DAP Kinase-Related Apoptosis-Inducing Protein Kinase 2 (DRAK2) Is a Key Regulator and Molecular Marker in Chronic Lymphocytic Leukemia

 , ,
, ,
Abstract
:1. Introduction
2. Results
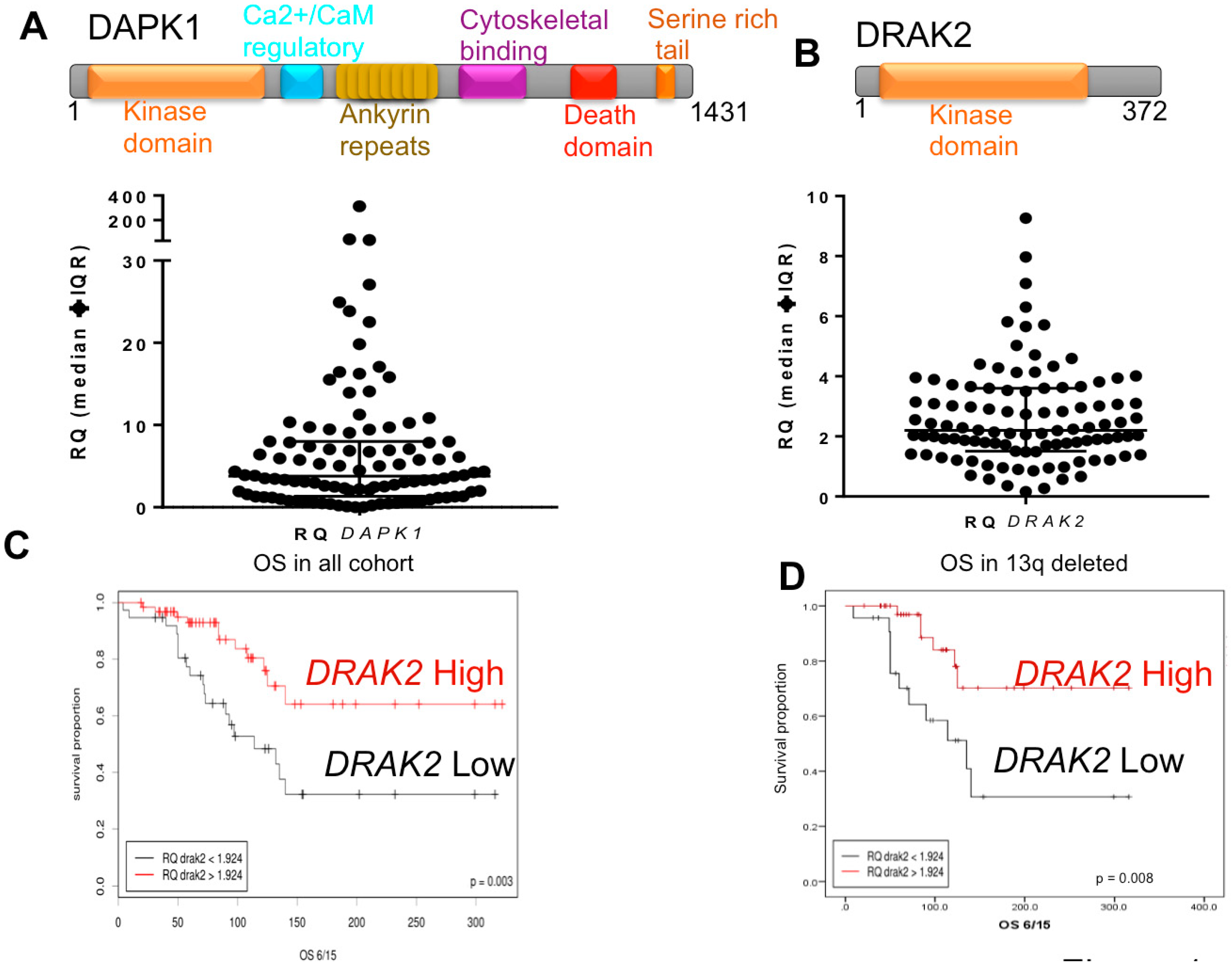
2.1. Low DRAK2 Expression Level Is Associated with Shorter Overall Survival in CLL Patients
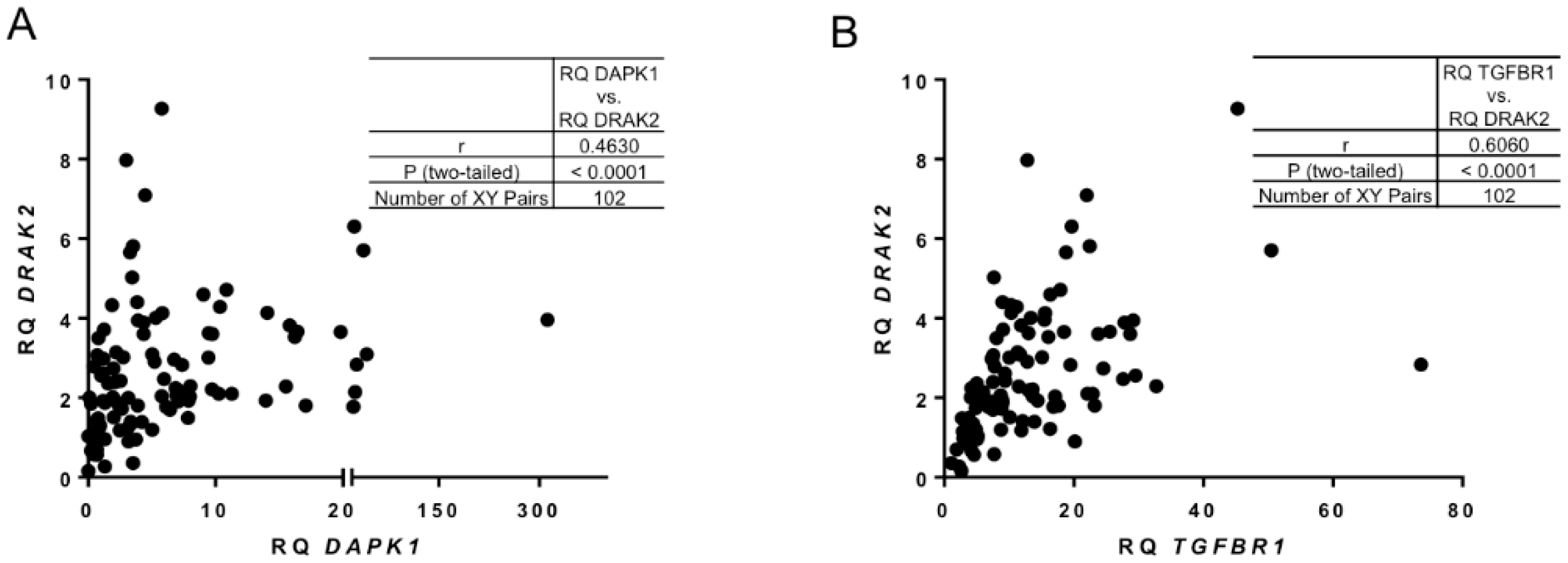
2.2. DRAK2 Expression Level Positively Correlates with DAPK1, and TGFBR1
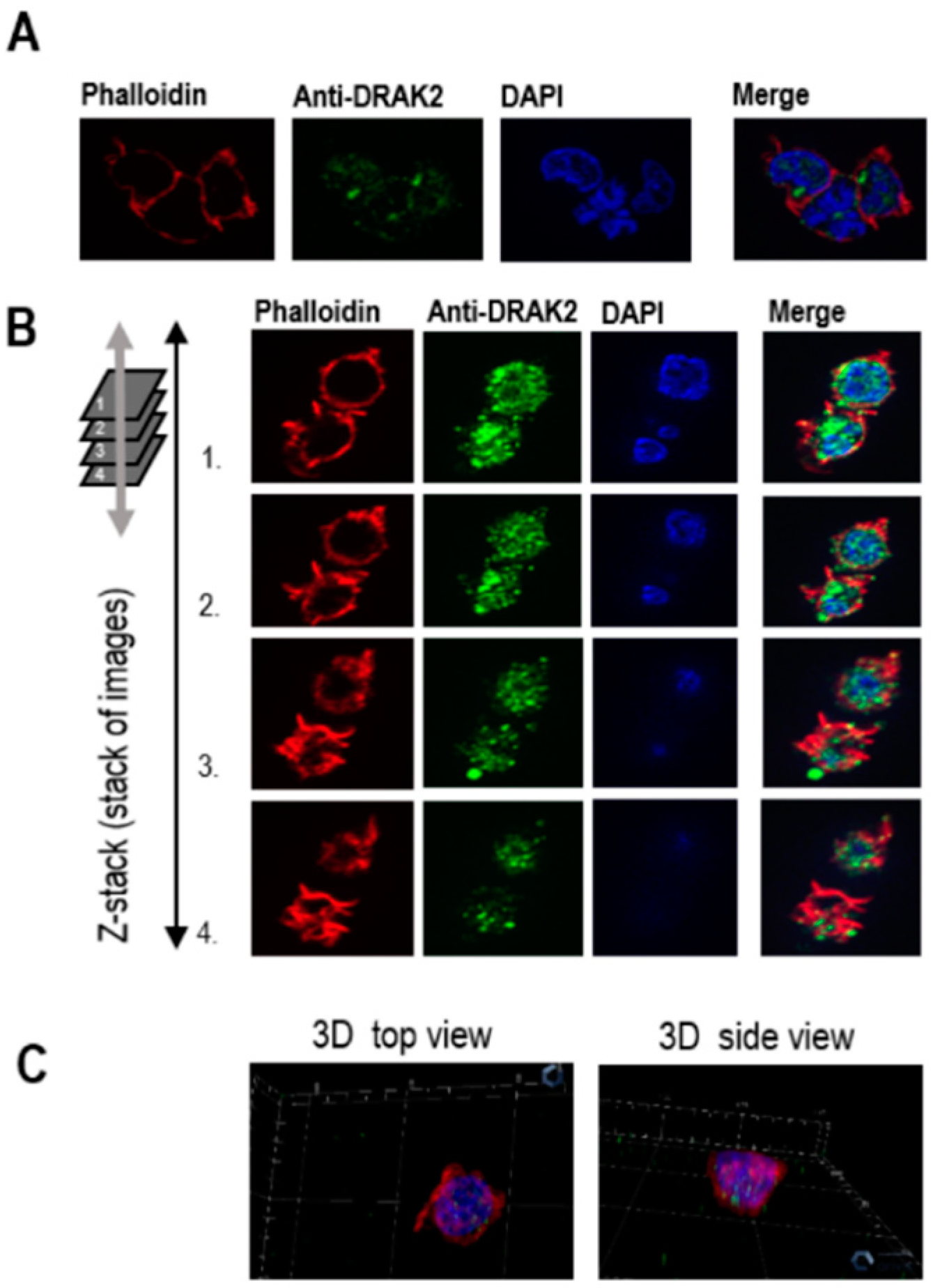
2.3. Subcellular Localization of DRAK2
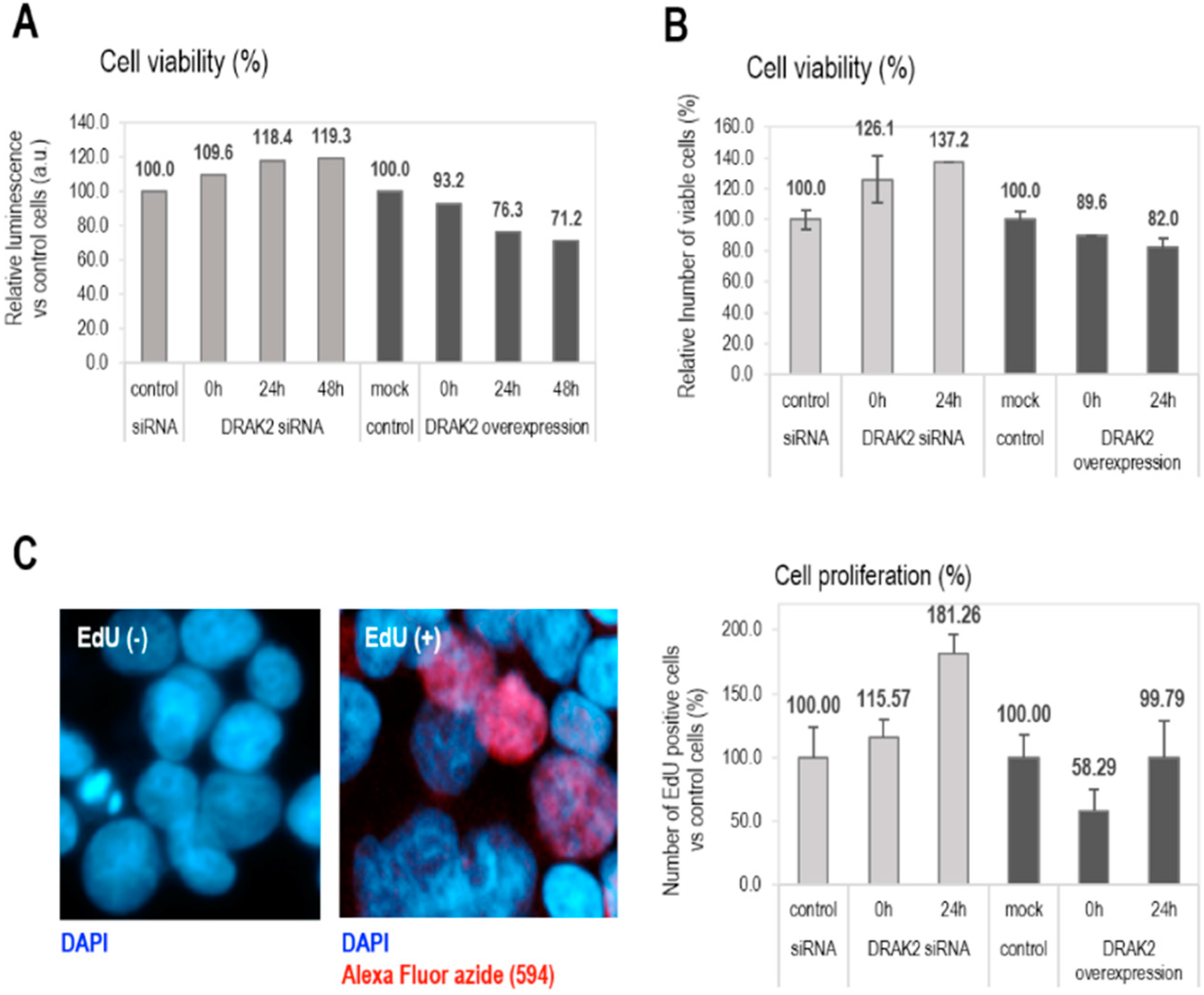
2.4. DRAK2 Impacts on Cell Viability and Proliferation
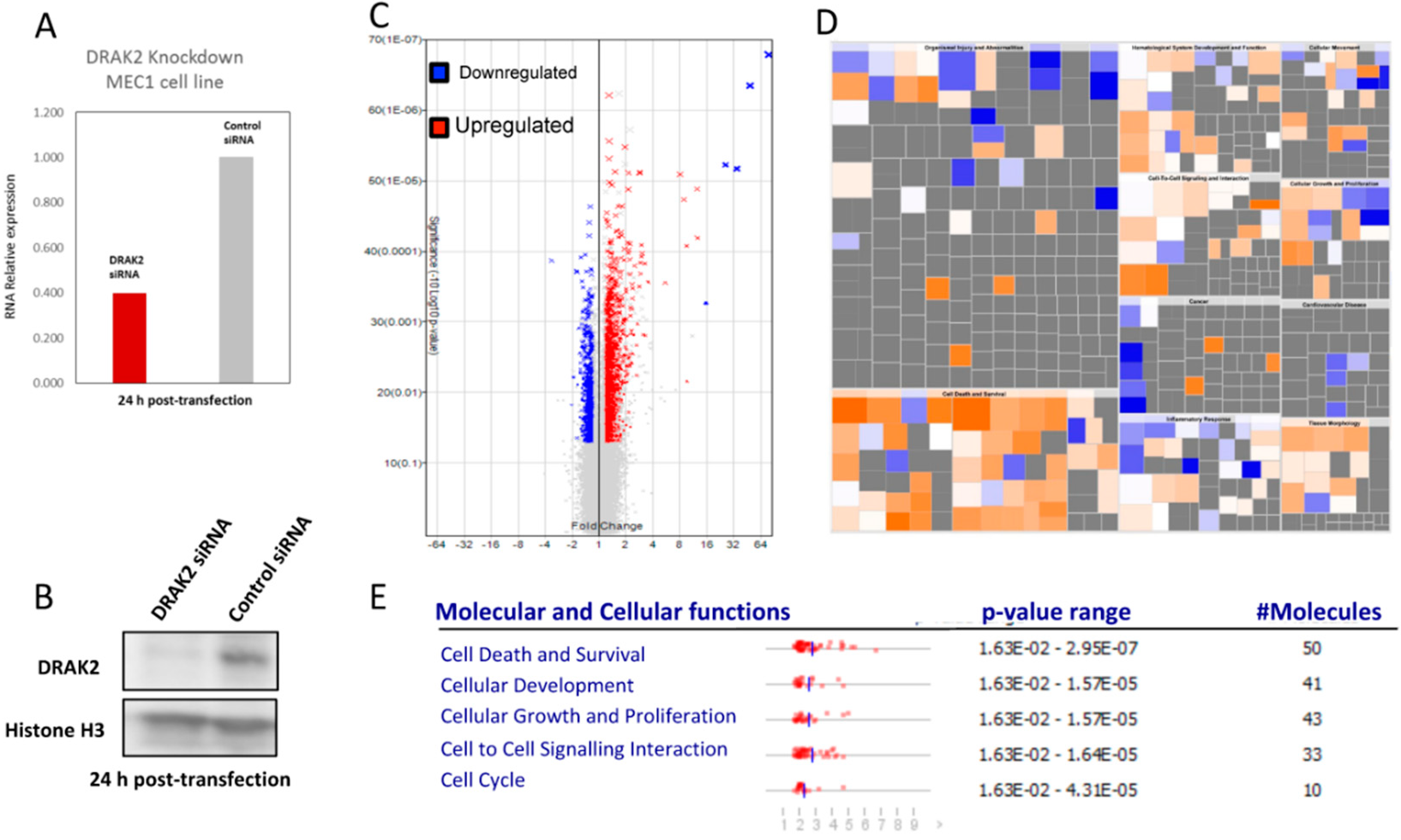
2.5. Impact of DRAK2 on Gene Expression in MEC-1
3. Discussion
4. Materials and Methods
4.1. Patients and Cell Isolation
4.2. Experimental Model and Nucleofection
4.3. RNA Extraction and qPCR
4.4. Expression and Data Analysis
4.5. Cell Viability Assay
4.6. EdU Cell Proliferation Assay
4.7. Trypan Blue Assay
4.8. Apoptosis Induction
4.9. Gene Expression Level Analyses: Microarrays and qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Hermelink, H.K.; Montserrat, E.; Catovsky, D.; Campo, E.; Harris, N.L.; Stein, H. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. In World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; Müller-Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J.W., Eds.; IARC: Lyon, France, 2008; pp. 180–182. [Google Scholar]
