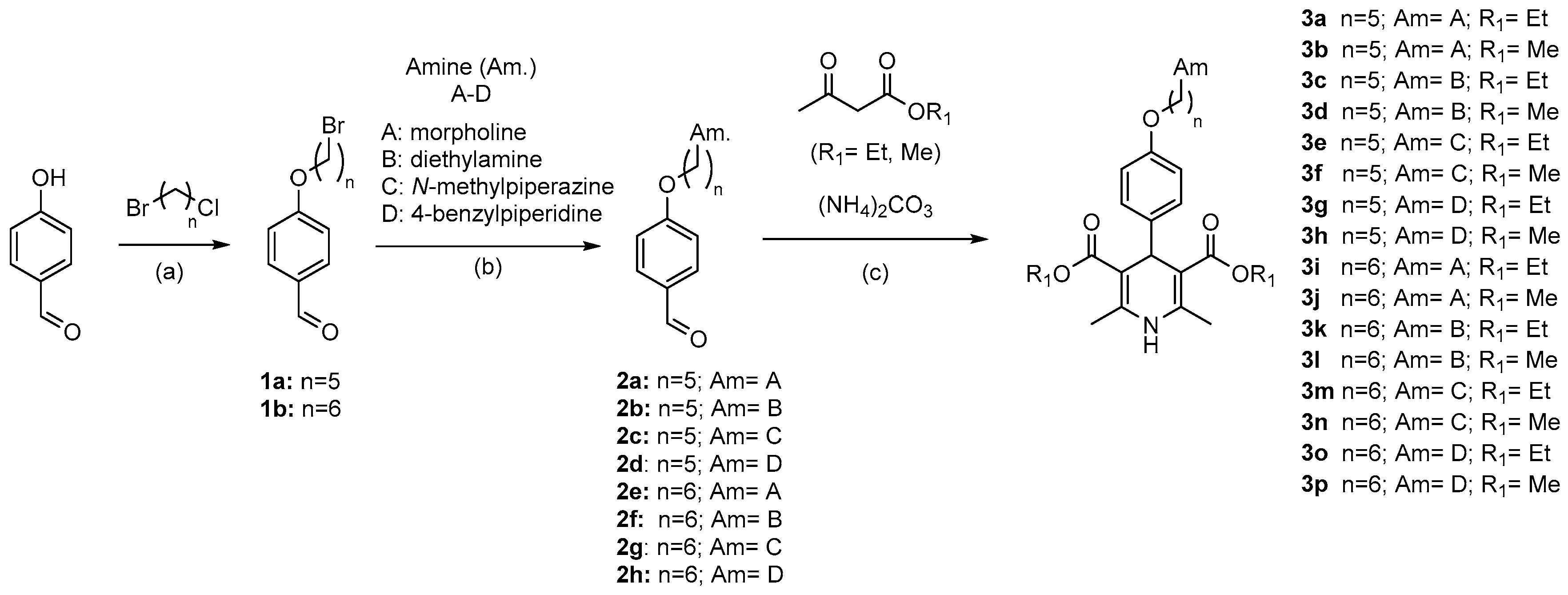
Synthesis of Compounds 2a-h
4-((5-Morpholinopentyl)oxy)benzaldehyde (2a). The crude was prepared according to general procedure B starting from 4-((5-bromopentyl)oxy)benzaldehyde 1a (1 equiv., 6.78 mmol, 1.84 g), commercially available morpholine (2 equiv., 13.56 mmol, 1.19 mL) and K2CO3 (3 equiv., 20.34 mmol, 2.81 g) in acetonitrile (25 mL) to afford 599.6 mg of the desired compound (32%) as a transparent oil. The crude was used without further purifications. 1H NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 7.89–7.75 (m, 2H), 7.05–6.94 (m, 2H), 4.05 (t, J = 6.4 Hz, 2H), 3.76 (s, 4H), 2.47 (d, J = 33.1 Hz, 6H), 1.94–1.77 (m, 2H), 1.62 (bs, 2H), 1.56–1.45 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 190.77, 164.15, 131.19, 129.82, 114.74, 68.97, 44.97, 32.48, 28.91, 26.61, 26.59, 25.34
4-((5-(Diethylamino)pentyl)oxy)benzaldehyde (2b). The crude was prepared according to general procedure B starting from 4-((5-bromopentyl)oxy)benzaldehyde 1a (1 equiv., 6.78 mmol, 1.84 g), commercially available diethylamine (2 equiv., 13.56 mmol, 1.404 mL) and K2CO3 (3 equiv., 20.34 mmol, 2.81 g) in acetonitrile (25 mL) to afford 510.50 mg of the desired compound (29%) as a transparent oil. The crude was used without further purifications. 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 1H), 7.85–7.78 (m, 2H), 7.03–6.92 (m, 2H), 4.03 (t, J = 6.5 Hz, 2H), 2.54 (q, J = 7.2 Hz, 5H), 2.45–2.36 (m, 2H), 1.82 (td, J = 13.3, 6.6 Hz, 3H), 1.56–1.43 (m, 4H), 1.39–1.31 (m, 2H), 1.03 (t, J = 7.2 Hz, 7H). 13C NMR (101 MHz, CDCl3) δ 190.91, 164.21, 131.99, 129.78, 114.75, 68.30, 52.80, 46.86, 29.09, 26.83, 24.10, 11.61.
