Head and Neck Paragangliomas—A Genetic Overview

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Classification Based on the Genetic and Molecular Background
2.2. Genetic Syndromes
2.3. Epigenetic Patterns in HNPGL
2.4. Next-Generation Sequencing (NGS)
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Koopman, K.; Gaal, J.; De Krijger, R.R. Pheochromocytomas and Paragangliomas: New Developments with Regard to Classification, Genetics, and Cell of Origin. Cancers 2019, 11, 1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asban, A.; Kluijfhout, W.P.; Drake, F.T.; Beninato, T.; Wang, E.; Chomsky-Higgins, K.; Shen, W.T.; Gosnell, J.E.; Suh, I.; Duh, Q.-Y. Trends of genetic screening in patients with pheochromocytoma and paraganglioma: 15-year experience in a high-volume tertiary referral center. J. Surg. Oncol. 2018, 117, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L. Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment. Hematol. Clin. 2016, 30, 135–150. [Google Scholar] [CrossRef]
- Burnichon, N.; Rohmer, V.; Amar, L.; Herman, P.; Leboulleux, S.; Darrouzet, V.; Niccoli, P.; Gaillard, D.; Chabrier, G.; Chabolle, F.; et al. The Succinate Dehydrogenase Genetic Testing in a Large Prospective Series of Patients with Paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 2817–2827. [Google Scholar] [CrossRef]
- Lenders, J.W.M.; Eisenhofer, G.; Mannelli, M.; Pacak, K. Phaeochromocytoma. Lancet 2005, 366, 665–675. [Google Scholar] [CrossRef]
- Boedeker, C.C. Paragangliomas and paraganglioma syndromes. GMS Curr. Top. Otorhinolaryngol. Head Neck Surg. 2011, 10. [Google Scholar] [CrossRef]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Yang, P.; Liu, Y.; Wang, S. Paraganglioma of the spermatic cord: Report of one case and review of literature. Int. J. Clin. Exp. Pathol. 2020, 13, 1779–1786. [Google Scholar]
- Dorobisz, K.; Dorobisz, T.; Temporale, H.; Zatoński, T.; Kubacka, M.; Chabowski, M.; Dorobisz, A.; Krecicki, T.; Janczak, D. Diagnostic and Therapeutic Difficulties in Carotid Body Paragangliomas, Based on Clinical Experience and a Review of the Literature. Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2016, 25, 1173–1177. [Google Scholar] [CrossRef] [Green Version]
- Else, T.; Greenberg, S.; Fishbein, L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Boedeker, C.C.; Hensen, E.F.; Neumann, H.P.; Maier, W.; Van Nederveen, F.H.; Suárez, C.; Kunst, H.P.; Rodrigo, J.P.; Takes, R.P.; Pellitteri, P.K.; et al. Genetics of hereditary head and neck paragangliomas. Head Neck 2013, 36, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Katabathina, V.S.; Rajebi, H.; Chen, M.M.; Restrepo, C.S.; Salman, U.; Vikram, R.; Menias, C.O.; Prasad, S.R. Genetics and imaging of pheochromocytomas and paragangliomas: Current update. Abdom. Radiol. N. Y. 2020, 45, 928–944. [Google Scholar] [CrossRef] [PubMed]