- Hallek, M. Chronic lymphocytic leukemia: 2015 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2015, 90, 446–460. [Google Scholar] [CrossRef]
- Zenz, T.; Mertens, D.; Kuppers, R.; Dohner, H.; Stilgenbauer, S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2010, 10, 37–50. [Google Scholar] [CrossRef]
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef]
- Baliakas, P.; Iskas, M.; Gardiner, A.; Davis, Z.; Plevova, K.; Nguyen-Khac, F.; Malcikova, J.; Anagnostopoulos, A.; Glide, S.; Mould, S.; et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: A systematic reappraisal of classic cytogenetic data. Am. J. Hematol. 2014, 89, 249–255. [Google Scholar] [CrossRef]
- Krober, A.; Seiler, T.; Benner, A.; Bullinger, L.; Bruckle, E.; Lichter, P.; Dohner, H.; Stilgenbauer, S. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood 2002, 100, 1410–1416. [Google Scholar] [CrossRef]
- Dohner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Krober, A.; Bullinger, L.; Dohner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.A.; Shanafelt, T.D. Prognostic factors and risk stratification in chronic lymphocytic leukemia. Semin. Oncol. 2016, 43, 233–240. [Google Scholar] [CrossRef]
- Chen, L.; Widhopf, G.; Huynh, L.; Rassenti, L.; Rai, K.R.; Weiss, A.; Kipps, T.J. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2002, 100, 4609–4614. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef]