4-((5-(4-Methylpiperazin-1-yl)pentyl)oxy)benzaldehyde (2c). The crude was prepared according to general procedure B starting from 4-((5-bromopentyl)oxy)benzaldehyde 1a (1 equiv., 5.08 mmol, 1.38 g), commercially available N-methylpiperazine (2 equiv., 10.16 mmol, 1.13 mL) and K2CO3 (3 equiv., 15.23 mmol, 2.11 g) in acetonitrile (25 mL) to afford 415.6 mg of the desired compound (28%) as a transparent oil. The crude was used without further purifications. 1H NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 7.87–7.75 (m, 2H), 7.01–6.90 (m, 2H), 4.04 (t, J = 6.4 Hz, 2H), 2.52 (bs, 8H), 2.39 (dd, J = 17.5, 9.7 Hz, 2H), 2.31 (d, J = 9.9 Hz, 3H), 1.92–1.76 (m, 2H), 1.62–1.54 (m, 2H), 1.53–1.43 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 190.81, 164.17, 132.00, 129.80, 114.97, 68.19, 58.49, 55.07, 53.18, 46.01, 28.97, 26.58, 24.00.
4-((5-(4-Benzylpiperidin-1-yl)pentyl)oxy)benzaldehyde (2d). The crude was prepared according to general procedure B starting from 4-((5-bromopentyl)oxy)benzaldehyde 1a (1 equiv., 1.48 mmol, 403.00 mg), commercially available 4-benzylpiperidine (2 equiv., 2.97 mmol, 0.521 mL) and K2CO3 (3 equiv., 4.45 mmol, 651.00 mg) in acetonitrile (8 mL) to afford 257.00 mg of the desired compound (43%) as a transparent oil. The crude was used without further purifications. 1H NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 7.85–7.73 (m, 2H), 7.28 (d, J = 7.1 Hz, 2H), 7.19 (dd, J = 8.4, 6.3 Hz, 1H), 7.15–7.10 (m, 2H), 7.02–6.92 (m, 2H), 4.04 (t, J = 6.4 Hz, 2H), 3.04 (bs, 2H), 2.55 (d, J = 6.6 Hz, 2H), 2.45 (s, 2H), 1.99 (s, 2H), 1.88–1.79 (m, 3H), 1.68 (d, J = 11.4 Hz, 4H), 1.55–1.42 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 190.82, 164.02, 140.70, 132.00, 129.79, 129.13, 128.16, 125.78,114.75, 68.22, 58.92, 54.02,, 43.22, 37.97, 43.22, 37.97, 32.13, 28.98, 26.75.
4-((6-Morpholinohexyl)oxy)benzaldehyde (2e). The crude was prepared according to general procedure B starting from 4-((6-bromohexyl)oxy)benzaldehyde 1b (1 equiv., 2.02 mmol, 500 mg), commercially available morpholine (2 equiv., 4.04 mmol, 0.352 mL) and K2CO3 (3 Equiv., 6.06 mmol, 837.55 mg) in acetonitrile (20 mL) to afford 600 mg of the desired compound (48%) as a transparent oil. The crude was used without further purifications. 1H NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 7.86–7.80 (m, 2H), 7.06–6.95 (m, 2H), 4.04 (t, J = 6.4 Hz, 2H), 3.77 (s, 3H), 2.46 (d, J = 37.6 Hz, 6H), 1.82 (dd, J = 14.7, 6.7 Hz, 2H), 1.55–1.45 (m, 3H), 1.41 (dt, J = 8.7, 2.7 Hz, 2H), 1.25 (d, J = 4.9 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 190.80, 164.20, 131.99, 129.79, 114.74, 66.94, 59.03, 44.97, 32.48, 29.01, 26.78, 26.43, 25.94
4-((6-(Diethylamino)hexyl)oxy)benzaldehyde (2f). The crude was prepared according to general procedure B starting from 4-((6-bromohexyl)oxy)benzaldehyde 1b (1 equiv., 2.02 mmol, 500 mg), commercially available diethylamine (2 equiv., 4.04 mmol, 0.416 mL) and K2CO3 (3 Equiv., 6.06 mmol, 837.55 mg) in acetonitrile (20 mL) to afford 237.4 mg of the desired compound (42%) as a transparent oil. The crude was used without further purifications. 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 1H), 7.85–7.78 (m, 2H), 7.03–6.92 (m, 2H), 4.03 (t, J = 6.5 Hz, 2H), 2.54 (q, J = 7.2 Hz, 5H), 2.45–2.36 (m, 2H), 1.82 (td, J = 13.3, 6.6 Hz, 3H), 1.56–1.43 (m, 4H), 1.39–1.31 (m, 2H), 1.03 (t, J = 7.2 Hz, 7H). 13C NMR (101 MHz, CDCl3) δ 190.82, 164.24, 132.00, 129.77, 114.76, 68.33, 52.82, 29.06, 27.44, 26.90, 25.98, 11.58
