Irisin and Autophagy: First Update


 , and
, and 
Abstract
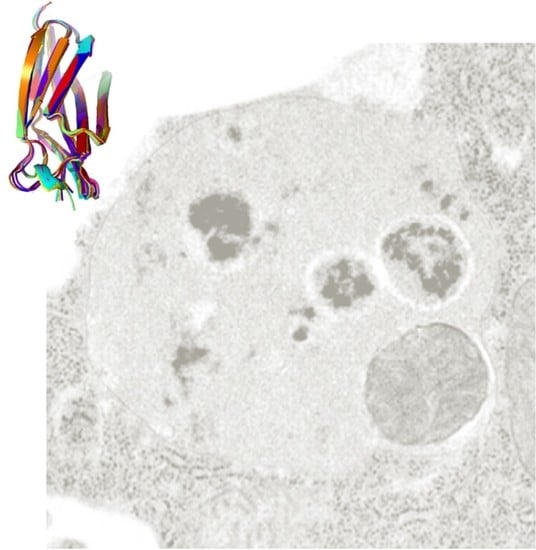
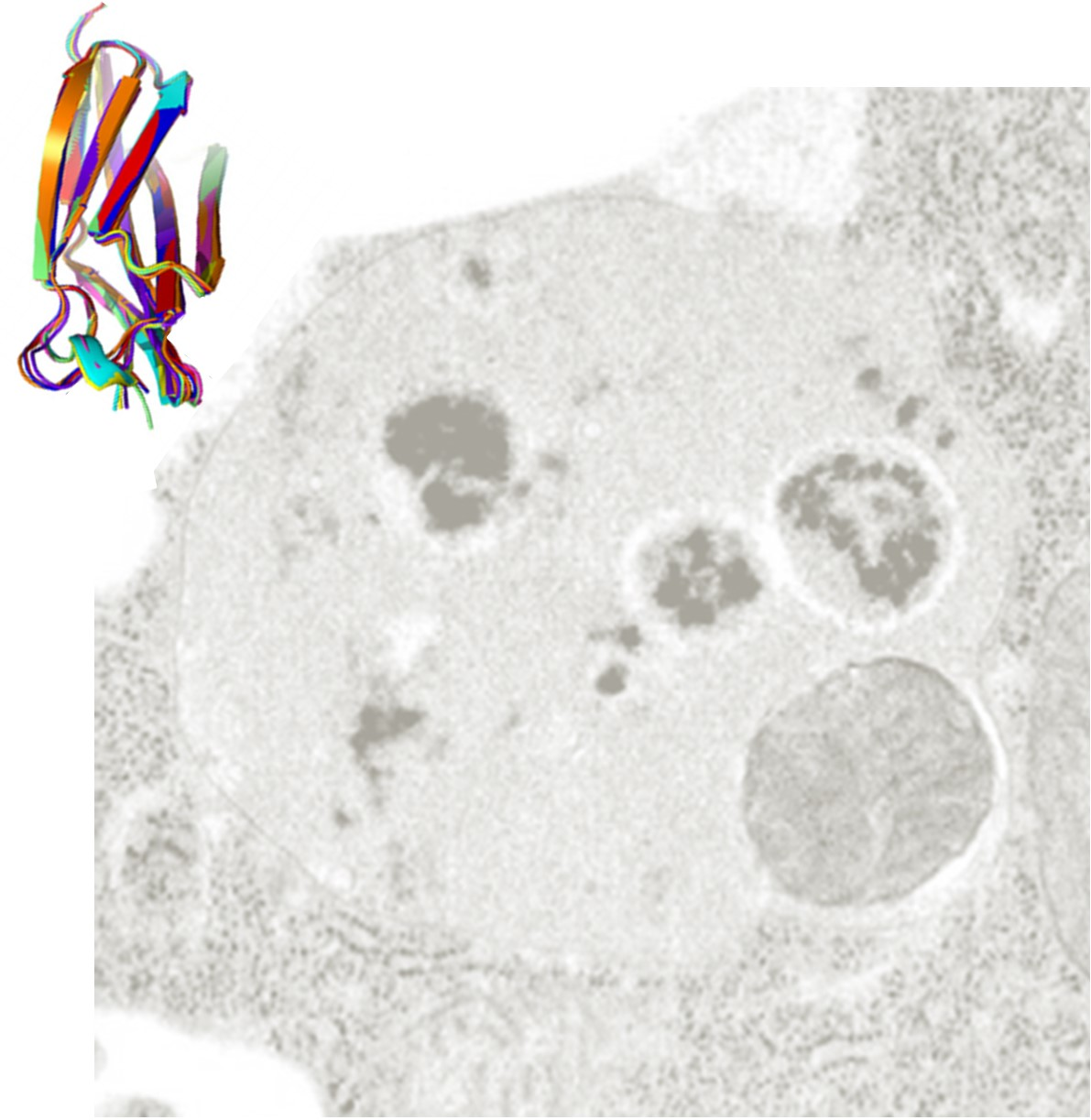
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Molecular Mechanism of Autophagy
3. Myokines
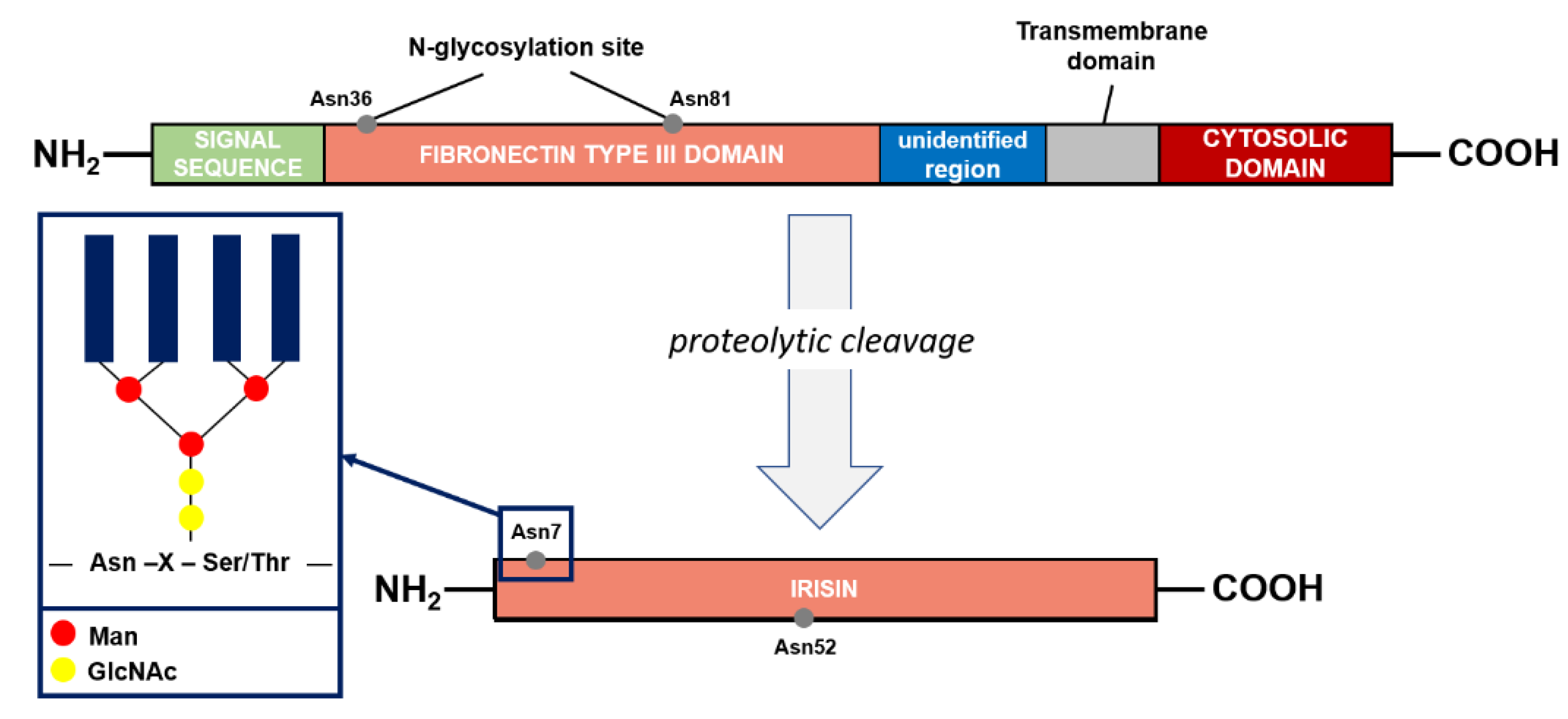
4. Irisin Structure and Function
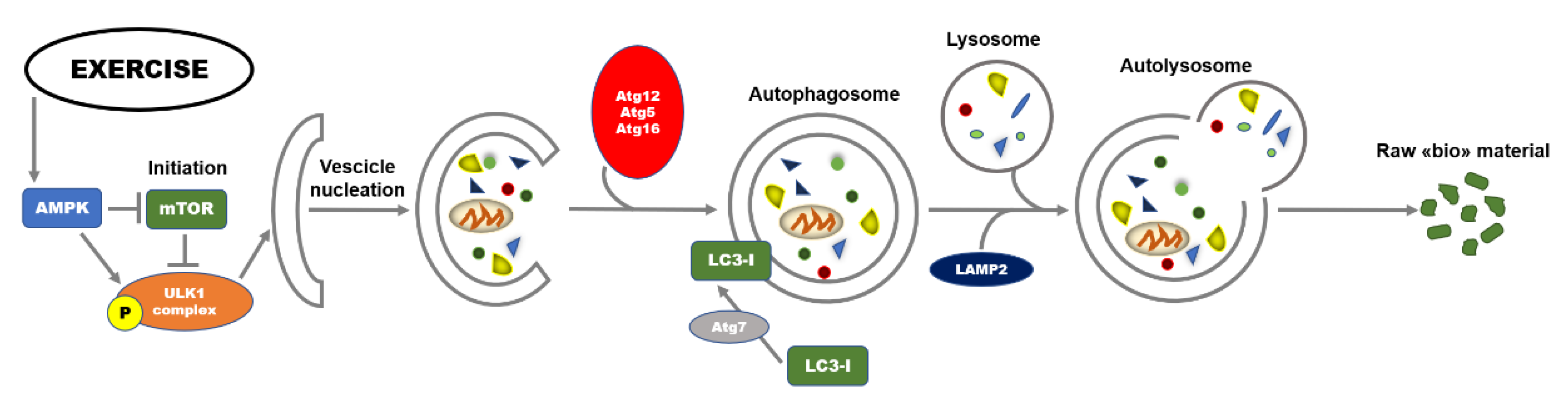
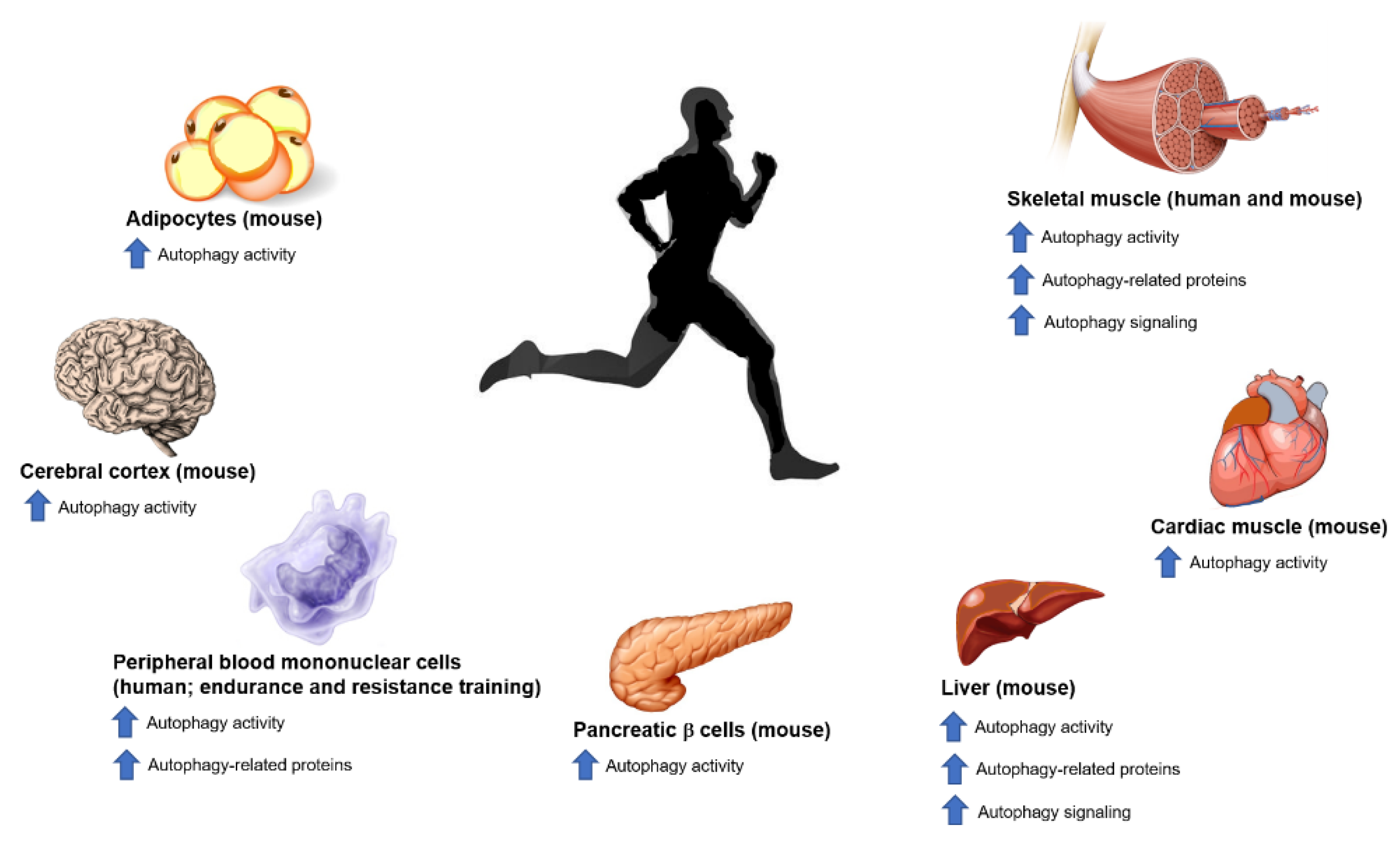
5. Exercise and Autophagy
6. Myokines and Autophagy
6.1. Myostatin
6.2. Decorin
6.3. IL-4
6.4. IL-6
6.5. IL-7
6.6. IL-15
6.7. Other Myokines
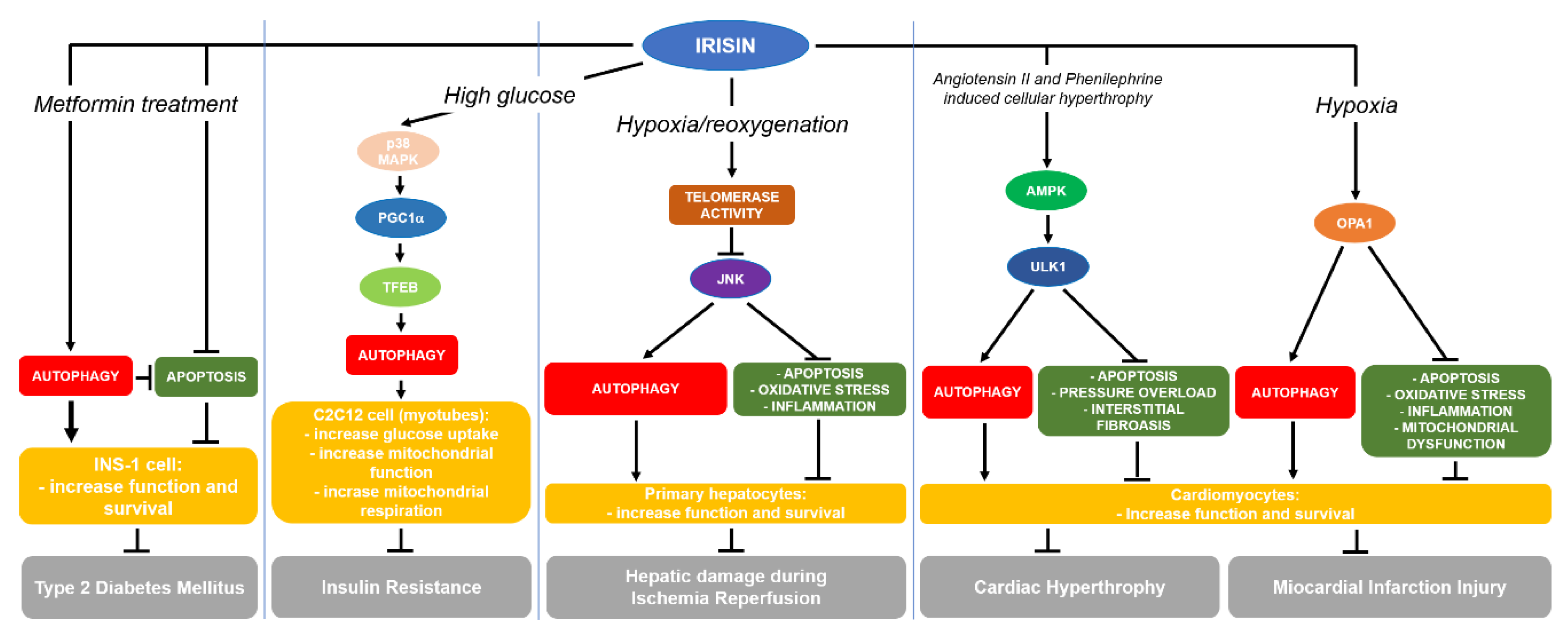
7. FNDC5/Irisin and Autophagy
7.1. Metabolic Disorders
7.2. Myocardial Ischemia/Reperfusion Injury
7.3. Myocardial Hypertrophy
7.4. Hepatic Ischemia/Reperfusion Injury
7.5. Chronic Kidney Disease
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Akerstrom, T.C.; Nielsen, A.R.; Fischer, C.P. Role of myokines in exercise and metabolism. J. Appl. Physiol. 2007, 103, 1093–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peake, J.M.; Della Gatta, P.; Suzuki, K.; Nieman, D.C. Cytokine expression and secretion by skeletal muscle cells: Regulatory mechanisms and exercise effects. Exerc. Immunol. Rev. 2015, 21, 8–25. [Google Scholar] [PubMed]
- Hoffmann, C.; Weigert, C. Skeletal muscle as an endocrine organ: The role of myokines in exercise adaptations. Cold Spring Harb. Perspect. Med. 2017, 19, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jun, H.S. Role of myokines in regulating skeletal muscle mass and function. Front. Physiol. 2019, 10, 42. [Google Scholar] [CrossRef]
- Steensberg, A.; Van Hall, G.; Osada, T. Production of interleukin-6 in contracting human skeletal muscles can account for the exercise-induced increase in plasma interleukin-6. J. Physiol. 2000, 529, 237–242. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar]
- Park, P.H. Autophagy induction: A critical event for the modulation of cell death/survival and inflammatory responses by adipokines. Arch. Pharmacal Res. 2018, 41, 1062–1073. [Google Scholar] [CrossRef]
- Moulis, M.; Vindis, C. Autophagy in metabolic age-related human diseases. Cells 2018, 7, 149. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Saltin, B. Exercise as medicine-evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand. J. Med. Sci. Sport. 2015, 25, 1–72. [Google Scholar]
- He, C.; Sumpter, R.; Levine, B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, O.C.; Estebanez, B.; Martinez-Florez, S.; de Paz, J.A.; Cuevas, M.J.; Gonzalez-Gallego, J. Mitochondrial function and mitophagy in the elderly: Effects of exercise. Oxid. Med. Cell. Longev. 2017, 2017, 2012798. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, N. Autophagy is a promoter for aerobic exercise performance during high altitude training. Oxid. Med. Cell. Longev. 2018, 2018, 3617508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.E.; Bareja, A.; Bartlett, D.B.; White, J.P. Autophagy as a Therapeutic Target to Enhance Aged Muscle Regeneration. Cells 2019, 8, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- Rautou, P.E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Choi, M.E. Autophagy in kidney health and disease. Antioxid. Redox Signal. 2014, 20, 519–537. [Google Scholar] [CrossRef] [Green Version]
- Park, S.S.; Seo, Y.K.; Kwon, K.S. Sarcopenia targeting with autophagy mechanism by exercise. BMB Rep. 2019, 52, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, A.M.; Bernardi, H.; Py, G.; Candau, R.B. Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R956–R969. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Sinha, S.C.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Morell, C.; Bort, A.; Vara-Ciruelos, D.; Ramos-Torres, A.; Altamirano-Dimas, M.; Diaz-Laviada, I.; Rodriguez-Henche, N. Up-Regulated Expression of LAMP2 and Autophagy Activity during Neuroendocrine Differentiation of Prostate Cancer LNCaP Cells. PLoS ONE 2016, 11, e0162977. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Frankel, L.B.; Wen, J.; Lees, M.; Hoyer-Hansen, M.; Farkas, T.; Krogh, A.; Jaattela, M.; Lund, A.H. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011, 30, 4628–4641. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K. Muscle as a secretory organ. Compr. Physiol. 2013, 3, 1337–1362. [Google Scholar]
- Bentzinger, C.F.; Lin, S.; Romanino, K.; Castets, P.; Guridi, M.; Summermatter, S. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet. Muscle 2013, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Salanova, M.; Gelfi, C.; Moriggi, M.; Vasso, M.; Vigano, A.; Minafra, L. Disuse deterioration of human skeletal muscle challenged by resistive exercise superimposed with vibration: Evidence from structural and proteomic analysis. FASEB J. 2014, 28, 4748–4763. [Google Scholar] [CrossRef]