- Rossi, D.; Khiabanian, H.; Spina, V.; Ciardullo, C.; Bruscaggin, A.; Fama, R.; Rasi, S.; Monti, S.; Deambrogi, C.; De Paoli, L.; et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014, 123, 2139–2147. [Google Scholar] [CrossRef] [Green Version]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Investig. 2005, 115, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, S.; Andersen, J.; Akar, S.; Zapata, J.M.; Takayama, S.; Krajewski, S.; Wang, H.G.; Zhang, X.; Bullrich, F.; Croce, C.M.; et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: Correlations with In vitro and In vivo chemoresponses. Blood 1998, 91, 3379–3389. [Google Scholar] [CrossRef] [Green Version]
- Loeder, S.; Zenz, T.; Schnaiter, A.; Mertens, D.; Winkler, D.; Dohner, H.; Debatin, K.M.; Stilgenbauer, S.; Fulda, S. A novel paradigm to trigger apoptosis in chronic lymphocytic leukemia. Cancer Res. 2009, 69, 8977–8986. [Google Scholar] [CrossRef] [Green Version]
- Billard, C. Apoptosis inducers in chronic lymphocytic leukemia. Oncotarget 2014, 5, 309–325. [Google Scholar] [CrossRef]
- Bialik, S.; Kimchi, A. The death-associated protein kinases: Structure, function, and beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, R.; Bialik, S.; Kimchi, A. The DAPK family: A structure-function analysis. Apoptosis 2014, 19, 286–297. [Google Scholar] [CrossRef]
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.Q.; Kwong, Y.L.; Wong, K.F.; Kho, C.S.; Jin, D.Y.; Tse, E.; Rosen, A.; Chim, C.S. Epigenetic inactivation of mir-34b/c in addition to mir-34a and DAPK1 in chronic lymphocytic leukemia. J. Transl. Med. 2014, 12, 52. [Google Scholar] [CrossRef] [Green Version]