4-((6-(4-Methylpiperazin-1-yl)hexyl)oxy)benzaldehyde (2g). The crude was prepared according to general procedure B starting from 4-((6-bromohexyl)oxy)benzaldehyde 1b (1 equiv., 4.03 mmol, 1 g), commercially available N-methylpiperazine (2 equiv., 8.06 mmol, 0.894 mL) and K2CO3 (3 Equiv., 12.09 mmol, 1.67 g) in acetonitrile (40 mL) to afford 632 mg of the desired compound (49%) as a transparent oil. The crude was used without further purifications. 1H NMR (400 MHz, CDCl3) δ 9.85 (s, 1H), 7.87–7.68 (m, 2H), 6.99–6.91 (m, 2H), 4.01 (t, J = 6.5 Hz, 2H), 2.44 (bs, 8H), 2.36–2.30 (m, 2H), 2.26 (s, 3H), 1.94–1.71 (m, 2H), 1.55–1.41 (m, 4H), 1.36 (td, J = 8.6, 1.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 190.84, 164.21, 132.00, 129.76, 114.7, 68.26, 58.59, 55.02, 53.14, 45.98, 28.99, 27.30, 26.75, 25.92.
4-((6-(4-Benzylpiperidin-1-yl)hexyl)oxy)benzaldehyde (2h). The crude was prepared according to general procedure B. starting 4-((6-bromohexyl)oxy)benzaldehyde 1b (1 equiv., 2.84 mmol, 705.47 mg), 4-benzylpiperidine (2 equiv., 5.69 mmol, 1.00 mL) and K2CO3 (3 Equiv., 8.52 mmol, 1177.55 mg) in acetonitrile (28.2 mL) to afford 752 mg of the desired compound as a transparent oil. The crude was used without further purifications.
Synthesis of Compounds 3a-p
Dimethyl-4-(4-((5-(diethylamino)pentyl)oxy)phenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (3d). 3d was prepared according to general procedure C starting from 4-((5-(diethylamino)pentyl)oxy)benzaldehyde 2b (1 equiv., 0.81 mmol, 214.00 mg), methyl acetoacetate (3.5 equiv., 2.84 mmol, 306.7 mL) and ammonium carbonate (2.5 equiv., 2.03 mmol, 195.00 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 90/10 + 1% Et3N to afford 104.00 mg (28%) of 3d as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.19–7.10 (m, 2H), 6.77–6.68 (m, 2H), 5.66 (s, 1H), 4.93 (s, 1H), 3.90 (t, J = 6.4 Hz, 2H), 3.64 (d, J = 2.5 Hz, 6H), 2.59 (q, J = 7.1 Hz, 4H), 2.51–2.40 (m, 2H), 2.33 (s, 6H), 1.82–1.70 (m, 2H), 1.59–1.50 (m, 2H), 1.49–1.40 (m, 2H), 1.06 (t, J = 7.2 Hz, 7H). 13C NMR (101 MHz, CDCl3) δ 168.25, 157.60, 144.08, 139.90, 128.69, 114.05, 104.23, 67.79, 52.84, 51.09, 46.97, 38.51, 29.82, 29.41, 26.66, 24.32, 19.70, 11.54. HRMS ESI-TOF [M]+ m/z calcd. for C26H38N2O5: 458.2770, found: 458.2781.
Diethyl-4-(4-((5-(diethylamino)pentyl)oxy)phenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (3c). 3c was prepared according to general procedure C starting from 4-((5-(diethylamino)pentyl)oxy)benzaldehyde 2b (1 equiv., 1.52 mmol, 400.00 mg), ethyl acetoacetate (3.5 Equiv., 5.32 mmol, 520.00 mL) and ammonium carbonate (2.5 equiv., 3.80 mmol, 365.14 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 90/10 + 1% Et3N to afford 54.00 mg (7%) of 3c as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.19–7.14 (m, 2H), 6.76–6.69 (m, 2H), 5.60 (s, 1H), 4.91 (s, 1H), 4.14–4.02 (m, 4H), 3.90 (t, J = 6.4 Hz, 2H), 2.61 (dd, J = 14.2, 7.1 Hz, 4H), 2.55–2.45 (m, 2H), 2.32 (s, 6H), 1.85–1.71 (m, 2H), 1.56 (dt, J = 10.1, 7.4 Hz, 2H), 1.45 (dd, J = 15.1, 8.0 Hz, 2H), 1.22 (t, J = 7.1 Hz, 6H), 1.07 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.83, 157.51, 143.64, 140.33, 129.08, 113.88, 104.56, 67.76, 59.81, 52.71, 46.95, 38.86, 29.36, 26.34, 24.28, 19.74, 14.41, 11.28. HRMS ESI-TOF [M]+ m/z calcd. for C28H42N2O5: 486,3071, found: 486,3094.