- Cavenagh, T.; Patel, J.; Nakhla, N.; Elstob, A.; Ingram, M.; Barber, B.; Snape, K.; Bano, G.; Vlahos, I. Succinate dehydrogenase mutations: Paraganglioma imaging and at-risk population screening. Clin. Radiol. 2019, 74, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Turchini, J.; Cheung, V.K.Y.; Tischler, A.S.; Krijger, R.R.D.; Gill, A.J. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology 2017, 72, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, I.; Young, W.F.; Kasperbauer, J.L.; Polonis, K.; Harmsen, W.S.; Colglazier, J.J.; DeMartino, R.R.; Oderich, G.S.; Kalra, M.; Bower, T.C. Tumor-specific prognosis of mutation-positive patients with head and neck paragangliomas. J. Vasc. Surg. 2020, 71, 1602–1612.e2. [Google Scholar] [CrossRef]
- Chase, W.H. Familial and bilateral tumours of the carotid body. J. Pathol. Bacteriol. 1933, 36, 1–12. [Google Scholar] [CrossRef]
- Martin, T.P.; Irving, R.M.; Maher, E.R. The genetics of paragangliomas: A review. Clin. Otolaryngol. 2007, 32, 7–11. [Google Scholar] [CrossRef]
- Burnichon, N.; Abermil, N.; Buffet, A.; Favier, J.; Gimenez-Roqueplo, A.-P. The genetics of paragangliomas. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2012, 129, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Calsina, B.; Currás-Freixes, M.; Buffet, A.; Pons, T.; Contreras, L.; Letón, R.; Méndez, I.C.; Remacha, L.; Calatayud, M.; Obispo, B.; et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 1652–1662. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhu, W.; Li, X.; Xue, L.; Wang, Z.-Y.; Wu, H. Genetic and epigenetic patterns in patients with the head-and-neck paragangliomas associate with differential clinical characteristics. J. Cancer Res. Clin. Oncol. 2017, 23, 8812–8960. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Khare, S.; Wubbenhorst, B.; Desloover, D.; D’Andrea, K.; Merrill, S.; Cho, N.W.; Greenberg, R.A.; Else, T.; Montone, K.; et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muth, A.; Crona, J.; Gimm, O.; Elmgren, A.; Filipsson, K.; Askmalm, M.S.; Sandstedt, J.; Tengvar, M.; Tham, E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 2018, 285, 187–204. [Google Scholar] [CrossRef]
- Niemann, S.; Becker-Follmann, J.; Nurnberg, G.; Rüschendorf, F.; Sieweke, N.; Hügens-Penzel, M.; Traupe, H.; Wienker, T.F.; Reis, A.; Muller, U. Assignment of PGL3 to chromosome 1 (q21-q23) in a family with autosomal dominant non-chromaffin paraganglioma. Am. J. Med. Genet. 2001, 98, 32–36. [Google Scholar] [CrossRef]
- Mannelli, M.; Castellano, M.; Schiavi, F.; Filetti, S.; Giacchè, M.; Mori, L.; Pignataro, V.; Bernini, G.; Giachè, V.; Bacca, A.; et al. Clinically Guided Genetic Screening in a Large Cohort of Italian Patients with Pheochromocytomas and/or Functional or Nonfunctional Paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 1541–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayley, J.-P.; Kunst, H.P.; Cascón, A.; Sampietro, M.L.; Gaal, J.; Korpershoek, E.; Hinojar-Gutiérrez, A.; Timmers, H.J.; Hoefsloot, L.H.; Hermsen, M.A.; et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010, 11, 366–372. [Google Scholar] [CrossRef]
- Kunst, H.P.M.; Rutten, M.H.; Hoefsloot, L.H.; Timmers, H.J.L.M.; Marres, H.A.; Jansen, J.C.; Kremer, H.; Bayley, J.-P.; Cremers, C.W.R.J.; De Mönnink, J.-P. SDHAF2 (PGL2-SDH5) and Hereditary Head and Neck Paraganglioma. Clin. Cancer Res. 2011, 17, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, R.; Garrahy, A.; Tuthill, A.; O’Halloran, D.; Joyce, C.; Casey, M.B.; O’Shea, P.M.; Bell, M. Universal Genetic Screening Uncovers a Novel Presentation of an SDHAF2 Mutation. J. Clin. Endocrinol. Metab. 2014, 99, E1392–E1396. [Google Scholar] [CrossRef] [Green Version]