- Sanjo, H.; Kawai, T.; Akira, S. DRAKs, novel serine/threonine kinases related to death-associated protein kinase that trigger apoptosis. J. Biol. Chem. 1998, 273, 29066–29071. [Google Scholar] [CrossRef] [Green Version]
- McGargill, M.A.; Wen, B.G.; Walsh, C.M.; Hedrick, S.M. A deficiency in Drak2 results in a T cell hypersensitivity and an unexpected resistance to autoimmunity. Immunity 2004, 21, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.H.; Leverrier, S.; Srikanth, S.; Gwack, Y.; Cahalan, M.D.; Walsh, C.M. Protein kinase D orchestrates the activation of DRAK2 in response to TCR-induced Ca2+ influx and mitochondrial reactive oxygen generation. J. Immunol. 2011, 186, 940–950. [Google Scholar] [CrossRef] [Green Version]
- Al-Qahtani, A.; Xu, Z.; Zan, H.; Walsh, C.M.; Casali, P. A role for DRAK2 in the germinal center reaction and the antibody response. Autoimmunity 2008, 41, 341–352. [Google Scholar] [CrossRef]
- Ye, P.; Zhao, L.; Gonda, T.J. The MYB oncogene can suppress apoptosis in acute myeloid leukemia cells by transcriptional repression of DRAK2 expression. Leuk Res. 2013, 37, 595–601. [Google Scholar] [CrossRef]
- Yang, K.M.; Kim, W.; Bae, E.; Gim, J.; Weist, B.M.; Jung, Y.; Hyun, J.S.; Hernandez, J.B.; Leem, S.H.; Park, T.; et al. DRAK2 participates in a negative feedback loop to control TGF-beta/Smads signaling by binding to type I TGF-beta receptor. Cell Rep. 2012, 2, 1286–1299. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, H.; Nakamura, N.; Kanazawa, H. Nuclear localization of the serine/threonine kinase DRAK2 is involved in UV-induced apoptosis. Biol. Pharm. Bull. 2006, 29, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.L.; McGargill, M.A. Drak2 Does Not Regulate TGF-beta Signaling in T Cells. PLoS ONE 2015, 10, e0123650. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Luo, H.; Wu, J. Drak2 overexpression results in increased beta-cell apoptosis after free fatty acid stimulation. J. Cell Biochem. 2008, 105, 1073–1080. [Google Scholar] [CrossRef]
- Cramer, P.; Langerbeins, P.; Eichhorst, B.; Hallek, M. Advances in first-line treatment of chronic lymphocytic leukemia: Current recommendations on management and first-line treatment by the German CLL Study Group (GCLLSG). Eur. J. Haematol. 2016, 96, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Gaidano, G.; Rossi, D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 2017, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Klein, U.; Lia, M.; Crespo, M.; Siegel, R.; Shen, Q.; Mo, T.; Ambesi-Impiombato, A.; Califano, A.; Migliazza, A.; Bhagat, G.; et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010, 17, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, S.J.; Hernandez, J.B.; Gatzka, M.; Walsh, C.M. Enhanced T cell apoptosis within Drak2-deficient mice promotes resistance to autoimmunity. J. Immunol. 