Dimethyl-2,6-dimethyl-4-(4-((5-morpholinopentyl)oxy)phenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3b). 3b was prepared according to general procedure C starting from 4-((5-morpholinopentyl)oxy)benzaldehyde 2a (1 equiv., 1.49 mmol, 413.00 mg), methyl acetoacetate (3.5 equiv., 5.96 mmol, 643.00 mL) and ammonium carbonate (2.5 equiv., 4.47 mmol, 429.00 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 97/3 + 1% Et3N to afford 147.00 mg (21%) of 3b as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.19–7.10 (m, 2H), 6.78–6.66 (m, 2H), 5.72 (s, 1H), 4.93 (s, 1H), 3.89 (t, J = 6.4 Hz, 2H), 3.76–3.67 (m, 4H), 3.63 (s, 6H), 2.46 (bs, 4H), 2.40–2.33 (m, 2H), 2.32 (s, 6H), 1.81–1.72 (m, 2H), 1.61–1.51 (m, 2H), 1.50–1.41 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 168.22, 157.32, 143.99, 140.24, 128.83, 114.05, 104.31, 67.08, 63.73, 58.08, 52.04, 51.14, 38.60, 28.72, 23.71, 19.79. Anal. Calcd. for C26H36N2O6: C, 66.08; H, 7.68; N, 5.93; found: C, 64.78; H, 7.81; N, 5.98.
Diethyl-2,6-dimethyl-4-(4-((5-morpholinopentyl)oxy)phenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3a). 3a was prepared according to general procedure C starting from 4-((5-morpholinopentyl)oxy)benzaldehyde 2a (1 equiv., 0.927 mmol, 257.00 mg), ethyl acetoacetate (3.5 equiv., 3.71 mmol, 473.00 mL) and ammonium carbonate (2.5 equiv., 2.78 mmol, 267.00 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 95/5 + 1% Et3N to afford 223.00 mg (48%) of 3a as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.21–7.08 (m, 2H), 6.78–6.65 (m, 2H), 5.57 (s, 1H), 4.92 (s, 1H), 4.21–4.00 (m, 4H), 3.90 (t, J = 6.4 Hz, 2H), 3.77 (bs, 4H), 2.51 (bs, 4H), 2.42 (bs, 2H), 2.32 (s, 6H), 1.88–1.68 (m, 2H), 1.60 (bs, 2H), 1.55–1.40 (m, 2H), 1.22 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.83, 157.48, 143.71, 140.34, 129.04, 113.84, 104.46, 67.69, 66.99, 59.78, 59.07, 53.83, 38.84, 29.33, 26.31, 24.13, 19.65, 14.39. Anal. Calcd. for C28H40N2O6: C, 67.18; H, 8.05; N, 5.60; found: C, 66.01; H, 8.15; N, 5.79.
Dimethyl-2,6-dimethyl-4-(4-((5-(4-methylpiperazin-1-yl)pentyl)oxy)phenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3f). 3f was prepared according to general procedure C starting from 4-((5-(4-methylpiperazin-1-yl)pentyl)oxy) benzaldehyde 2c (1 equiv., 0.837 mmol, 243.00 mg), methyl acetoacetate (3.5 equiv., 3.35 mmol, 362.00 mL) and ammonium carbonate (2.5 equiv., 2.51 mmol, 241.00 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 92/8 + 1% Et3N to afford 46.05 mg (11%) of 3f as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.17–7.10 (m, 2H), 6.72 (d, J = 8.6 Hz, 2H), 5.89 (s, 1H), 4.92 (s, 1H), 4.08 (t, J = 6.5 Hz, 2H), 3.89 (t, J = 6.3 Hz, 2H), 3.63 (s, 6H), 3.53–3.47 (m, 4H), 2.38 (t, J = 2.4 Hz, 4H), 2.31 (d, J = 1.9 Hz, 9H), 1.81–1.72 (m, 2H), 1.72–1.63 (m, 2H), 1.56–1.46 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 168.26, 157.52, 155.58, 144.17, 140.00, 128.68, 114.02, 104.12, 67.57, 65.52, 54.74, 51.08, 46.13, 43.55, 38.49, 29.09, 28.85, 22.75, 19.63. HRMS ESI-TOF [M]+ m/z calcd. for C27H39N3O5: 485,2869, found: 485,2890.