- Williamson, D.L.; Gallagher, P.M.; Carroll, C.C.; Raue, U.; Trappe, S.W. Reduction in hybrid single muscle fiber proportions with resistance training in humans. J. Appl. Physiol. 2001, 91, 1955–1961. [Google Scholar] [CrossRef]
- Speranza, L.; Grilli, A.; Patruno, A. Plasmatic markers of muscular stress in isokinetic exercise. J. Biol. Regul. Homeost. Agents 2007, 21, 21–29. [Google Scholar] [PubMed]
- Goldstein, M.S. Humoral nature of the hypoglycemic factor of muscular work. Diabetes 1961, 10, 232–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K. Muscles and their myokines. J. Exp. Biol. 2011, 214, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaer, M.; Pollack, S.F.; Mohr, T.; Weiss, H.; Gleim, G.W.; Bach, F.W. Regulation of glucose turnover and hormonal responses during electrical cycling in tetraplegic humans. Am. J. Physiol. 1996, 271, R191–R199. [Google Scholar] [CrossRef]
- Misra, A.; Bloomgarden, Z. Metabolic memory: Evolving concepts. J. Diabetes 2018, 10, 186–187. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, C.P.; Graham, Z.A. Muscle-bone interactions: Movement in the field of mechano–humoral coupling of muscle and bone. Ann. N. Y. Acad. Sci. 2017, 1402, 10–17. [Google Scholar] [CrossRef]
- Huh, J.Y. The role of exercise-induced myokines in regulating metabolism. Arch. Pharmacal Res. 2018, 41, 14–29. [Google Scholar] [CrossRef]
- Bauman, W.A.; Cardozo, C.P. Spinal cord injury: Skeletal pathophysiology and clinical issues. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 8th ed.; Rosen, C.J., Ed.; Wiley-Blackwell: Oxford, UK, 2013. [Google Scholar]
- Karstoft, K.; Pedersen, B.K. Skeletal muscle as a gene regulatory endocrine organ. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 270–275. [Google Scholar] [CrossRef]
- Leal, L.G.; Lopes, M.A.; Batista, M.L. Physical exercise-induced myokines and muscle-adipose tissue crosstalk: A review of current knowledge and the implications for health and metabolic diseases. Front. Physiol. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Panati, K.; Narala, V.R.; Narasimha, V.R.; Derangula, M.; Arva Tatireddigari, V.R.R.; Yeguvapalli, S. Expression, purification and biological characterisation of recombinant human irisin (12.5 kDa). J. Genet. Eng. Biotechnol. 2018, 16, 459–466. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Chinnam, N.; Ohashi, T.; Shah, R.S.; Erickson, H.P. The structure of irisin reveals a novel intersubunit β-sheet fibronectin type III (FNIII) dimer: Implications for receptor activation. Biol. Chem. 2013, 288, 33738–33744. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.; Liu, D. N-Glycosylation is required for FDNC5 stabilization and irisin secretion. Biochem. J. 2017, 474, 3167–3177. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.Y.; Bailey, U.M.; Jamaluddin, M.F.; Mahmud, S.H.; Raman, S.C.; Schulz, B.L. Sequence-based protein stabilization in the absence of glycosylation. Nat. Commun. 2014, 5, 3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Wrann, C.D.; Jedrychowski, M.; Vidoni, S.; Kitase, Y.; Nagano, K.; Zhou, C.; Chou, J.; Parkman, V.A.; Novick, S.J.; et al. Irisin Mediates Effects on Bone and Fat via αV Integrin Receptors. Cell 2018, 175, 1756–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [Green Version]
- Martinez Munoz, I.Y.; Camarillo Romero, E.D.S.; Garduno Garcia, J.J. Irisin a Novel Metabolic Biomarker: Present Knowledge and Future Directions. Int. J. Endocrinol. 2018, 2018, 7816806. [Google Scholar] [CrossRef] [Green Version]
- Huh, J.Y.; Siopi, A.; Mougios, V.; Park, K.H.; Mantzoros, C.S. Irisin in response to exercise in humans with and without metabolic syndrome. J. Clin. Endocrinol. Metab. 2015, 100, E453–E457. [Google Scholar] [CrossRef]
- Moreno, M.; Moreno-Navarrete, J.M.; Serrano, M.; Ortega, F.; Delgado, E.; Sanchez-Ragnarsson, C.; Valdés, S.; Botas, P.; Ricart, W.; Fernández-Real, J.M. Circulating irisin levels are positively associated with metabolic risk factors in sedentary subjects. PLoS ONE 2015, 10, e0124100. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Ando, D.; Takamatsu, K.; Goto, K. Resistance exercise induces a greater irisin response than endurance exercise. Metabolism 2015, 64, 1042–1050. [Google Scholar] [CrossRef]