2008, 181, 7606–7616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinhardt, G.; Wendtner, C.M.; Hallek, M. Molecular pathogenesis of chronic lymphocytic leukemia: Factors and signaling pathways regulating cell growth and survival. J. Mol. Med. 1999, 77, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Guerra, M.; Colomer, D. NF-kappaB as a therapeutic target in chronic lymphocytic leukemia. Expert. Opin. Targets 2010, 14, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Petlickovski, A.; Laurenti, L.; Li, X.; Marietti, S.; Chiusolo, P.; Sica, S.; Leone, G.; Efremov, D.G. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood 2005, 105, 4820–4827. [Google Scholar] [CrossRef] [Green Version]
- Limon, J.J.; Fruman, D.A. Akt and mTOR in B Cell Activation and Differentiation. Front. Immunol. 2012, 3, 228. [Google Scholar] [CrossRef] [Green Version]
- Krysov, S.; Steele, A.J.; Coelho, V.; Linley, A.; Sanchez Hidalgo, M.; Carter, M.; Potter, K.N.; Kennedy, B.; Duncombe, A.S.; Ashton-Key, M.; et al. Stimulation of surface IgM of chronic lymphocytic leukemia cells induces an unfolded protein response dependent on BTK and SYK. Blood 2014, 124, 3101–3109. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | N | Percentage |
|---|---|---|
| Age ≥ 65 | 65 | 63.7 |
| Sex: Male | 71 | 69.6 |
| Binet stage n = 90 | ||
| A | 49 | 54.4 |
| B | 16 | 17.8 |
| C | 25 | 27.8 |
| IGHV status n = 63 | ||
| Mutated | 38 | 60.3 |
| Unmutated | 25 | 39.7 |
| TP53 status n = 100 | ||
| Mutated | 12 | 12 |