Diethyl-2,6-dimethyl-4-(4-((5-(4-methylpiperazin-1-yl)pentyl)oxy)phenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3e). 3e was prepared according to general procedure C starting from 4-((5-(4-methylpiperazin-1-yl)pentyl)oxy) benzaldehyde 2c (1 equiv., 0.689 mmol, 200.00 mg), ethyl acetoacetate (3.5 equiv., 2.40 mmol, 308.12 mL) and ammonium carbonate (2.5 equiv., 1.72 mmol, 165.00 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 98/2 + 1% Et3N to afford 114.7 mg (32%) of 3e as a light brown solid. 1H NMR (400 MHz, CDCl3) δ 7.22–7.09 (m, 2H), 6.79–6.62 (m, 2H), 5.54 (s, 1H), 4.92 (s, 1H), 4.18–4.00 (m, 4H), 3.89 (t, J = 6.4 Hz, 2H), 2.59 (bs, 8H), 2.45–2.39 (m, 2H), 2.36 (s, 3H), 2.32 (s, 6H), 1.81–1.71 (m, 2H), 1.63–1.53 (m, 2H), 1.50–1.40 (m, 2H), 1.22 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.82, 157.51, 143.58, 140.33, 129.09, 113.88, 104.61, 77.48, 77.16, 76.84, 67.68, 59.83, 58.43, 52.73, 45.78, 38.88, 29.32, 24.18, 19.78, 14.43. HRMS ESI-TOF [M]+ m/z calcd. for C29H43N3O5: 513,3197, found: 513,3203.
Dimethyl-4-(4-((5-(4-benzylpiperidin-1-yl)pentyl)oxy)phenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (3h). 3h was prepared according to general procedure C starting from 4-((5-(4-benzylpiperidin-1-yl)pentyl)oxy)benzaldehyde 2d (1 equiv., 0.783 mmol, 286.00 mg), methyl acetoacetate (3.5 equiv., 3.13 mmol, 338.10 mL) and ammonium carbonate (2.5 equiv., 2.35 mmol, 226.00 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 98/2 + 1% Et3N to afford 64.32 mg (15%) of 3h as ayellow oil. 1H NMR (400 MHz, CDCl3) δ 7.30–7.23 (m, 4H), 7.20–7.06 (m, 5H), 6.75–6.65 (m, 2H), 5.76–5.67 (m, 1H), 4.93 (s, 1H), 3.88 (t, J = 6.5 Hz, 2H), 3.63 (d, J = 2.5 Hz, 6H), 2.92 (d, J = 11.4 Hz, 2H), 2.53 (d, J = 7.0 Hz, 2H), 2.36–2.27 (m, 6H), 1.87 (t, J = 11.2 Hz, 2H), 1.79–1.70 (m, 2H), 1.63 (d, J = 12.1 Hz, 2H), 1.58–1.48 (m, 4H), 1.46–1.39 (m, 3H), 1.37–1.30 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 168.23, 157.59, 144.06, 140.80, 139.88, 129.23, 128.68, 128.27, 125.89, 114.03, 104.21, 67.73, 59.06, 54.07, 51.08, 43.29, 38.50, 38.03, 32.14, 29.36, 26.81, 24.34, 19.68. HRMS ESI-TOF [M]+ m/z calcd. for C34H44N2O5: 560,3238, found: 560,3250.
Diethyl-4-(4-((5-(4-benzylpiperidin-1-yl)pentyl)oxy)phenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (3g). 3g was prepared according to general procedure C starting from 4-((5-(4-benzylpiperidin-1-yl)pentyl)oxy)-benzaldehyde 2d (1 equiv., 1.88 mmol, 687.00 mg), ethyl acetoacetate (3.5 equiv., 7.52 mmol, 1.03 mL) and ammonium carbonate (2.5 Equiv., 5.64 mmol, 542.14 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 97/3 + 1% Et3N to afford 227.00 mg (21%) of 3g as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 7.8 Hz, 1H), 7.24 (s, 1H), 7.15 (dt, J = 8.1, 6.6 Hz, 5H), 6.74–6.67 (m, 2H), 5.88 (bs, 1H), 4.91 (s, 1H), 4.14–4.00 (m, 4H), 3.88 (t, J = 6.4 Hz, 2H), 2.95 (d, J = 11.3 Hz, 2H), 2.53 (d, J = 7.0 Hz, 2H), 2.38–2.32 (m, 2H), 2.31 (s, 6H), 1.91 (t, J = 11.2 Hz, 2H), 1.79–1.70 (m, 2H), 1.64 (d, J = 13.0 Hz, 2H), 1.60–1.48 (m, 3H), 1.47–1.34 (m, 4H), 1.21 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.83, 157.42, 143.82, 140.63, 140.32, 129.17, 128.98, 128.23, 125.87, 113.79, 104.30, 77.48, 77.16, 76.84, 67.66, 59.72, 58.90, 53.94, 43.16, 38.78, 37.88, 31.88, 29.27, 26.56, 24.25, 19.54, 14.35. HRMS ESI-TOF [M]+ m/z calcd. for C36H48N2O5: 588,3535, found: 588,3563.