- Murawska-Cialowicz, E.; Wolanski, P.; Zuwala-Jagiello, J.; Feito, Y.; Petr, M.; Kokstejn, J.; Stastny, P.; Goliński, D. Effect of HIIT with Tabata Protocol on Serum Irisin, Physical Performance, and Body Composition in Men. Int. J. Environ. Res. Public Health 2020, 17, 3589. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Navarrete, J.M.; Ortega, F.; Serrano, M.; Guerra, E.; Pardo, G.; Tinahones, F.; Ricart, W.; Fernández-Real, J.M. Irisin is expressed and produced by human muscle and adipose tissue in association with obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2013, 98, E769–E778. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Liu, S.; Wong, M.D.; Tan, C.S.; Tavintharan, S.; Sum, C.F.; Lim, S.C. Relationship between circulating irisin, renal function and body composition in type 2 diabetes. J. Diabetes Complicat. 2014, 28, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Zybek-Kocik, A.; Sawicka-Gutaj, N.; Wrotkowska, E.; Sowi’nski, J.; Ruchała, M. Time-dependent irisin concentration changes in patients affected by overt hypothyroidism. Endokrynol. Pol. 2016, 67, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Gouveia, M.C.; Vella, J.P.; Cafeo, F.R.; Affonso Fonseca, F.L.; Bacci, M.R. Association between irisin and major chronic diseases: A review. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4072–4077. [Google Scholar]
- Zhang, Y.; Xie, C.; Wang, H.; Foss, R.M.; Clare, M.; George, E.V.; Li, S.; Katz, A.; Cheng, H.; Ding, Y.; et al. Irisin exerts dual effects on browning and adipogenesis of human white adipocytes. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E530–E541. [Google Scholar] [CrossRef]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Spagnuolo, R.; Labarbuta, R.; Porreca, I.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; Perrini, S.; et al. The myokine irisin is released in response to saturated fatty acids and promotes pancreatic betacell survival and insulin secretion. Diabetes 2017, 66, 2849–2856. [Google Scholar] [CrossRef] [Green Version]
- Meilianna, A.; Dewi, N.M.; Wijaya, A. Adipose tissue, inflammation (Meta-inflammation) and Obesity management. Indones. Biomed. J. 2015, 7, 129–146. [Google Scholar] [CrossRef]
- Sahin-Efe, A.; Upadhyay, J.; Ko, B.J.; Dincer, F.; Park, K.H.; Migdal, A.; Vokonas, P.; Mantzoros, C. Irisin and leptin concentrations in relation to obesity, and developing type 2 diabetes: A cross sectional and a prospective case-control study nested in the Normative Aging Study. Metabolism 2018, 79, 24–32. [Google Scholar] [CrossRef]
- De Meneck, F.; de Souza, L.V.; Oliveira, V.; do Franco, M.C. High irisin levels in overweight/obese children and its positive correlation with metabolic profile, blood pressure, and endothelial progenitor cells. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 756–764. [Google Scholar] [CrossRef]
- Yan, B.; Shi, X.; Zhang, H. Association of serum irisin with metabolic syndrome in obese Chinese adults. PLoS ONE 2014, 9, e94235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Daghri, N.M.; Alkharfy, K.M.; Rahman, S. Irisin as a predictor of glucose metabolism in children: Sexually dimorphic effects. Eur. J. Clin. Investig. 2014, 44, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Panagiotou, G.; Mougios, V.; Brinkoetter, M.; Vamvini, M.T.; Schneider, B.E.; Mantzoros, C.S. FNDC5 and irisin in humans: I. Predictors of circulating concentrations in serum and plasma and II. mRNA expression and circulating concentrations in response to weight loss and exercise. Metabolism 2012, 61, 1725–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, M.; Crujeiras, A.B.; Amil, M. Association of irisin with fat mass, resting energy expenditure, and daily activity in conditions of extreme body mass index. Int. J. Endocrinol. 2014, 2014, 857270. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, Y.; Kurose, S.; Shinno, H. Relationships between serum irisin levels and metabolic parameters in Japanese patients with obesity. Obes. Sci. Pr. 2016, 2, 203–209. [Google Scholar] [CrossRef]
- Park, K.H.; Zaichenko, L.; Brinkoetter, M. Circulating irisin in relation to insulin resistance and the metabolic syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 4899–4907. [Google Scholar] [CrossRef]
- Bi, J.; Yang, L.; Wang, T.; Zhang, J.; Li, T.; Ren, Y.; Wang, M.; Chen, X.; Lv, Y.; Wu, R. Irisin Improves Autophagy of Aged Hepatocytes via Increasing Telomerase Activity in Liver Injury. Oxid. Med. Cell. Longev. 2020, 2020, 6946037. [Google Scholar] [CrossRef]
- Xin, T.; Lu, C. Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging 2020, 12, 4474–4488. [Google Scholar] [CrossRef]
- Moon, H.S.; Dincer, F.; Mantzoros, C.S. Pharmacological concentrations of irisin increase cell proliferation without influencing markers of neurite outgrowth and synaptogenesis in mouse H19-7 hippocampal cell lines. Metabolism 2013, 62, 1131–1136. [Google Scholar] [CrossRef] [Green Version]
