Structural Insights into β-arrestin/CB1 Receptor Interaction: NMR and CD Studies on Model Peptides



Abstract
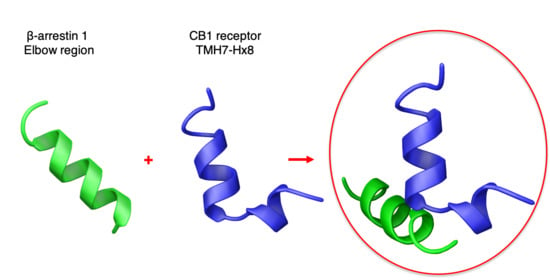
:
1. Introduction
2. Results and Discussion
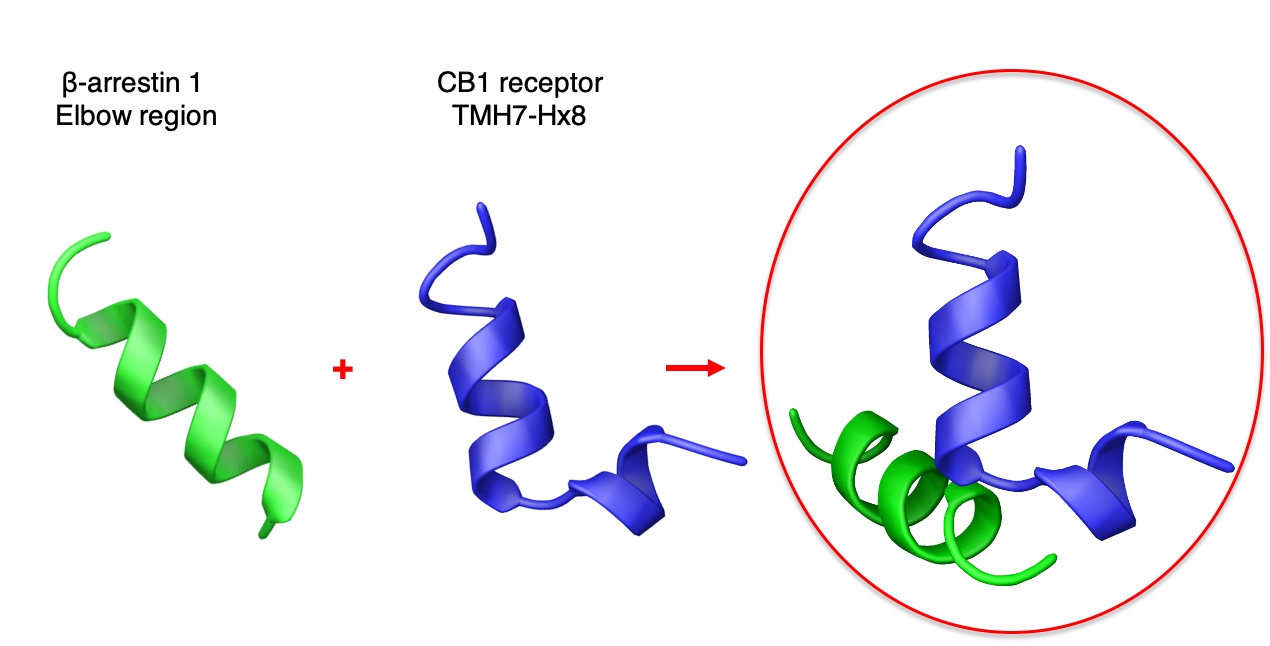
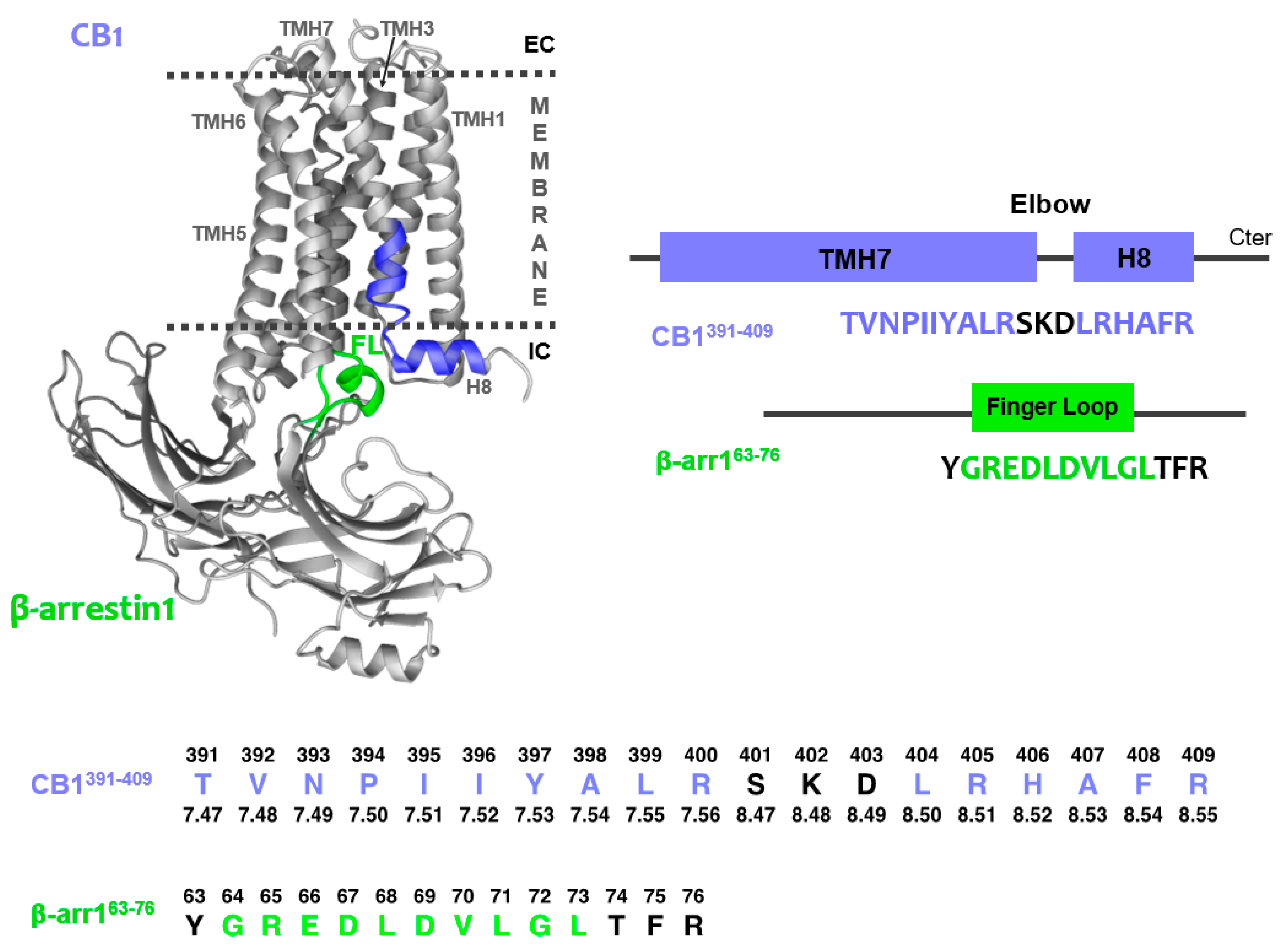
2.1. Peptide Design
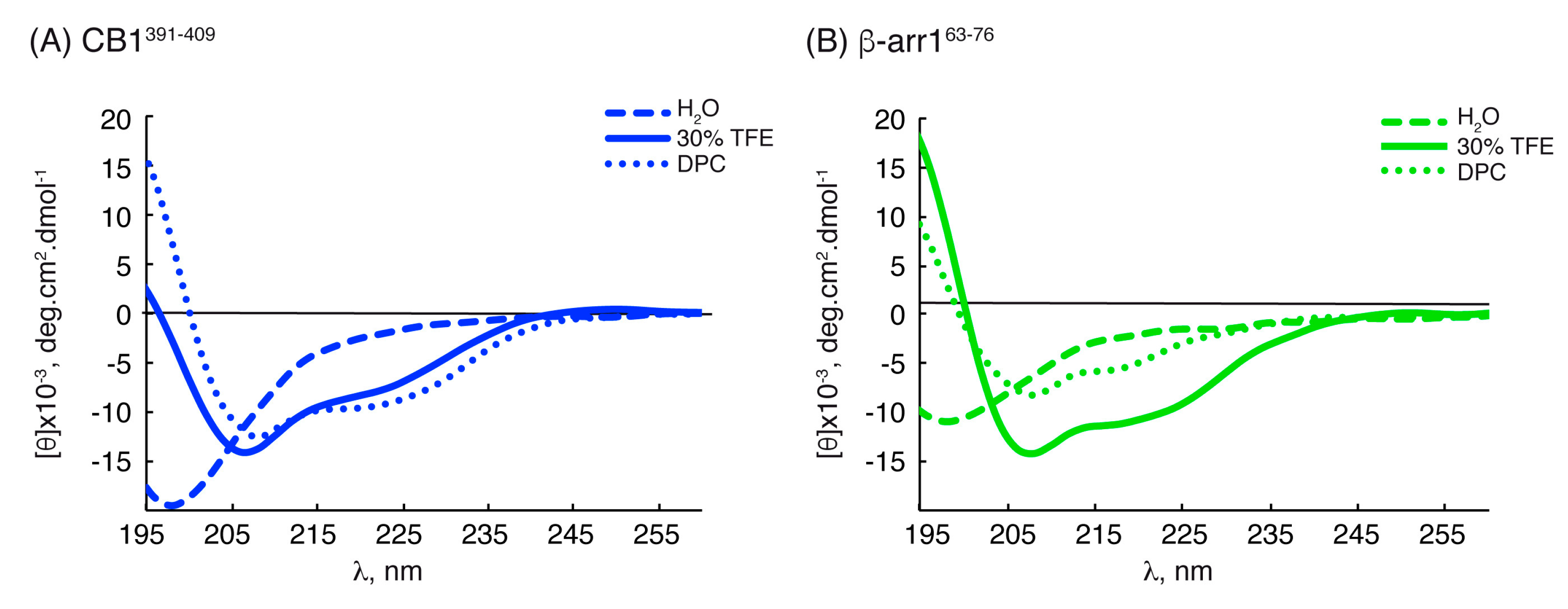
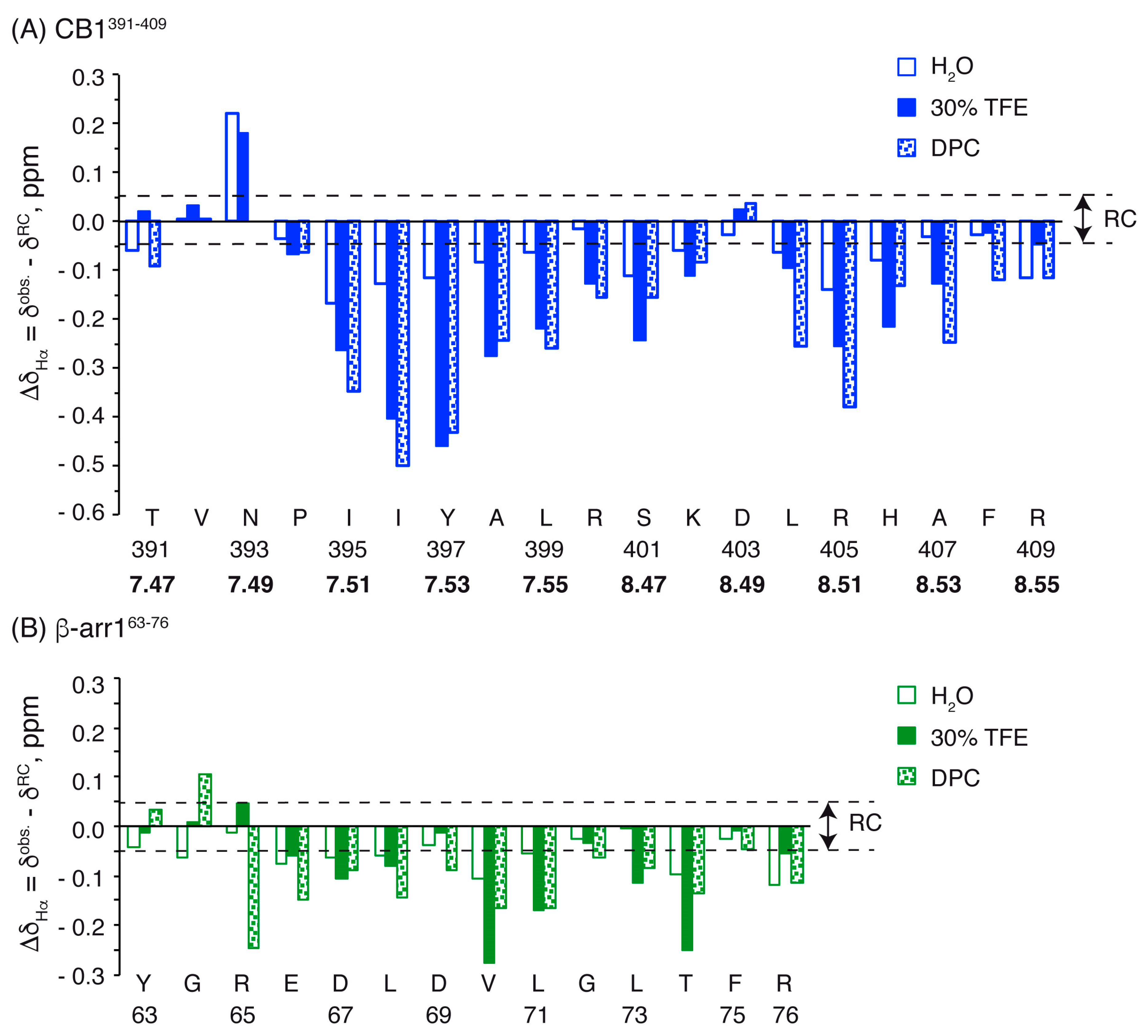
