycemia in youth with type 1 diabetes. Diabetes Care 2007, 30, 2331–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullmann, S.; Callaghan, M.F.; Heni, M.; Weiskopf, N.; Scheffler, K.; Häring, H.-U.; Fritsche, A.; Veit, R.; Preissl, H. Specific white matter tissue microstructure changes associated with obesity. Neuroimage 2016, 125, 36–44. [Google Scholar] [CrossRef]
- Abbruzzese, G.; Schenone, A.; Scramuzza, G.; Caponnetto, C.; Gasparetto, B.; Adezati, L.; Abbruzzese, M.; Viviani, G.L. Impairment of central motor conduction in diabetic patients. Electroencephalogr. Clin. Neurophysiol. 1993, 89, 335–340. [Google Scholar] [CrossRef]
- Dolu, H.; Ulas, U.H.; Bolu, E.; Ozkardes, A.; Odabasi, Z.; Ozata, M.; Vural, O. Evaluation of central neuropathy in type II diabetes mellitus by multimodal evoked potentials. Acta Neurol. Belg. 2003, 103, 206–211. [Google Scholar]
- Goldenberg, Z.; Kucera, P.; Brezinova, M.; Kurca, E.; Barak, L.; Traubner, P. Clinically unapparent central motor pathways lesion in patients with type I diabetes mellitus. A transcranial magnetic stimulation study. Bratisl. Lek. Listy. 2004, 105, 400–403. [Google Scholar]
- El Bardawil, M.M.; El Hamid, M.; El Sawy, N.; Megallaa, M.; El Emary, W. Postural control and central motor pathway involvement in type 2 diabetes mellitus: Dynamic posturographic and electrophysiologic studies. Alex. Med. J. 2013, 49, 299–307. [Google Scholar] [CrossRef]
- Uccioli, L.; Giacomini, P.G.; Pasqualetti, P.; Di Girolamo, S.; Ferrigno, P.; Monticone, G.; Bruno, E.; Boccasena, P.; Magrini, A.; Parisi, L.; et al. Contribution of central neuropathy to postural instability in IDDM patients with peripheral neuropathy. Diabetes Care 1997, 20, 929–934. [Google Scholar] [CrossRef]
- Biessels, G.J.; Cristino, N.A.; Rutten, G.J.; Hamers, F.P.; Erkelens, D.W.; Gispen, W.H. Neurophysiological changes in the central and peripheral nervous system of streptozotocin-diabetic rats. Course of development and effects of insulin treatment. Brain 1999, 122 Pt 4, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Madsen, J.G.; Østergaard, J.A.; Andersen, H.; Pedersen, M. Attenuation of Cortically Evoked Motor-Neuron Potential in Streptozotocin-Induced Diabetic Rats: A Study about the Effect of Diabetes upon Cortical-Initiated Movement. Biomed. Res. Int. 2020, 2020, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekiner, C.; McLean, W.G. Neurofilament Protein Phosphorylation in Spinal Cord of Experimentally Diabetic Rats. J. Neurochem. 1991, 56, 1362–1367. [Google Scholar] [CrossRef]
- Pekiner, C.; Cullum, N.A.; Hughes, J.N.; Hargreaves, A.J.; Mahon, J.; Casson, I.F.; McLean, W.G. Glycation of Brain Actin in Experimental Diabetes. J. Neurochem. 1993, 61, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Hnaway, J.; Young, R.R. Localization of the pyramidal tract in the internal capsule of man. J. Neurol. Sci. 1977, 34, 63–70. [Google Scholar] [CrossRef]
- Passingham, R.E.; Perry, V.H.; Wilkinson, F. The long-term effects of removal of sensorimotor cortex in infant and adult rhesus monkeys. Brain 1983, 106, 675–705. [Google Scholar] [CrossRef] [PubMed]
- Courtine, G.; Roy, R.R.; Raven, J.; Hodgson, J.; Brain, H.M. Performance of locomotion and foot grasping following a unilateral thoracic corticospinal tract lesion in monkeys (Macaca mulatta). Brain 2005, 128, 2338–2358. [Google Scholar] [CrossRef] [Green Version]
- Liddell, E.G.T.; Phillips, C.G. Pyramidal section in the cat. Brain 1944, 67, 1–9. [Google Scholar] [CrossRef]
- Benitez, S.U.; Carneiro, E.M.; de Oliveira, A.L.R. Synaptic input changes to spinal cord motoneurons correlate with motor control impairments in a type 1 diabetes mellitus model. Brain Behav. 2015, 5, e00372. [Google Scholar] [CrossRef]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Xie, F.; Zheng, B. White matter inhibitors in CNS axon regeneration failure. Exp. Neurol. 2008, 209, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.M.; Guénard, V.; Kleitman, N.; Aebischer, P.; Bunge, M.B. A Combination of BDNF and NT-3 Promotes Supraspinal Axonal Regeneration into Schwann Cell Grafts in Adult Rat Thoracic Spinal Cord. Exp. Neurol. 