- Li, D.J.; Li, Y.H.; Yuan, H.B.; Qu, L.F.; Wang, P. The novel exercise-induced hormone irisin protects against neuronal injury via activation of the Akt and ERK1/2 signaling pathways and contributes to the neuroprotection of physical exercise in cerebral ischemia. Metabolism 2017, 68, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Küster, O.C.; Laptinskaya, D.; Fissler, P.; Schnack, C.; Zügel, M.; Nold, V.; Thurm, F.; Pleiner, S.; Karabatsiakis, A.; von Einem, B.; et al. Novel Blood-Based Biomarkers of Cognition, Stress, and Physical or Cognitive Training in Older Adults at Risk of Dementia: Preliminary Evidence for a Role of BDNF, Irisin, and the Kynurenine Pathway. J. Alzheimers Dis. 2017, 59, 1097–1111. [Google Scholar] [CrossRef]
- De Oliveira Bristot, V.J.; de Bem Alves, A.C.; Cardoso, L.R.; da Luz Scheffer, D.; Aguiar, A.S., Jr. The Role of PGC-1α/UCP2 Signaling in the Beneficial Effects of Physical Exercise on the Brain. Front. Neurosci. 2019, 13, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaianni, G.; Cuscito, C.; Mongelli, T.; Pignataro, P.; Buccoliero, C.; Liu, P.; Lu, P.; Sartini, L.; Di Comite, M.; Mori, G.; et al. The myokine irisin increases cortical bone mass. Proc. Natl. Acad. Sci. USA 2015, 112, 12157–12162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocchi, A.; He, C. Regulation of Exercise-Induced Autophagy in Skeletal Muscle. Curr. Pathobiol. Rep. 2017, 5, 177–186. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; Levine, B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokladny, K.; Zuhl, M.N.; Mandell, M.; Bhattacharya, D.; Schneider, S.; Deretic, V.; Moseley, P.L. Regulatory coordination between two major intracellular homeostatic systems: Heat shock response and autophagy. J. Biol. Chem. 2013, 288, 14959–14972. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Hood, D.A. The regulation of autophagy during exercise in skeletal muscle. J. Appl. Physiol. 2016, 120, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Ghareghani, P.; Shanaki, M.; Ahmadi, S.; Khoshdel, A.R.; Rezvan, N.; Meshkani, R. Gorgani-Firuzjaee, S. Aerobic endurance training improves nonalcoholic fatty liver disease (NAFLD) features via miR-33 dependent autophagy induction in high fat diet fed mice. Obes. Res. Clin. Pr. 2018, 12, 80–89. [Google Scholar] [CrossRef]
- Salminen, A.; Vihko, V. Autophagic response to strenuous exercise in mouse skeletal muscle fibers. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1984, 45, 97–106. [Google Scholar] [CrossRef]
- Dohm, G.L.; Tapscott, E.B.; Kasperek, G.J. Protein degradation during endurance exercise and recovery. Med. Sci. Sports Exerc. 1987, 19, S166–S171. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Coletto, L.; Schiavinato, A.; Castagnaro, S.; Bertaggia, E.; Sandri, M.; Bonaldo, P. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 2011, 7, 1415–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, B.T.; Siu, P.M. Autophagic cellular responses to physical exercise in skeletal muscle. Sports Med. 2014, 44, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, A.; He, C. Activating autophagy by aerobic exercise in mice. J. Vis. Exp. 2017, 120, e55099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooren, F.C.; Kruger, K. Exercise, autophagy, and apoptosis. Prog. Mol. Biol. Trans. Sci. 2015, 135, 407–422. [Google Scholar]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Ballabio, A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.S.; Jeon, S.I.; Park, J.Y.; Lee, J.Y.; Lee, S.C.; Cho, K.J.; Jeong, J.M. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J. Physiol. Sci. 2016, 66, 417–430. [Google Scholar] [CrossRef]
- Ogura, Y.; Iemitsu, M.; Naito, H.; Kakigi, R.; Kakehashi, C.; Maeda, S.; Akema, T. Single bout of running exercise changes LC3-II expression in rat cardiac muscle. Biochem. Biophys. Res. Commun. 2011, 414, 756–760. [Google Scholar] [CrossRef]
- Li, H.; Miao, W.; Ma, J.; Xv, Z.; Bo, H.; Li, J.; Ji, L.L. Acute exercise-induced mitochondrial stress triggers an inflammatory response in the myocardium via NLRP3 inflammasome activation with mitophagy. Oxid. Med. Cell. Longev. 2016, 2016, 1987149. [Google Scholar] [CrossRef] [Green Version]
- Mejias-Pena, Y.; Estebanez, B.; Rodriguez-Miguelez, P.; Fernandez-Gonzalo, R.; Almar, M.; de Paz, J.A.; Cuevas, M.J. Impact of resistance training on the autophagy-inflammation-apoptosis crosstalk in elderly subjects. Aging 2017, 9, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Mejias-Pena, Y.; Rodriguez-Miguelez, P.; Fernandez-Gonzalo, R.; Martinez Florez, S.; Almar, M.; de Paz, J.A.; Gonzalez-Gallego, J. Effects of aerobic training on markers of autophagy in the elderly. Age 2017, 38, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, C.D.; Lee, M.S.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.I.; Wiley, J.W. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chu, X.; Yin, M.; Liu, X.; Yuan, H.; Niu, Y.; Fu, L. mTOR and autophagy in normal brain aging and caloric restriction ameliorating age-related cognition deficits. Behav. Brain Res. 2014, 264, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Ren, J. Autophagy and cardiovascular aging: Lesson learned from rapamycin. Cell Cycle 2012, 11, 2092–2099. [Google Scholar] [CrossRef]