- Luchetti, A.; Walsh, D.; Rodger, F.; Clark, G.; Martin, T.; Irving, R.; Sanna, M.; Yao, M.; Robledo, M.; Neumann, H.P.H.; et al. Profiling of Somatic Mutations in Phaeochromocytoma and Paraganglioma by Targeted Next Generation Sequencing Analysis. Int. J. Endocrinol. 2015, 2015, 138573. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef]
- Bausch, B.; Schiavi, F.; Ni, Y.; Welander, J.; Patocs, A.; Ngeow, J.; Wellner, U.; Malinoc, A.; Taschin, E.; Barbon, G.; et al. Clinical Characterization of the Pheochromocytoma and Paraganglioma Susceptibility Genes SDHA, TMEM127, MAX, and SDHAF2 for Gene-Informed Prevention. JAMA Oncol. 2017, 3, 1204–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Feng, Y.; Wells, M.; Huang, Z.; Chen, X. SDHx gene detection and clinical Phenotypic analysis of multiple paraganglioma in the head and neck. Laryngoscope 2018, 129, E67–E71. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.J.; Miller, J.H.; Suzuki, D.T.; Lewontin, R.C.; Gelbart, W.M. Somatic Versus Germinal Mutation. In An Introduction to Genetic Analysis, 7th ed.; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Guha, A.; Musil, Z.; Vicha, A.; Zelinka, T.; Pacak, K.; Astl, J.; Chovanec, M. A systematic review on the genetic analysis of paragangliomas: Primarily focused on head and neck paragangliomas. Neoplasma 2019, 66, 671–680. [Google Scholar] [CrossRef]
- Rattenberry, E.; Vialard, L.; Yeung, A.; Bair, H.; McKay, K.; Jafri, M.; Canham, N.; Cole, T.R.; Denes, J.; Hodgson, S.V.; et al. A Comprehensive Next Generation Sequencing–Based Genetic Testing Strategy to Improve Diagnosis of Inherited Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2013, 98, E1248–E1256. [Google Scholar] [CrossRef]
- Crona, J.; Taïeb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Taïeb, D.; Pacak, K. New Insights into the Nuclear Imaging Phenotypes of Cluster 1 Pheochromocytoma and Paraganglioma. Trends Endocrinol. Metab. 2017, 28, 807–817. [Google Scholar] [CrossRef]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2018, 4, 6–9. [Google Scholar] [CrossRef]
- Gimenez-Roqueplo, A.-P.; Dahia, P.L.; Robledo, M. An Update on the Genetics of Paraganglioma, Pheochromocytoma, and Associated Hereditary Syndromes. Horm. Metab. Res. 2012, 44, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Gunawardane, P.T.K.; Grossman, A.B. The clinical genetics of phaeochromocytoma and paraganglioma. Arch. Endocrinol. Metab. 2017, 61, 490–500. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Strome, S.E.; Hatten, K.M. Is routine genetic testing warranted in head and neck paragangliomas? Laryngoscope 2018, 129, 1491–1493. [Google Scholar] [CrossRef] [Green Version]