1995, 134, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Blits, B.; Dijkhuizen, P.A.; Boer, G.J.; Verhaagen, J. Intercostal Nerve Implants Transduced with an Adenoviral Vector Encoding Neurotrophin-3 Promote Regrowth of Injured Rat Corticospinal Tract Fibers and Improve Hindlimb Function. Exp. Neurol. 2000, 164, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-M.; Guénard, V.; Kleitman, N.; Bunge, M.B. Axonal regeneration into Schwann cell-seeded guidance channels grafted into transected adult rat spinal cord. J. Comp. Neurol. 2004, 351, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Vavrek, R.; Pearse, D.D.; Fouad, K. Neuronal Populations Capable of Regeneration following a Combined Treatment in Rats with Spinal Cord Transection. J. Neurotrauma 2007, 24, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.N.; Zaaimi, B.; Fisher, K.M.; Edgley, S.A.; Soteropoulos, D.S. Pathways mediating functional recovery. Prog. Brain Res. 2015, 218, 389–412. [Google Scholar]
- Zaaimi, B.; Edgley, S.A.; Soteropoulos, D.S.; Brain, S.B. Changes in descending motor pathway connectivity after corticospinal tract lesion in macaque monkey. Brain 2012, 135, 2277–2289. [Google Scholar] [CrossRef]
- Umeda, T.; Takahashi, M.; Isa, K.; Isa, T. Formation of Descending Pathways Mediating Cortical Command to Forelimb Motoneurons in Neonatally Hemidecorticated Rats. J. Neurophysiol. 2010, 104, 1707–1716. [Google Scholar] [CrossRef] [Green Version]
- Bareyre, F.M.; Kerschensteiner, M.; Raineteau, O.; Mettenleiter, T.C.; Weinmann, O.; Schwab, M.E. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat. Neurosci. 2004, 7, 269–277. [Google Scholar] [CrossRef]
- Tohyama, T.; Kinoshita, M.; Kobayashi, K.; Isa, K.; Watanabe, D.; Kobayashi, K.; Liu, M.; Isa, T. Contribution of propriospinal neurons to recovery of hand dexterity after corticospinal tract lesions in monkeys. Proc. Natl. Acad. Sci. USA 2017, 114, 604–609. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Hayashi, T.; Murata, Y.; Ose, T.; Higo, N. Premotor Cortical-Cerebellar Reorganization in a Macaque Model of Primary Motor Cortical Lesion and Recovery. J. Neurosci. 2019, 39, 8484–8496. [Google Scholar] [CrossRef]
- Jaillard, A.; Martin, C.D.; Garambois, K.; Brain, J.L. Vicarious function within the human primary motor cortex? Brain 2005, 128, 1122–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, Y.; Higo, N.; Oishi, T.; Isa, T. Increased expression of the growth-associated protein-43 gene after primary motor cortex lesion in macaque monkeys. Neurosci. Res. 2015, 98, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, Y.; Higo, N.; Hayashi, T.; Nishimura, Y.; Sugiyama, Y.; Oishi, T.; Tsukada, H.; Isa, T.; Onoe, H. Temporal plasticity involved in recovery from manual dexterity deficit after motor cortex lesion in macaque monkeys. J. Neurosci. 2015, 35, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, Y.; Higo, N.; Oishi, T.; Yamashita, A.; Matsuda, K.; Hayashi, M.; Yamane, S. Effects of motor training on the recovery of manual dexterity after primary motor cortex lesion in macaque monkeys. J. Neurophysiol. 2008, 99, 773–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, Y.; Higo, N.; Yoshino-Saito, K.; Murata, Y.; Nishimura, Y.; Oishi, T.; Isa, T. Effects of early versus late rehabilitative training on manual dexterity after corticospinal tract lesion in macaque monkeys. J. Neurophysiol. 2013, 109, 2853–2865. [Google Scholar] [CrossRef] [Green Version]
- Allred, R.P.; Maldonado, M.A.; Hsu And, J.E.; Jones, T.A. Training the “less-affected” forelimb after unilateral cortical infarcts interferes with functional recovery of the impaired forelimb in rats. Restor. Neurol. Neurosci. 2005, 23, 297–302. [Google Scholar]
- Girgis, J.; Merrett, D.; Kirkland, S.; Metz, G.A.S.; Verge, V.; Fouad, K. Reaching training in rats with spinal cord injury promotes plasticity and task specific recovery. Brain 2007, 130, 2993–3003. [Google Scholar] [CrossRef] [Green Version]
- Carmel, J.B.; Kimura, H.; Berrol, L.J.; Martin, J.H. Motor cortex electrical stimulation promotes axon outgrowth to brain stem and spinal targets that control the forelimb impaired by unilateral corticospinal injury. Eur. J. Neurosci. 2013, 37, 1090–1102. [Google Scholar] [CrossRef] [Green Version]