- Jo, E.K.; Shin, D.M.; Choi, A.M. Autophagy: Cellular defense to excessive inflammation. Microbes Infect. 2012, 14, 119–125. [Google Scholar] [CrossRef]
- Fan, J.; Kou, X.; Jia, S.; Yang, X.; Yang, Y.; Chen, N. Autophagy as a potential target for sarcopenia. J. Cell. Physiol. 2016, 231, 1450–1459. [Google Scholar] [CrossRef]
- Tachtsis, B.; Smiles, W.J.; Lane, S.C.; Hawley, J.A.; Camera, D.M. Acute endurance exercise induces nuclear p53 abundance in human skeletal muscle. Front. Physiol. 2016, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Bai, L.; Yan, J.; Li, Y.; Shen, W.; Wang, Y.; Liu, J. Mitochondrial dynamic remodeling in strenuous exercise-induced muscle and mitochondrial dysfunction: Regulatory effects of hydroxytyrosol. Free. Radic. Biol. Med. 2011, 50, 1437–1446. [Google Scholar] [CrossRef]
- Wohlgemuth, S.E.; Lees, H.A.; Marzetti, E.; Manini, T.M.; Aranda, J.M.; Daniels, M.J.; Anton, S.D. An exploratory analysis of the effects of a weight loss plus exercise program on cellular quality control mechanisms in older overweight women. Rejuvenation Res. 2011, 14, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp. Gerontol. 2010, 45, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Bareja, A.; Lee, D.E.; White, J.P. Maximizing Longevity and Healthspan: Multiple Approaches All Converging on Autophagy. Front. Cell. Dev. Biol. 2019, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Hopkinson, N.S.; Kemp, P.R. Myostatin induces autophagy in skeletal muscle in vitro. Biochem. Biophys. Res. Commun. 2011, 415, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Seiliez, I.; Taty, G.C.; Bugeon, J.; Dias, K.; Sabin, N.; Gabillard, J.C. Myostatin induces atrophy of trout myotubes through inhibiting the TORC1 signaling and promoting ubiquitin-proteasome and autophagy–lysosome degradative pathways. Gen. Comp. Endocrinol. 2013, 186, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.T.; Yang, Y.J.; Huang, R.H.; Zhang, Z.H.; Lin, X. Myostatin Activates the Ubiquitin-Proteasome and Autophagy-Lysosome Systems Contributing to Muscle Wasting in Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2015, 2015, 684965. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, L.H.; Paula-Gomes, S.; Zanon, N.M.; Kettelhut, I.C. Myostatin promotes distinct responses on protein metabolism of skeletal and cardiac muscle fibers of rodents. Braz. J. Med. Biol. Res. 2017, 50, e6733. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, L.H.; Ang, J.; Peker, N.; Dagda, R.K.; McFarlane, C. G protein-coupled receptor kinase 2 regulates mitochondrial bioenergetics and impairs myostatin-mediated autophagy in muscle cells. Am. J. Physiol. Cell. Physiol. 2019, 317, C674–C686. [Google Scholar] [CrossRef] [Green Version]
- Gallot, Y.S.; Durieux, A.C.; Castells, J.; Desgeorges, M.M.; Vernus, B.; Plantureux, L.; Rémond, D.; Jahnke, V.E.; Lefai, E.; Dardevet, D.; et al. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res. 2014, 74, 7344–7356. [Google Scholar] [CrossRef] [Green Version]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T. Decorin causes autophagy in endothelial cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef] [Green Version]
- Yao, T.; Zhang, C.G.; Gong, M.T.; Zhang, M.; Wang, L.; Ding, W. Decorin-mediated inhibition of the migration of U87MG glioma cells involves activation of autophagy and suppression of TGF-beta signaling. FEBS Open Bio 2016, 6, 707–719. [Google Scholar] [CrossRef]
- Zhao, H.; Xi, H.; Wei, B.; Cai, A.; Wang, T.; Wang, Y. Expression of decorin in intestinal tissues of mice with inflammatory bowel disease and its correlation with autophagy. Exp. Ther. Med. 2016, 12, 3885–3892. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Photopulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2013, 6, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Gubbiotti, M.A.; Iozzo, R.V. Decorin-inducible Peg3 Evokes Beclin 1-mediated Autophagy and Thrombospondin 1-mediated Angiostasis. J. Biol. Chem. 2017, 292, 5055–5069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neill, T.; Sharpe, C.; Owens, R.T.; Iozzo, R.V. Decorin-Evoked Paternally Expressed Gene 3 (PEG3) is an Upstream Regulator of the Transcription Factor EB (TFEB) in Endothelial Cell Autophagy. J. Biol. Chem. 2017, 292, 16211–16220. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.A.; Arroyo, D.S.; Gaviglio, E.A.; Rodriguez-Galan, M.C.; Wang, J.M.; Iribarren, P. Interleukin 4 induces the apoptosis of mouse microglial cells by a caspase-dependent mechanism. Neurobiol. Dis. 2011, 43, 616–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, R.H.; Qi, R.Q.; Liu, H.Y. Interleukin-4 affects microglial autophagic flux. Neural Regen. Res. 2019, 14, 1594–1602. [Google Scholar] [PubMed]