- Khatami, F.; Mohammadamoli, M.; Tavangar, S.M. Genetic and epigenetic differences of benign and malignant pheochromocytomas and paragangliomas (PPGLs). Endocr. Regul. 2018, 52, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Welander, J.; Söderkvist, P.; Gimm, O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr. Relat. Cancer 2011, 18, R253–R276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahia, P.L.M. Pheochromocytomas and Paragangliomas, Genetically Diverse and Minimalist, All at Once! Cancer Cell 2017, 31, 159–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Li, M.; Guan, X.; Yu, A.; Xiao, Q.; Wang, C.; Hu, Y.; Zhu, F.; Yin, H.; Yi, X.; et al. Clinical Syndromes and Genetic Screening Strategies of Pheochromocytoma and Paraganglioma. J. Kidney Cancer VHL 2018, 5, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Opocher, G.; Schiavi, F. Genetics of pheochromocytomas and paragangliomas. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Kantorovich, V.; King, K.S.; Pacak, K. SDH-related pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.D. Paragangliomas of the Head and Neck: An Overview from Diagnosis to Genetics. Head Neck Pathol. 2017, 11, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Ong, R.K.S.; Flores, S.K.; Reddick, R.L.; Dahia, P.L.M.; Shawa, H. A Unique Case of Metastatic, Functional, Hereditary Paraganglioma Associated with anSDHC Germline Mutation. J. Clin. Endocrinol. Metab. 2018, 103, 2802–2806. [Google Scholar] [CrossRef]
- Weinhold, B. Epigenetics: The Science of Change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Chiu, T.-P.; Kribelbauer, J.F.; Mann, R.S.; Bussemaker, H.J.; Rohs, R. Systematic prediction of DNA shape changes due to CpG methylation explains epigenetic effects on protein–DNA binding. Epigenetics Chromatin 2018, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Cronin, M. Whole Cancer Genome Sequencing by Next-Generation Methods. Am. J. Clin. Pathol. 2011, 136, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currás-Freixes, M.; Inglada-Pérez, L.; Mancikova, V.; Montero-Conde, C.; Letón, R.; Comino-Méndez, I.; Apellániz-Ruiz, M.; Sánchez-Barroso, L.; Sánchez-Covisa, M.A.; Alcázar, V.; et al. Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J. Med. Genet. 2015, 52, 647–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roose, L.M.; Rupp, N.J.; Röösli, C.; Valcheva, N.; Weber, A.; Beuschlein, F.; Tschopp, O. Tinnitus with Unexpected Spanish Roots: Head and Neck Paragangliomas Caused by SDHAF2 Mutation. J. Endocr. Soc. 2020, 4. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, F.M.; Eisenhofer, G.; Tao, J.J.; Kant, J.A.; Adams, K.T.; Linehan, W.M.; Pacak, K. High Frequency ofSDHBGermline Mutations in Patients with Malignant Catecholamine-Producing Paragangliomas: Implications for Genetic Testing. J. Clin. Endocrinol. Metab. 2006, 91, 4505–4509. [Google Scholar] [CrossRef] [Green Version]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.-P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef]


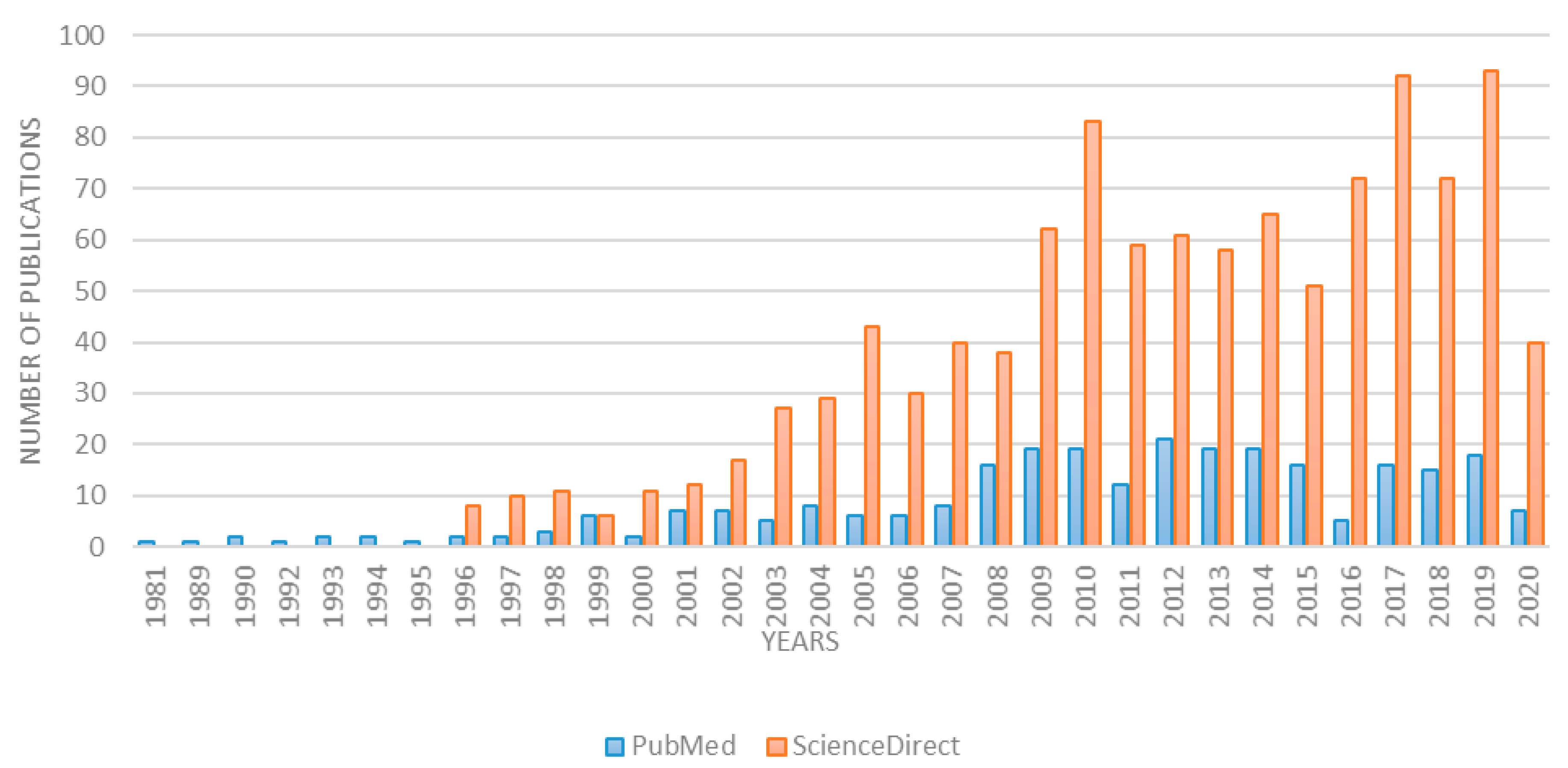{kind=link}
| Author, Year | No. of Patients | Genes | Findings |
|---|---|---|---|
| Niemann et al. (2001) [25] | Five patients with histologically proven paraganglioma (single family members) and one patient (of this family) with imaging findings consistent with a PGL. 33 family members were clinically unaffected. | SDHC gene location | The disease locus in PGL3 was determined to be located at 1q21-q23. |