| Unmutated | 88 | 88 |
| Cytogenetics n = 101 | ||
| 13q | 62 | 61.4 |
| 11q | 20 | 19.8 |
| 12+ | 6 | 5.9 |
| 17p | 8 | 7.9 |
| Normal karyotype | 24 | 23.8 |
| ZAP70 status n = 19 | ||
| >20% | 9 | 47.4 |
| <20% | 10 | 52.6 |
| CD38 status n = 36 | ||
| >20% | 13 | 36.1 |
| <20% | 23 | 63.9 |
| Treatment status n = 102 | ||
| Treated | 35 | 34.3 |
| Untreated | 67 | 65.7 |
| Covariate | HR | STD Err | p-Value | Conf. Interval | HR | STD Err | p-Value | Conf. Interval | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Univariate Analysis | Multivariate Analysis | |||||||||
| Age > 70 years | 1.05 | 0.02 | 0.014 | 1.01 | 1.091 | 1.094 | 0.026 | 0.001 | 1.04 | 1.151 |
| 17p status | 3.681 | 0.542 | 0.016 | 1.273 | 10.65 | 3.188 | 0.619 | 0.06 | 0.947 | 10.729 |
| IGHV status | 3.093 | 0.418 | 0.007 | 1.363 | 7.02 | 5.514 | 0.473 | 0.0003 | 2.183 | 13.925 |
| DRAK2 expression | 0.343 | 0.379 | 0.005 | 0.163 | 0.722 | 0.428 | 0.427 | 0.04 | 0.185 | 0.987 |
| 13q Status | 11q Status | +12 Status | 17p Status | Age | Gender | ZAP70 | CD38 | IGHV | TP53 Mut | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RQ DRAK2 | Pearson r | 0.014 | −0.178 | −0.064 | −0.075 | −0.033 | −0.068 | −0.267 | −0.129 | −0.111 | −0.028 |
| p | 0.892 | 0.075 | 0.524 | 0.456 | 0.741 | 0.495 | 0.270 | 0.453 | 0.387 | 0.783 | |