- Xia, F.; Deng, C.; Jiang, Y.; Qu, Y.; Deng, J.; Cai, Z.; Ding, Y.; Guo, Z.; Wang, J. IL4 (interleukin 4) induces autophagy in B cells leading to exacerbated asthma. Autophagy 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Dickinson, J.D.; Sweeter, J.M.; Warren, K.J.; Ahmad, I.M.; De Deken, X.; Zimmerman, M.C.; Brody, S.L. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol. 2018, 14, 272–284. [Google Scholar] [CrossRef]
- Terawaki, S.; Camosseto, V.; Prete, F.; Wenger, T.; Papadopoulos, A.; Rondeau, C.; Combes, A.; Rodriguez Rodrigues, C.; Vu Manh, T.P.; Fallet, M.; et al. RUN and FYVE domain-containing protein 4 enhances autophagy and lysosome tethering in response to Interleukin-4. J. Cell. Biol. 2015, 210, 1133–1152. [Google Scholar] [CrossRef] [Green Version]
- Terawaki, S.; Camosseto, V.; Pierre, P.; Gatti, E. RUFY4: Immunity piggybacking on autophagy? Autophagy 2016, 12, 598–600. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh, H.J., 3rd. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL-6-pSTAT3 pathway. Autophagy 2012, 8, 989–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delk, N.A.; Farach-Carson, M.C. Interleukin-6: A bone marrow stromal cell paracrine signal that induces neuroendocrine differentiation and modulates autophagy in bone metastatic PCa cells. Autophagy 2012, 8, 650–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.; Yuan, G.; Guo, X.; Liu, Q.; Zhang, J.; Gao, X.; Guo, X.; Xu, S.; Li, T.; Shao, Q.; et al. A novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: Hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy 2016, 12, 1129–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, A. The absence of interleukin-6 enhanced arsenite-induced renal injury by promoting autophagy of tubular epithelial cells with aberrant extracellular signal-regulated kinase activation. Am. J. Pathol. 2010, 176, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Z.; Sui, C.Y.; Chen, Q.; Chen, X.P.; Zhang, H.; Zhou, X.P. Promotion of autophagy at the maturation step by IL-6 is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Mol. Cell. Biochem. 2013, 380, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Zhou, Z.; He, J.; Yan, C.; Ding, S. IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Sci. Rep. 2015, 5, 15701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Z.; Qiao, F.; Lu, Q.; Ma, Y.; Liu, Y.; Lu, F.; Xu, Z. Interleukin-6 downregulated vascular smooth muscle cell contractile proteins via ATG4B-mediated autophagy in thoracic aortic dissection. Heart Vessels. 2017, 32, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, A.K.; Blumer, J.; Marasco, M.R. Interleukin 6 protects pancreatic b cells from apoptosis by stimulation of autophagy. FASEB J. 2017, 31, 4140–4152. [Google Scholar] [CrossRef] [Green Version]
- Marasco, M.R.; Conteh, A.M.; Reissaus, C.A.; Cupit, J.E.; Appleman, E.M.; Mirmira, R.G.; Linnemann, A.K. Interleukin-6 Reduces β-Cell Oxidative Stress by Linking Autophagy With the Antioxidant Response. Diabetes 2018, 67, 1576–1588. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.P.; da Rocha, A.L.; Cabrera, E.M.B.; Marafon, B.B.; Kohama, E.B.; Rovina, R.L.; Simabuco, F.M.; Bueno Junior, C.R.; de Moura, L.P.; Pauli, J.R.; et al. Role of interleukin-6 in inhibiting hepatic autophagy markers in exercised mice. Cytokine 2020, 130, 155085. [Google Scholar] [CrossRef]
- Ruppert, S.M.; Li, W.; Zhang, G.; Carlson, A.L.; Limaye, A.; Durum, S.K.; Khaled, A.R. The major isoforms of Bim contribute to distinct biological activities that govern the processes of autophagy and apoptosis in interleukin-7 dependent lymphocytes. Biochim. Biophys. Acta 2012, 1823, 1877–1893. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, W.; Zhang, L.; Xu, L.; Chen, X.; Zhou, S.; Xu, Z.; Xiao, M.; Bai, H.; Liu, F.; et al. IL-7 suppresses macrophage autophagy and promotes liver pathology in Schistosoma japonicum-infected mice. J. Cell. Mol. Med. 2018, 22, 3353–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, M.; Yunjia, Z.; Zhiying, D.; Yanduo, J.; Guocheng, J. Interleukin 7 receptor activates PI3K/Akt/mTOR signaling pathway via downregulation of Beclin-1 in lung cancer. Mol. Carcinog. 2019, 58, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Huang, G.; Zhu, P.; Liu, J.; Ye, B.; Du, Y.; Fan, Z. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 2016, 7, 11023. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Xie, X.; Zhang, L.; Wang, H.; Jie, Z.; Zhou, X.; Shi, J.; Zhao, S.; Zhang, B.; Cheng, X.; et al. TBK-binding protein 1 regulates IL-15-induced autophagy and NKT cell survival. Nat. Commun. 2018, 9, 2812. [Google Scholar] [CrossRef]