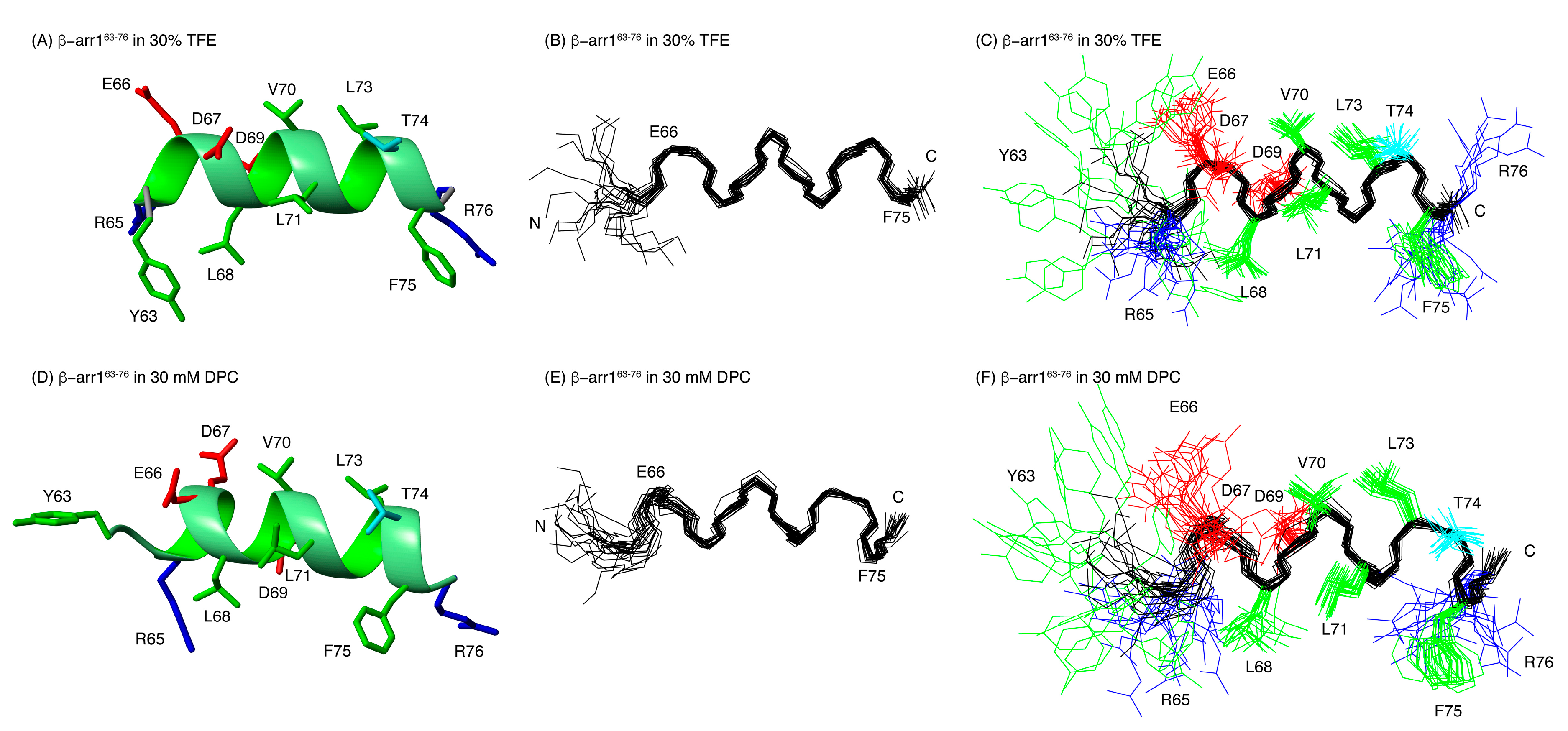
2.2. Structural Behavior of the Free CB1 and β-Arrestin1 Peptides
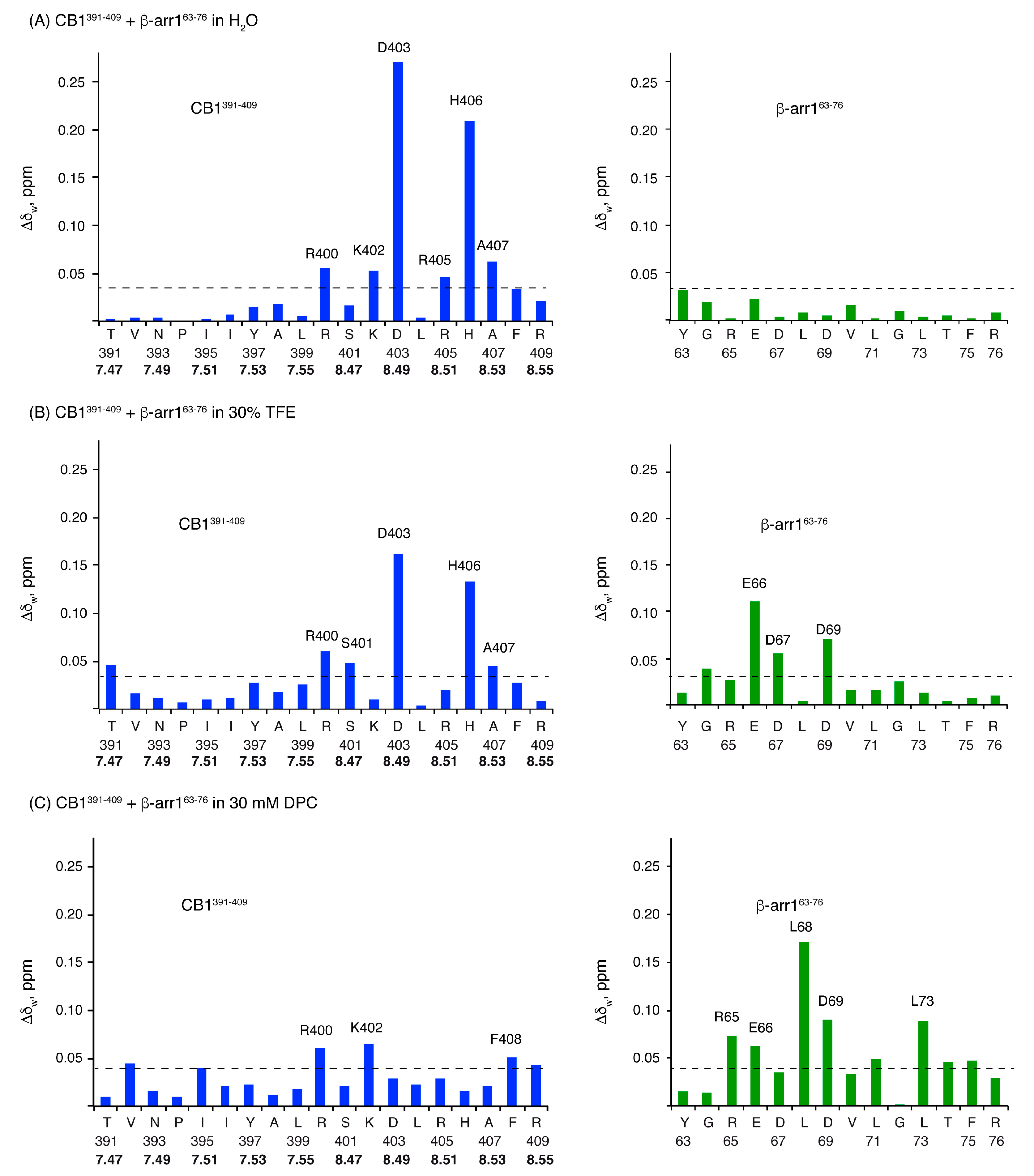
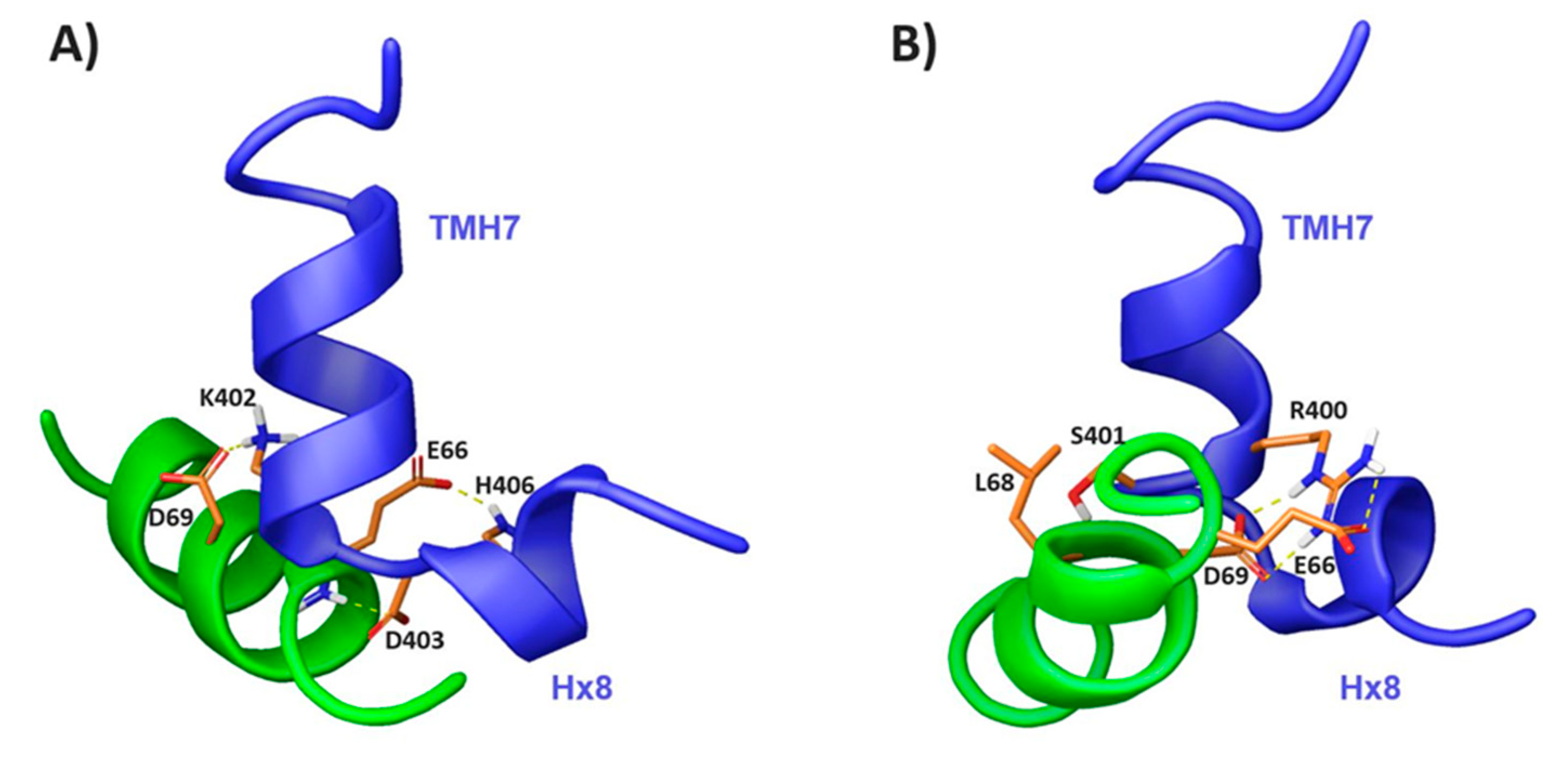
2.3. Characterization of the CB1 and β-Arrestin1 Interface
3. Materials and Methods
3.1. Chemicals and Peptides
- -
- CB1 peptide (CB1391-409; Ac-TVNPIIYALRSKDLRHAFR-NH2): HPLC: tR = 17.2 min; 95.3% (gradient: 18-36% B in 23 min; buffer A: 0.05% TFA + 2% CH3CN; buffer B: 0.05% TFA + 90% CH3CN). MALDI-TOF: Theoretical MW = 2311.74; Found [M+H]+ = 2312.17.