3,5-Dimethyl-4-(4-{[6-(diethylamino)hexyl]oxy}phenyl)-2,6–dimethyl-1,4–dihydropyridine-3,5- dicarboxylate (3l). 3l was prepared according to general procedure C starting from 4-((6-(diethylamino)hexyl)oxy)benzaldehyde 2f (1 equiv., 0.55 mmol, 160.7 mg), methyl acetoacetate (3.5 equiv., 1.93 mmol, 0.279 mL) and ammonium carbonate (2.5 equiv., 1.38 mmol, 132.60 mg) at 75 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using AE/MeOH 96/4 + 1% Et3N to afford 25.10 mg (3%) of 3l as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.19–7.11 (m, 2H), 6.72 (d, J = 8.6 Hz, 2H), 5.79 (s, 1H), 4.92 (s, 1H), 3.88 (t, J = 6.4 Hz, 2H), 2.69 (dd, J = 14.0, 6.9 Hz, 4H), 2.63–2.50 (m, 2H), 2.33 (s, 6H), 1.75 (dd, J = 14.0, 7.2 Hz, 2H), 1.62–1.53 (m, 2H), 1.50–1.41 (m, 2H), 1.36 (dd, J = 14.9, 7.8 Hz, 2H), 1.12 (t, J = 7.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.24, 157.59, 144.04, 139.94, 128.73, 114.05, 104.28, 67.67, 51.11, 46.84, 38.54, 29.35, 27.28, 26.06, 19.76. HRMS ESI-TOF [M]+ m/z calcd. for C27H40N2O5: 472,2927, found: 472,2937.
3,5-Diethyl-4-(4-{[6-(diethylamino)hexyl]oxy}phenyl)–2,6–dimethyl-1,4–dihydropyridine-3,5-dicarboxylate (3k). 3k was prepared according to general procedure C starting from 4-((6-(diethylamino)hexyl)oxy)benzaldehyde 2f (1 equiv., 0.90 mmol, 249 mg), ethyl acetoacetate (3 equiv., 2.69 mmol, 0.263 mL) and ammonium carbonate (2 equiv., 1.80 mmol, 172.58 mg) at 70 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using AE/MeOH 98/2 + 1% Et3N to afford 129.2 mg (25%) of 3k as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.20–7.10 (m, 2H), 6.77–6.67 (m, 2H), 5.87 (s, 1H), 4.91 (s, 1H), 4.13–3.98 (m, 4H), 3.88 (t, J = 6.4 Hz, 2H), 2.67 (q, J = 7.2 Hz, 4H), 2.55 (dd, J = 9.3, 6.7 Hz, 2H), 2.31 (s, 6H), 1.78–1.68 (m, 2H), 1.50 (dddd, J = 21.2, 15.5, 10.0, 7.3 Hz, 5H), 1.38–1.29 (m, 2H), 1.21 (t, J = 7.1 Hz, 6H), 1.08 (t, J = 7.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.81, 157.55, 143.58, 140.28, 129.08, 113.89, 104.61, 67.78, 59.83, 46.93, 38.88, 29.43, 27.46, 26.15, 19.79, 14.43. HRMS ESI-TOF [M]+ m/z calcd. for C29H44N2O5: 500,3236, found: 500,3250.
3,5-Dimethyl-2,6-dimethyl-4-(4-{[6-(morpholin-4-yl)hexyl]oxy}phenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3j). 3j was prepared according to general procedure C starting from 4-((6-morpholinohexyl)oxy)benzaldehyde 2e (1 equiv., 0.69 mmol, 200 mg), methyl acetoacetate (3.5 equiv., 2.40 mmol, 0.259 mL) and ammonium carbonate (2.5 equiv., 1.72 mmol, 165.03 mg) at 60 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 95/5 + 1% NH3 to afford 53 mg (7%) of 3j as birght orange oil. 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 8.7 Hz, 2H), 6.73 (d, J = 8.7 Hz, 2H), 5.63 (s, J = 14.2 Hz, 1H), 4.93 (s, J = 4.1 Hz, 1H), 3.89 (t, J = 6.4 Hz, 2H), 3.80 (s, 4H), 3.64 (d, J = 2.4 Hz, 6H), 2.66–2.51 (m, J = 9.6, 7.8 Hz, 4H), 2.47 (bs, J = 15.5 Hz, 2H), 2.33 (s, 6H), 1.80–1.68 (m, 2H), 1.60 (bs, 2H), 1.52–1.42 (m, 2H), 1.41–1.31 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 168.23, 157.61, 143.99, 139.92, 128.91, 128.73, 114.06, 113.97, 104.31, 67.70, 58.99, 53.52, 51.12, 38.55, 29.35, 27.23, 26.09, 19.77. HRMS ESI-TOF [M]+ m/z calcd. for C27H38N2O6: 486,2711, found: 486,2730.