- Xu, A.; Bhanumathy, K.K.; Wu, J.; Ye, Z.; Freywald, A.; Leary, S.C.; Li, R.; Xiang, J. IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci. 2016, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Swadling, L.; Pallett, L.J.; Diniz, M.O.; Baker, J.M.; Amin, O.E.; Stegmann, K.A.; Burton, A.R.; Schmidt, N.M.; Jeffery-Smith, A.; Zakeri, N.; et al. Human Liver Memory CD8(+) T Cells Use Autophagy for Tissue Residence. Cell Rep. 2020, 30, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Lee, J.O.; Lee, Y.W.; Kim, S.A.; Seo, I.H.; Han, J.A.; Kang, M.J.; Kim, S.J.; Cho, Y.H.; Park, J.J.; et al. LIF, a Novel Myokine, Protects Against Amyloid-Beta-Induced Neurotoxicity via Akt-Mediated Autophagy Signaling in Hippocampal Cells. Int. J. Neuropsychopharmacol. 2019, 22, 402–414. [Google Scholar] [CrossRef]
- Seldin, M.M.; Lei, X.; Tan, S.Y.; Stanson, K.P.; Wei, Z.; Wong, G.W. Skeletal muscle-derived myonectin activates the mammalian target of rapamycin (mTOR) pathway to suppress autophagy in liver. J. Biol. Chem. 2013, 288, 36073–36082. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Li, Z.Y.; Wang, D.S.; Xu, T.Y.; Fan, M.B.; Cheng, M.H.; Miao, C.Y. Aggravated ulcerative colitis caused by intestinal Metrnl deficiency is associated with reduced autophagy in epithelial cells. Acta Pharmacol. Sin. 2020. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Mathew, H.; Mantzoros, C.S. Irisin: A true, circulating hormone. Metabolism 2015, 64, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Du, F.; Li, X.; Wang, M.; Duan, R.; Zhang, J.; Wu, J.; Zhang, Q. Effects and underlying mechanisms of irisin on the proliferation and apoptosis of pancreatic beta cells. PLoS ONE 2017, 12, e0175498. [Google Scholar]
- Huerta, A.E.; Prieto-Hontoria, P.L.; Fernandez-Galilea, M.; Sainz, N.; Cuervo, M.; Martinez, J.A.; Moreno-Aliaga, M.J. Circulating irisin and glucose metabolism in overweight/obese women: Effects of alpha-lipoic acid and eicosapentaenoic acid. J. Physiol. Biochem. 2015, 71, 547–558. [Google Scholar] [CrossRef]
- Duran, I.D.; Gulcelik, N.E.; Unal, M.; Topcuoglu, C.; Sezer, S.; Tuna, M.M.; Berker, D.; Guler, S. Irisin levels in the progression of diabetes in sedentary women. Clin. Biochem. 2015, 48, 1268–1272. [Google Scholar] [CrossRef]
- Akour, V.; Kasabri, N.; Boulatova, Y.; Bustanji, R.; Naffa, D.; Hyasat, N.; Khawaja, H.; Bustanji, A.; Zayed, M.; Momani, M. Levels of metabolic markers in drug-naive prediabetic and type 2 diabetic patients. Acta Diabetol. 2017, 54, 163–170. [Google Scholar] [CrossRef]
- Li, D.J.; Huang, F.; Lu, W.J.; Jiang, G.J.; Deng, Y.P.; Shen, F.M. Metformin promotes irisin release from murine skeletal muscle independently of AMP-activated protein kinase activation. Acta Physiol. 2015, 213, 711–721. [Google Scholar] [CrossRef]
- Duan, H.; Ma, B.; Ma, X.; Wang, H.; Ni, Z.; Wang, B.; Li, X.; Jiang, P.; Umar, M.; Li, M. Anti-diabetic activity of recombinant irisin in STZ-induced insulin-deficient diabetic mice. Int. J. Biol. Macromol. 2016, 84, 457–463. [Google Scholar] [CrossRef]
- Xin, C.; Liu, J.; Zhang, J.; Zhu, D.; Wang, H.; Xiong, L.; Lee, Y.; Ye, J.; Lian, K.; Xu, C.; et al. Irisin improves fatty acid oxidation and glucose utilization in type 2 diabetes by regulating the AMPK signaling pathway. Int. J. Obes. 2016, 40, 443–451. [Google Scholar] [CrossRef]
- Ye, X.; Shen, Y.; Ni, C.; Ye, J.; Xin, Y.; Zhang, W.; Ren, Y. Irisin reverses insulin resistance in C2C12 cells via the p38-MAPK-PGC-1α pathway. Peptides 2019, 119, 170120. [Google Scholar] [CrossRef]
- Li, Q.; Jia, S.; Xu, L.; Li, B.; Chen, N. Metformin-induced autophagy and irisin improves INS-1 cell function and survival in high-glucose environment via AMPK/SIRT1/PGC-1α signal pathway. Food Sci. Nutr. 2019, 7, 1695–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, S.; Kuloglu, T.; Aydin, S.; Kalayci, M.; Yilmaz, M.; Cakmak, T.; Albayrak, S.; Gungor, S.; Colakoglu, N.; Ozercan, I.H. A comprehensive immunohistochemical examination of the distribution of the fat-burning protein irisin in biological tissues. Peptides 2014, 61, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Shi, C.X.; Gao, R.; Sun, H.J.; Xiong, X.Q.; Ding, L.; Chen, Q.; Li, Y.H.; Wang, J.J.; Kang, Y.M.; et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin. Sci. 2015, 129, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.; Shen, J.; Liu, Q.; Zhang, Y.; Kuang, J.; Pu, S.; Cheng, S.; Zou, M.; Jiang, W.; Jiang, C.; et al. Irisin is regulated by CAR in liver and is a mediator of hepatic glucose and lipid metabolism. Mol. Endocrinol. 2016, 30, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonalcoholic fatty liver disease: The pathogenetic roles of insulin resistance and adipocytokines. Curr. Mol. Med. 2009, 9, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Batirel, S.; Bozaykut, P.; Mutlu Altundag, E.; Kartal Ozer, N.; Mantzoros, C.S. The effect of irisin on antioxidant system in liver. Free Radic. Biol. Med. 2014, 75, S16. [Google Scholar] [CrossRef]