- -
- β-arrestin1 peptide (β-arr163-76; Ac-YGREDLDVLGLTFR-NH2): HPLC: tR = 7.9 min; 95.0% (gradient: 30-44% B in 14 min; buffer A: 0.05% TFA + 2% CH3CN; buffer B: 0.05% TFA + 90% CH3CN). MALDI-TOF: Theoretical MW = 1694.93; Found [M+H]+ = 1696.22.
3.2. Peptide Numbering
3.3. CD Spectroscopy
3.4. NMR Studies
3.4.1. Sample Preparation
3.4.2. Spectra Acquisition
3.4.3. Spectra Assignment
3.4.4. Estimation of Helix Populations
3.5. Structure Calculation
3.6. NMR-Driven Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Adenylyl cyclase |
| cAMP | Cyclic adenosine monophosphate |
| CB1 | Cannabinoid receptor type 1 |
| CD | Circular dichroism |
| COSY | Phase sensitive correlated spectroscopy |
| DPC | Dodecylphosphocholine |
| FL | Finger loop |
| pERK1/2 | Extracellular signal-regulated kinase 1/2 |
| GIRKs | G protein-coupled inwardly-rectifying potassium channels |
| GPCR | G protein-coupled receptor |
| GRKs | G protein-coupled receptor kinases |
| MAPK | Mitogen-activated protein kinases |
| NOESY | Nuclear Overhauser enhancement spectroscopy |
| PI3K | Phosphatidylinositide-3-kinase |
| TFE | Trifluoroethanol |
| TMH | Transmembrane helix |
| TOCSY | Total correlated spectroscopy |
| VGCC | Voltage-gated calcium channels |
References
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharm. 1988, 34, 605–613. [Google Scholar]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S. Cannabinoid Receptors in Brain: Pharmacogenetics, neuropharmacology, neurotoxicology, and potential therapeutic applications. Int. Rev. Neurobiol. 2009, 88, 335–369. [Google Scholar] [PubMed]
- Pertwee, R.G. The pharmacology of cannabinoid receptors and their ligands: An overview. Int. J. Obes. (Lond.) 2006, 30, S13–S18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urits, I.; Borchart, M.; Hasegawa, M.; Kochanski, J.; Orhurhu, V.; Viswanath, O. An Update of Current Cannabis-Based Pharmaceuticals in Pain Medicine. Pain 2019, 8, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.S.; Lourenço, D.M.; Paulo, S.L.; Mateus, J.M.; Ferreira, M.F.; Mouro, F.M.; Moreira, J.B.; Ribeiro, F.F.; Sebastião, A.M.; Xapelli, S. Cannabinoid actions on neural stem cells: Implications for pathophysiology. Molecules 2019, 24, 1350. [Google Scholar] [CrossRef] [Green Version]
- Billakota, S.; Devinsky, O.; Marsh, E. Cannabinoid therapy in epilepsy. Curr. Opin. Neurol. 2019, 32, 220–226. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharm. 2019, 176, 1384–1394. [Google Scholar] [CrossRef]
- Moreno, E.; Cavic, M.; Krivokuca, A.; Casadó, V.; Canela, E. The endocannabinoid system as a target in cancer diseases: Are we there yet? Front. Pharm. 2019, 10, 339. [Google Scholar] [CrossRef] [Green Version]
- Sherman, C.; Ruthirakuhan, M.; Vieira, D.; Lanctôt, K.L.; Herrmann, N. Cannabinoids for the treatment of neuropsychiatric symptoms, pain and weight loss in dementia. Curr. Opin. Psychiatry 2018, 31, 140–146. [Google Scholar] [CrossRef]
- Poleszak, E.; Wośko, S.; Sławińska, K.; Szopa, A.; Wróbel, A.; Serefko, A. Cannabinoids in depressive disorders. Life Sci. 2018, 213, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.M.; Maurya, N.; Velmurugan, B.K. Medicinal Use of Synthetic Cannabinoids—a Mini Review. Curr. Pharm. Rep. 2019, 5, 1–13. [Google Scholar] [CrossRef]
- Morales, P.; Jagerovic, N. Novel approaches and current challenges with targeting the endocannabinoid system. Expert Opin. Drug Discov. 2020, 15, 917–930. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Peigneur, S.; Hendrickx, L.A.; Tytgat, J. Targeting Cannabinoid Receptors: Current Status and Prospects of Natural Products. Int. J. Mol. Sci. 2020, 21, 5064. [Google Scholar] [CrossRef] [PubMed]
- Brunt, T.M.; Bossong, M.G. The neuropharmacology of cannabinoid receptor ligands in central signaling pathways. Eur. J. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Dupré, D.J.; Denovan-Wright, E.M. Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J. Biol. Chem. 2014, 289, 24845–24862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, M.; Felder, C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.B.; Cawston, E.E.; Grimsey, N.L.; Hunter, M.R.; Korde, A.; Vemuri, V.K.; Makriyannis, A.; Glass, M. Gα s signalling of the CB 1 receptor and the influence of receptor number. Br. J. Pharm. 2017, 174, 2545–2562. [Google Scholar] [CrossRef] [Green Version]