3,5-Ethyl-2,6-dimethyl-4-(4-{[6-(morpholin-4-yl)hexyl]oxy}phenyl)-1,4-dihydropyridine-3,5-dicarboxylate (3i). 3i was prepared according to general procedure C starting from 4-((6-morpholinohexyl)oxy)benzaldehyde 2e (1 equiv., 0.412 mmol, 120 mg), ethyl acetoacetate (2.5 equiv., 1.03 mmol, 0.089 mL) and ammonium carbonate (1.5 equiv., 0.62 mmol, 59.38 mg) at 70 °C over 15h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 96/4 + 1% NH3 to afford 37.00 mg (19%) of 3i as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.21–7.12 (m, 2H), 6.77–6.69 (m, 2H), 5.54 (s, 1H), 4.92 (s, 1H), 4.16–4.02 (m, 4H), 3.89 (t, J = 6.4 Hz, 2H), 3.76 (bs, 4H), 2.49 (bs, 4H), 2.38 (bs, 2H), 2.32 (s, 6H), 1.80–1.68 (m, 2H), 1.50–1.41 (m, 3H), 1.40–1.31 (m, 3H), 1.22 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.82, 157.55, 143.57, 140.31, 129.09, 113.89, 104.61, 67.77, 59.83, 59.11, 53.69, 38.88, 29.40, 27.32, 26.14, 19.78, 14.42. HRMS ESI-TOF [M]+ m/z calcd. for C29H42N2O6: 514,3028, found: 514,3043.
3,5-Dimethyl-2,6-dimethyl-4-(4-{[6-(4-methylpiperazin-1-yl)hexyl]oxy}phenyl)-1,4-dihydro-pyridine-3,5-dicarboxylate (3n). 3n was prepared according to general procedure C starting from 4-((6-(4-methylpiperazin-1-yl)hexyl)oxy)benzaldehyde 2g (1 equiv., 0.82 mmol, 250 mg), methyl acetoacetate (4 equiv., 3.29 mmol, 0.355 mL) and ammonium carbonate (3 equiv., 2.46 mmol, 236.38 mg) at 70 °C over 15h. After beingworked up, the crude was finally purified by flash column chromatography using EA/MeOH 95/5 + 1% Et3N to afford 61.6 mg of 3n as white powder (15%). 1H NMR (400 MHz, CDCl3) δ 7.15 (d, J = 8.6 Hz, 2H), 6.72 (d, J = 8.6 Hz, 2H), 5.66 (s, 1H), 4.93 (s, 1H), 3.88 (t, J = 6.4 Hz, 2H), 3.64 (sJ, 6H), 2.54 (bs, 8H), 2.43–2.36 (m, 2H), 2.33 (s, 9H), 1.80–1.66 (m, 2H), 1.59–1.50 (m, 2H), 1.48–1.39 (m, 2H), 1.35 (dd, J = 14.0, 7.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 168.23, 157.63, 144.01, 139.88, 128.71, 114.06, 104.29, 67.76, 58.60, 54.86, 52.95, 51.11, 45.92, 38.52, 29.83, 29.39, 27.40, 26.70, 26.14, 19.76. Anal. Calcd. for C28H41N3O5: C, 64.48; H, 8.27; N, 8.41; found: C, 65.48; H, 8.31; N, 8.53. HRMS ESI-TOF [M]+ m/z calcd. for C28H41N3O5: 499,3039, found: 499,3046.
3,5-Diethyl-2,6-dimethyl-4-(4-{[6-(4-methylpiperazin-1-yl)hexyl]oxy}phenyl)-1,4-dihydro-pyridine-3,5-dicarboxylate (3m). 3m was prepared according to general procedure C starting from 4-((6-(4-methylpiperazin-1-yl)hexyl) oxy)benzaldehyde 2g (1 equiv., 0.70 mmol, 212 mg), ethyl acetoacetate (3 equiv., 2.09 mmol, 0.204 mL) and ammonium carbonate (2 equiv., 1.39 mmol, 133.95 mg) at 70 °C over 15h. After was worked up, the crude was finally purified by flash column chromatography using EA/MeOH 97/3 + 1% Et3N to afford 144.4 mg (20%) of 3m as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 8.7 Hz, 2H), 6.71 (d, J = 8.7 Hz, 2H), 5.62 (s, 1H), 4.91 (s, 1H), 4.14–3.99 (m, 4H), 3.88 (t, J = 6.4 Hz, 2H), 2.59 (bs, 8H), 2.49–2.39 (m, 2H), 2.36 (s, 3H), 2.32 (s, 6H), 1.78–1.68 (m, 2H), 1.60–1.50 (m, 2H), 1.50–1.41 (m, 2H), 1.40–1.31 (m, 2H), 1.22 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.82, 157.53, 143.63, 140.31, 129.07, 113.88, 104.56, 67.74, 59.81, 58.41, 54.51, 52.60, 45.69, 38.86, 29.37, 27.31, 26.09, 19.76, 14.42. HRMS ESI-TOF [M]+ m/z calcd. for C30H45N3O5: 527,3341, found: 527,3359.