- Liu, T.Y.; Xiong, X.Q.; Ren, X.S.; Zhao, M.X.; Shi, C.X.; Wang, J.J.; Zhou, Y.B.; Zhang, F.; Han, Y.; Gao, X.Y.; et al. FNDC5 Alleviates Hepatosteatosis by Restoring AMPK/mTOR-Mediated Autophagy, Fatty Acid Oxidation, and Lipogenesis in Mice. Diabetes 2016, 65, 3262–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, S.; Kuloglu, T.; Aydin, S.; Eren, M.N.; Celik, A.; Yilmaz, M.; Kalayci, M.; Sahin, I.; Gungor, O.; Gurel, A.; et al. Cardiac, skeletal muscle and serum irisin responses to with or without water exercise in young and old male rats: Cardiac muscle produces more irisin than skeletal muscle. Peptides 2014, 52, 68–73. [Google Scholar] [CrossRef]
- Anastasilakis, A.D.; Koulaxis, D.; Kefala, N.; Polyzos, S.A.; Upadhyay, J.; Pagkalidou, E.; Economou, F.; Anastasilakis, C.D.; Mantzoros, C.S. Circulating irisin levels are lower in patients with either stable coronary artery disease (CAD) or myocardial infarction (MI) versus healthy controls, whereas follistatin and activin A levels are higher and can discriminate MI from CAD with similar to CK-MB accuracy. Metabolism 2017, 73, 1–8. [Google Scholar]
- Kuloglu, T.; Aydin, S.; Eren, M.N.; Yilmaz, M.; Sahin, I.; Kalayci, M.; Sarman, E.; Kaya, N.; Yilmaz, O.F.; Turk, A.; et al. Irisin: A potentially candidate marker for myocardial infarction. Peptides 2014, 55, 85–91. [Google Scholar] [CrossRef]
- Deng, W. Association of serum irisin concentrations with presence and severity of coronary artery disease. Med. Sci. Monit. 2016, 22, 4193–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhao, Y.T.; Zhang, S.; Dubielecka, P.M.; Du, J.; Yano, N.; Chin, Y.E.; Zhuang, S.; Qin, G.; Zhao, T.C. Irisin plays a pivotal role to protect the heart against ischemia and reperfusion injury. J. Cell. Physiol. 2017, 232, 3775–3785. [Google Scholar] [CrossRef] [PubMed]
- Aronis, K.N.; Moreno, M.; Polyzos, S.A. Circulating irisin levels and coronary heart disease: Association with future acute coronary syndrome and major adverse cardiovascular events. Int. J. Obes. 2015, 39, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Li, R.L.; Wu, S.S.; Wu, Y.; Wang, X.X.; Chen, H.Y.; Xin, J.J.; Li, H.; Lan, J.; Xue, K.Y.; Li, X.; et al. Irisin alleviates pressure overload-induced cardiac hypertrophy by inducing protective autophagy via mTOR-independent activation of the AMPK-ULK1 pathway. J. Mol. Cell. Cardiol. 2018, 121, 242–255. [Google Scholar] [CrossRef]
- Li, R.; Wang, X.; Wu, S.; Wu, Y.; Chen, H.; Xin, J.; Li, H.; Lan, J.; Xue, K.; Li, X.; et al. Irisin ameliorates angiotensin II-induced cardiomyocyte apoptosis through autophagy. J. Cell. Physiol. 2019, 234, 17578–17588. [Google Scholar] [CrossRef]
- Shelbaya, S.; Abushady, M.M.; Nasr, M.S.; Bekhet, M.M.; Mageed, Y.A.; Abbas, M. Study of irisin hormone level in type 2 diabetic patients and patients with diabetic nephropathy. Curr. Diabetes Rev. 2018, 14, 481–486. [Google Scholar] [CrossRef]
- Pan, Y.J.; Zhou, S.J.; Feng, J.; Bai, Q.; La-Ta, A.; Zhang, A.H. Urotensin II Induces Mice Skeletal Muscle Atrophy Associated with Enhanced Autophagy and Inhibited Irisin Precursor (Fibronectin Type III Domain Containing 5) Expression in Chronic Renal Failure. Kidney Blood Press. Res. 2019, 44, 479–495. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pesce, M.; Ballerini, P.; Paolucci, T.; Puca, I.; Farzaei, M.H.; Patruno, A. Irisin and Autophagy: First Update. Int. J. Mol. Sci. 2020, 21, 7587. https://doi.org/10.3390/ijms21207587
Pesce M, Ballerini P, Paolucci T, Puca I, Farzaei MH, Patruno A. Irisin and Autophagy: First Update. International Journal of Molecular Sciences. 2020; 21(20):7587. https://doi.org/10.3390/ijms21207587
Chicago/Turabian StylePesce, Mirko, Patrizia Ballerini, Teresa Paolucci, Iris Puca, Mohammad Hosein Farzaei, and Antonia Patruno. 2020. "Irisin and Autophagy: First Update" International Journal of Molecular Sciences 21, no. 20: 7587. https://doi.org/10.3390/ijms21207587
APA StylePesce, M., Ballerini, P., Paolucci, T., Puca, I., Farzaei, M. H., & Patruno, A. (2020). Irisin and Autophagy: First Update. International Journal of Molecular Sciences, 21(20), 7587. https://doi.org/10.3390/ijms21207587