| Mannelli et al. (2009) [26] | 501 patients with PCC and/or PGL 160 patients under 50 years of age whose DNA sequencing results revealed wild-type RET, VHL, SDHB, SDHC, and SDHD were subsequently analyzed for genomic rearrangements involving the VHL gene or one of the SDH genes. | RET VHL SDHD SDHB SDHC Genomic rearrangements (total deletion of the SDHD gene) | Detection of germinal mutations (such as VHL, RET, NF1, SDHB, SDHC and SDHD) in 32.1% of cases. From 100% in patients with associated lesions to 11.6% in patients with a single tumor. Genomic rearrangements were found in two of 160 patients (1.2%), both involving total deletion of the SDHD gene. |
| Bayley et al. (2010) [27] | 443 patients with apparently sporadic PCC/PGL who did not have mutations in SDHD, SDHC, or SDHB. Examination of a Spanish family with HNPGL presenting with a young age of onset. | SDHAF2 | No germinal (315 patients) or somatic (128 patients) mutations, and no germinal deletions of the SDHAF2 gene were found. After pedigree analysis of a Spanish family with HNPGL a pathogenic mutation in SDHAF2 was found that resulted in an amino acid substitution (p.Gly78Arg). The same mutation was noted previously in a Dutch kindred. |
| Kunst et al. (2011) [28] | 57 family members. | SDHAF2 | Establishing a correlation between HNPGL occurrence (based on phenotypic analysis) and SDHAF2 mutation. The mutation carriers showed early onset of the disease and high levels of multifocality. |
| Casey et al. (2014) [29] | 31 patients with confirmed PCC/PGL. | TMEM127 SDHAF2 RET | The occurrence of TMEM127, SDHAF2 and RET mutations was found in patients without indications for genetic testing based on phenotypic evaluation. |
| Fishbein et al. (2015) [23] | Stage 1: whole exome sequencing on a discovery set of 21 patients with PCC/PGL. Stage 2: targeted sequencing of a separate validation set of 103 patients withPCC/PGL. | NF1 ATRX | Mutations in NF1 were detected in 42% of tumors. In 28% of SDHB-related tumors, deleterious variants of ATRX were found (PP119F1 p.W2275* and PP098F2 p.R2197H). ATRX protein was not detected in tumor cells by immunohistochemistry. The study found somatic mutation of ATRX in 12.6% of cases; 30% of them had truncating mutations and 69% missense mutations, classified as deleterious. |
| Luchetti et al. (2015) [30] | 85 patients: PCC 60, PGL 5, HNPGL 20. | HRAS BRAF | Missense mutation was found in six cases (PCC = 6/60, PGL = 0/5, and HNPGL = 0/20) in HRAS in the hotspot region of codon 13 and 61. In one case of PCC, an activating BRAF mutation was found. In two patients a missense mutation was identified in the tetramerization domain of TP53 protein. |
| Fishbein et al. (2017) [31] | 173 patients with PCCs/PGLs. | SDHB, RET, WHL, NF1, SDHD, MAX EGLN1 (PHD2), TMEM127, CSDE1, HRAS, EPAS1, MAML3, BRAF, NGFR | 27% of patients had germinal mutations (including SDHB 9%, RET 6%, VHL 4%, and NF1 3%). SDHD, MAX, EGLN1 (PHD2), and TMEM127 mutations were found in less than 2% each. CSDE1 was identified as a somatically mutated driver gene complementary to the other four known drivers (HRAS, RET, EPAS1, and NF1). MAML3, BRAF, NGFR, and NF1 fusion genes were discovered. |