3,5-Dimethyl-4-(4-{[6-(4-benzylpiperidin-1-yl)hexyl]oxy}phenyl)-2,6-dimethyl-1,4-dihydro-pyridine-3,5-dicarboxylate (3p). 3p was prepared according to general procedure C starting from 4-((6-(4-benzylpiperidin-1-yl)hexyl)oxy)benzaldehyde 2h (1 equiv., 0.79 mmol, 300 mg), methyl acetoacetate (3 equiv., 2.37 mmol, 0.256 mL) and ammonium carbonate (2 equiv., 1.58 mmol, 152.02 mg) at 70 °C over 24h. After being worked up, the crude was finally purified by flash column chromatography using DCM/MeOH 92/8 + 1% NH3 to afford 29.91 mg (7%) of 3p as light yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.30–7.26 (m, 1H), 7.25 (s, 1H), 7.22–7.11 (m, 5H), 6.79–6.66 (m, 2H), 5.72 (d, J = 13.5 Hz, 1H), 4.93 (s, 1H), 3.87 (t, J = 6.4 Hz, 2H), 3.64 (s, 6H), 3.03 (bs, 2H), 2.55 (d, J = 6.3 Hz, 2H), 2.42 (bs, 2H), 2.33 (s, 6H), 2.02 (bs, 2H), 1.77–1.63 (m, 5H), 1.63–1.52 (m, 4H), 1.49–1.40 (m, 3H), 1.34 (dd, J = 13.7, 6.2 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 168.25, 157.47, 144.12, 140.06, 129.14, 128.93, 128.75, 128.66, 126.53, 114.03, 113.94, 104.21, 67.39, 59.84, 57.72, 53.12, 51.10, 42.12, 38.54, 36.84, 29.12, 29.03, 26.72, 25.71, 23.63, 19.73. HRMS ESI-TOF [M]+ m/z calcd. for C35H46N2O5: 574,3391, found: 574,3407.
3,5-Dimethyl-4-(4-{[6-(4-benzylpiperidin-1-yl)hexyl]oxy}phenyl)-2,6-dimethyl-1,4-dihydro-pyridine-3,5-dicarboxylate (3o). 3o was prepared according to general procedure C starting from 4-((6-(4-benzylpiperidin-1-yl)hexyl)oxy)benzaldehyde 2h (1 equiv., 0.79 mmol, 300 mg), ethyl acetoacetate (2.5 equiv., 1.97 mmol, 0.251 mL) and ammonium carbonate (1.5 equiv., 1.19 mmol, 114.01 mg) at 75 °C over 20h. The crudewas dissolved in MeOH and solvent was evaporated in vacuo. The residue was dissolved in DCM and washed with water and brine. The organic layer was then dried over Na2SO4 and finally purified by flash column chromatography using DCM/MeOH 96/4 + 1% NH3 to afford 144.4 mg (31%) of 3o as dark yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.30–7.26 (m, 1H), 7.25 (s, 1H), 7.21–7.10 (m, 5H), 6.76–6.64 (m, 2H), 5.68 (s, 1H), 4.91 (s, 1H), 4.14–4.03 (m, 4H), 3.88 (t, J = 6.4 Hz, 2H), 2.95 (d, J = 11.2 Hz, 2H), 2.53 (d, J = 7.0 Hz, 2H), 2.32 (s, 7H), 1.90 (t, J = 11.2 Hz, 2H), 1.78–1.69 (m, 2H), 1.64 (d, J = 12.8 Hz, 2H), 1.58–1.49 (m, 4H), 1.47–1.29 (m, 7H), 1.22 (t, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.82, 157.55, 143.66, 140.77, 140.27, 129.24, 129.05, 128.28, 125.91, 113.87, 104.52, 67.81, 59.79, 59.11, 43.27, 38.84, 38.01, 32.05, 29.41, 27.57, 26.89, 26.15, 19.70, 14.40. Anal. Calcd. for C37H50N2O5: C, 73.72; H, 8.36; N, 4.65; found: C, 71.09; H, 8.47; N, 4.58. HRMS ESI-TOF [M]+ m/z calcd. for C37H50N2O5: 602,3705, found: 602,3720.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}