- D’Antona, A.M.; Ahn, K.H.; Wang, L.; Mierke, D.F.; Lucas-Lenard, J.; Kendall, D.A. A cannabinoid receptor 1 mutation proximal to the DRY motif results in constitutive activity and reveals intramolecular interactions involved in receptor activation. Brain Res. 2006, 1108, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadji, V.; Lucas-Lenard, J.M.; Chin, C.N.; Kendall, D.A. Involvement of the carboxyl terminus of the third intracellular loop of the cannabinoid CB1 receptor in constitutive activation of G(s). J. Neurochem. 1999, 72, 2032–2038. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Hille, B.; Mackie, K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB(1) receptor coupling to G(q/11) G proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 19144–19149. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.M.; Sung, J.Y.; Hébert, T.E. Gβγ subunits—Different spaces, different faces. Pharm. Res. 2016, 111, 434–441. [Google Scholar] [CrossRef]
- Howlett, A.C. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharm. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, M.S.; Finlay, D.B.; Patel, M.; Javitch, J.A.; Glass, M.; Grimsey, N.L. Cannabinoid CB1 and CB2 Receptor-Mediated Arrestin Translocation: Species, Subtype, and Agonist-Dependence. Front. Pharm. 2019, 10, 350. [Google Scholar] [CrossRef] [PubMed]
- Flores-Otero, J.; Ahn, K.H.; Delgado-Peraza, F.; Mackie, K.; Kendall, D.A.; Yudowski, G.A. Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Peraza, F.; Ahn, K.H.; Nogueras-Ortiz, C.; Mungrue, I.N.; Mackie, K.; Kendall, D.A.; Yudowski, G.A. Mechanisms of Biased beta-Arrestin-Mediated Signaling Downstream from the Cannabinoid 1 Receptor. Mol. Pharm. 2016, 89, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Wouters, E.; Walraed, J.; Banister, S.D.; Stove, C.P. Insights into biased signaling at cannabinoid receptors: Synthetic cannabinoid receptor agonists. Biochem. Pharm. 2019, 169, 113623. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of beta-blocker action: Carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.K.; Violin, J.D.; Whalen, E.J.; Gesty-Palmer, D.; Shenoy, S.K.; Lefkowitz, R.J. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 9988–9993. [Google Scholar] [CrossRef] [Green Version]
- Khajehali, E.; Malone, D.T.; Glass, M.; Sexton, P.M.; Christopoulos, A.; Leach, K. Biased Agonism and Biased Allosteric Modulation at the CB 1 Cannabinoid Receptor s. Mol. Pharm. 2015, 88, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Homan, K.T.; Vishnivetskiy, S.A.; Manglik, A.; Tesmer, J.J.G.; Gurevich, V.V.; Gurevich, E.V. G Protein-coupled Receptor Kinases of the GRK4 Protein Subfamily Phosphorylate Inactive G Protein-coupled Receptors (GPCRs). J. Biol. Chem. 2015, 290, 10775–10790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Williams, A.H.; Penthala, N.R.; Prather, P.L.; Crooks, P.A.; Zhan, C.G. Binding Modes and Selectivity of Cannabinoid 1 (CB1) and Cannabinoid 2 (CB2) Receptor Ligands. ACS Chem. Neurosci. 2020, 30, 3455–3463. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Jing, Y.; Wang, L.; Ma, S.; Jun, J.J.; Xie, X.Q. Prediction of Orthosteric and Allosteric Regulations on Cannabinoid Receptors Using Supervised Machine Learning Classifiers. Mol. Pharm. 2019, 16, 2605–2615. [Google Scholar] [CrossRef]
- Jenkinson, S.; Goody, S.M.G.; Bassyouni, A.; Jones, R.; Otto-Bruc, A.; Duquennoy, S.; DaSilva, J.K.; Butler, P.; Mead, A. Translation of in vitro cannabinoid 1 receptor agonist activity to in vivo pharmacodynamic endpoints. J. Pharm. Toxicol. Methods 2020, 104, 106899. [Google Scholar] [CrossRef] [PubMed]
- Al-zoubi, R.; Morales, P.; Reggio, P.H. Structural Insights into CB1 Receptor Biased Signaling. Int. J. Mol. Sci. 2019, 20, 1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, P.; Goya, P.; Jagerovic, N. Emerging strategies targeting CB2 cannabinoid receptor: Biased agonism and allosterism. Biochem. Pharm. 2018, 157, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Functional selectivity and biased receptor signaling. J. Pharm. Exp. 2011, 336, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Kenakin, T.; Christopoulos, A. Measurements of ligand bias and functional affinity. Nat. Rev. Drug Discov. 2013, 12, 483. [Google Scholar] [CrossRef]