| Bausch et al. (2017) [32] | 972 unrelated patients without mutations in the classic PCC/PGL associated genes. | SDHA, TMEM127, MAX, SDHAF2 | Six percent of patients were mutation carriers (including SDHA, TMEM127, MAX, and SDHAF2). 91% of patients had familial, multiple, extra-adrenal, and/or malignant tumors and/or had younger age of onset. Extra-adrenal tumors occurred in 48% of mutation carriers and in 79% of carriers with HNPGL. |
| Chen et al. (2017) [22] | 37 patients with HNPGLs. | SDHD SDHB SDHAF2 | SDHD gene mutations were found in: the Chinese founder mutation (c.3G>C, p.Met1Ile) in six cases, a missense mutation (c.284T>C, p.L95P) in one case, an in-frame deletion (c.278–280delATT, p.Y93S) in one case. A missense SDHB mutation (c.647A>G) and a nonsense SDHAF2 mutation (c.130C>T, p.Gln44Ter) were found in two cases. Frequent methylation was observed in six of the TSGs tested (HIC1, DcR1, DcR2, DR4, DR5, and CASPS8). Four of them (HIC1, DcR1, DcR2 and CASPS8) showed more frequent mutations in SDH-associated HNPGL than in non-mutated ones. |
| Calsina et al. (2018) [21] | 830 patients with PPGLs, negative for the main PPGL driver genes. | MDH2 | Twelve heterozygous variants of MDH2 were found (five of the 12 were missense (41.7%), one synonymous (8.3%), four were located in the intronic region (33.3%), one was an in-frame deletion (8.3%), and one affected a donor splice-site (8.3%). Five of these were unreported variants. The study showed the functional impact of two variants (p.Arg104Gly and p.Lys314del) and suggests altered molecular function of two variants (p.Val160Met and p.Ala256Thr). |
| Ding et al. (2019) [33] | 23 cases of multiple HNPGL. | SDHD, SDHB, SDHC, SDHAF2, VHL, RET | Family 1: 12 SDHD mutations (8 bilateral carotid body tumor (CBT) with 1 bilateral malignant CBT) Family 2: 3 SDHD mutations (1 bilateral CBT, 2 unilateral CBT) Family 3: 2 cases of SDHD mutations (vagus PGL and pheochromocytoma) Other patients: sporadic manifestations (5 cases SDHD gene mutation, 1 case RET gene mutation). Two novel mutations were found: c.387–393del7 mutation of SDHD gene and c.3247A>G mutation of RET gene. More frequent occurrence of SDHD mutations was found in patients and family members with multiple HNPGL. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majewska, A.; Budny, B.; Ziemnicka, K.; Ruchała, M.; Wierzbicka, M. Head and Neck Paragangliomas—A Genetic Overview. Int. J. Mol. Sci. 2020, 21, 7669. https://doi.org/10.3390/ijms21207669
Majewska A, Budny B, Ziemnicka K, Ruchała M, Wierzbicka M. Head and Neck Paragangliomas—A Genetic Overview. International Journal of Molecular Sciences. 2020; 21(20):7669. https://doi.org/10.3390/ijms21207669
Chicago/Turabian StyleMajewska, Anna, Bartłomiej Budny, Katarzyna Ziemnicka, Marek Ruchała, and Małgorzata Wierzbicka. 2020. "Head and Neck Paragangliomas—A Genetic Overview" International Journal of Molecular Sciences 21, no. 20: 7669. https://doi.org/10.3390/ijms21207669
APA StyleMajewska, A., Budny, B., Ziemnicka, K., Ruchała, M., & Wierzbicka, M. (2020). Head and Neck Paragangliomas—A Genetic Overview. International Journal of Molecular Sciences, 21(20), 7669. https://doi.org/10.3390/ijms21207669