- Carmel, J.B.; Martin, J.H. Motor cortex electrical stimulation augments sprouting of the corticospinal tract and promotes recovery of motor function. Front. Integr. Neurosci. 2014, 8, 4935. [Google Scholar] [CrossRef] [Green Version]
- Carmel, J.B.; Kimura, H.; Martin, J.H. Electrical stimulation of motor cortex in the uninjured hemisphere after chronic unilateral injury promotes recovery of skilled locomotion through ipsilateral control. J. Neurosci. 2014, 34, 462–466. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, H.; Miyatake, N.; Kitayama, N.; Murao, S.; Tanaka, S. A pilot study of short-term toe resistance training in patients with type 2 diabetes mellitus. Diabetol. Int. 2017, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Miyatake, N.; Murao, S.; Tanaka, S. A randomized controlled trial of short-term toe resistance training to improve toe pinch force in patients with type 2 diabetes. Acta Med. Okayama 2018, 72, 9–15. [Google Scholar] [PubMed]
- Zhou, S. Chronic neural adaptations to unilateral exercise: Mechanisms of cross education. Exerc. Sport Sci. Rev. 2000, 28, 177–184. [Google Scholar] [PubMed]
- Frontera, W.R.; Hughes, V.A.; Krivickas, L.S.; Kim, S.-K.; Foldvari, M.; Roubenoff, R. Strength training in older women: Early and late changes in whole muscle and single cells. Muscle Nerve 2003, 28, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Jitsuki, S.; Nakajima, W.; Murata, Y.; Jitsuki-Takahashi, A.; Katsuno, Y.; Tada, H.; Sano, A.; Suyama, K.; Mochizuki, N.; et al. CRMP2-binding compound, edonerpic maleate, accelerates motor function recovery from brain damage. Science 2018, 360, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, T.; Chopp, M.; Ye, X.; Liu, Z.; Zacharek, A.; Cui, Y.; Roberts, C.; Buller, B.; Chen, J. Niaspan increases axonal remodeling after stroke in type 1 diabetes rats. Neurobiol. Dis. 2012, 46, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Biophys. Rev. 2020, 18, 62. [Google Scholar] [CrossRef] [Green Version]
- Nagayach, A.; Patro, N.; Patro, I. Experimentally induced diabetes causes glial activation, glutamate toxicity and cellular damage leading to changes in motor function. Front. Cell. Neurosci. 2014, 8, 224. [Google Scholar] [CrossRef] [Green Version]
- Mark, L.P.; Prost, R.W.; Ulmer, J.L.; Smith, M.M.; Daniels, D.L.; Strottmann, J.M.; Brown, W.D.; Hacein-Bey, L. Pictorial review of glutamate excitotoxicity: Fundamental concepts for neuroimaging. Am. J. Neuroradiol. 2001, 22, 1813–1824. [Google Scholar]
- Baydas, G.; Nedzvetskii, V.S.; Tuzcu, M.; Yasar, A.; Kirichenko, S.V. Increase of glial fibrillary acidic protein and S-100B in hippocampus and cortex of diabetic rats: Effects of vitamin E. Eur. J. Pharmacol. 2003, 462, 67–71. [Google Scholar] [CrossRef]
- Baydas, G.; Reiter, R.J.; Yasar, A.; Tuzcu, M.; Akdemir, S.; Nedzvetskii, V.S. Melatonin reduces glial reactivity in the hippocampus, cortex, and cerebellum of streptozotocin-induced diabetic rats. Free Radic. Biol. Med. 2003, 35, 797–804. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Induction Mechanism | Type of Diabetes |
|---|---|---|
| STZ rats | Chemical induction | Type 1 diabetes |
| Bio-Breeding (BB) rats | Spontaneous | Type 1 diabetes |
| Zucker rats | Spontaneous | Obesity model of type 2 diabetes |
| Goto–Kakizaki rats | Spontaneous | Lean model of type 2 diabetes |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muramatsu, K. Diabetes Mellitus-Related Dysfunction of the Motor System. Int. J. Mol. Sci. 2020, 21, 7485. https://doi.org/10.3390/ijms21207485
Muramatsu K. Diabetes Mellitus-Related Dysfunction of the Motor System. International Journal of Molecular Sciences. 2020; 21(20):7485. https://doi.org/10.3390/ijms21207485
Chicago/Turabian StyleMuramatsu, Ken. 2020. "Diabetes Mellitus-Related Dysfunction of the Motor System" International Journal of Molecular Sciences 21, no. 20: 7485. https://doi.org/10.3390/ijms21207485
APA StyleMuramatsu, K. (2020). Diabetes Mellitus-Related Dysfunction of the Motor System. International Journal of Molecular Sciences, 21(20), 7485. https://doi.org/10.3390/ijms21207485