| N | 101 | 101 | 101 | 101 | 102 | 102 | 19 | 36 | 63 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szoltysek, K.; Ciardullo, C.; Zhou, P.; Walaszczyk, A.; Willmore, E.; Rand, V.; Marshall, S.; Hall, A.; J. Harrison, C.; Eswaran, J.; et al. DAP Kinase-Related Apoptosis-Inducing Protein Kinase 2 (DRAK2) Is a Key Regulator and Molecular Marker in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2020, 21, 7663. https://doi.org/10.3390/ijms21207663
Szoltysek K, Ciardullo C, Zhou P, Walaszczyk A, Willmore E, Rand V, Marshall S, Hall A, J. Harrison C, Eswaran J, et al. DAP Kinase-Related Apoptosis-Inducing Protein Kinase 2 (DRAK2) Is a Key Regulator and Molecular Marker in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences. 2020; 21(20):7663. https://doi.org/10.3390/ijms21207663
Chicago/Turabian StyleSzoltysek, Katarzyna, Carmela Ciardullo, Peixun Zhou, Anna Walaszczyk, Elaine Willmore, Vikki Rand, Scott Marshall, Andy Hall, Christine J. Harrison, Jeyanthy Eswaran, and et al. 2020. "DAP Kinase-Related Apoptosis-Inducing Protein Kinase 2 (DRAK2) Is a Key Regulator and Molecular Marker in Chronic Lymphocytic Leukemia" International Journal of Molecular Sciences 21, no. 20: 7663. https://doi.org/10.3390/ijms21207663
APA StyleSzoltysek, K., Ciardullo, C., Zhou, P., Walaszczyk, A., Willmore, E., Rand, V., Marshall, S., Hall, A., J. Harrison, C., Eswaran, J., & Soundararajan, M. (2020). DAP Kinase-Related Apoptosis-Inducing Protein Kinase 2 (DRAK2) Is a Key Regulator and Molecular Marker in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences, 21(20), 7663. https://doi.org/10.3390/ijms21207663