- Wisler, J.W.; Xiao, K.; Thomsen, A.R.B.; Lefkowitz, R.J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 2014, 27, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR-G protein binding. Nature 2017, 545, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C. Structural Basis for G Protein-Coupled Receptor Activation. Biochemistry 2017, 56, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Heydenreich, F.M.; Flock, T.; Miljus, T.; Balaji, S.; Bouvier, M.; Veprintsev, D.B.; Tate, C.G.; et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 2016, 40, 383–388. [Google Scholar] [CrossRef]
- Mahoney, J.P.; Sunahara, R.K. Mechanistic insights into GPCR-G protein interactions. Curr. Opin. Struct. Biol. 2016, 41, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.; Makriyannis, A.; Stevens, R.C.; Liu, Z. Crystal Structure of the Human Cannabinoid CB 1. Cell 2016, 167, 750–762. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.-H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Krishna Kumar, K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.D.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.E.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458.e12. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Johnston, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170, 457–469.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Sente, A.; Peer, R.; Srivastava, A.; Baidya, M.; Lesk, A.M.; Balaji, S.; Shukla, A.K.; Babu, M.M.; Flock, T. Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol. 2018, 25, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Lacroix, E.; Viguera, A.R.; Serrano, L. Elucidating the folding problem of α-helices: Local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters. J. Mol. Biol. 1998, 284, 173–191. [Google Scholar] [CrossRef]
- Chaturvedi, M.; Maharana, J.; Shukla, A.K. Terminating G-Protein Coupling: Structural Snapshots of GPCR-β-Arrestin Complexes. Cell 2020, 180, 1041–1043. [Google Scholar] [CrossRef]
- Buck, M. Trifluoroethanol and colleagues: Cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 1998, 31, 297–355. [Google Scholar] [CrossRef]
- Tyukhtenko, S.; Tiburu, E.K.; Deshmukh, L.; Vinogradova, O.; Janero, D.R.; Makriyannis, A. NMR solution structure of human cannabinoid receptor-1 helix 7/8 peptide: Candidate electrostatic interactions and microdomain formation. Biochem. Biophys. Res. Commun. 2009, 390, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Elgeti, M.; Kazmin, R.; Rose, A.S.; Szczepek, M.; Hildebrand, P.W.; Bartl, F.J.; Scheerer, P.; Hofmann, K.P. The arrestin-1 finger loop interacts with two distinct conformations of active rhodopsin. J. Biol. Chem. 2018, 293, 4403–4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesteros, J.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Stuart, C.S., Ed.; Elsevier: San Diego, CA, USA, 1995; Volume 25, pp. 366–428. ISBN 9780121852955. [Google Scholar]
- Piotto, M.; Saudek, V.; Sklenář, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. Nmr 1992, 2, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wüthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB inter-union task group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. Eur. J. Biochem. 1998, 256, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K.; Billeter, M.; Braun, W. Polypeptide secondary structure determination by nuclear magnetic resonance observation of short proton-proton distances. J. Mol. Biol. 1984, 180, 715–740. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [Green Version]
- Chaves-Arquero, B.; Pérez-Cañadillas, J.M.; Jiménez, M.A. Effect of Phosphorylation on the Structural Behaviour of Peptides Derived from the Intrinsically Disordered C-Terminal Domain of Histone H1.0. Chem. A Eur. J. 2020, 26, 5970–5981. [Google Scholar] [CrossRef]
- Güntert, P. Automated NMR Structure Calculation With CYANA. In Protein NMR Techniques; Humana Press: Hoboken, NJ, USA, 2004; Volume 278, pp. 353–378. ISBN 1064-3745. [Google Scholar]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. Nmr 2009, 44, 213–223. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Roccatano, D.; Colombo, G.; Fioroni, M.; Mark, A.E. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: A molecular dynamics study. Proc. Natl. Acad. Sci. USA 2002, 99, 12179–12184. [Google Scholar] [CrossRef] [Green Version]
- Zamora-Carreras, H.; Maestro, B.; Strandberg, E.; Ulrich, A.S.; Sanz, J.M.; Jiménez, M.Á. Micelle-triggered β-hairpin to α-helix transition in a 14-residue peptide from a choline-binding repeat of the pneumococcal autolysin LytA. Chem. A Eur. J. 2015, 21, 8076–8089. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Wang, J.; Plouffe, B.; Smith, J.S.; Yamani, L.; Kaur, S.; Jean-Charles, P.Y.; Gauthier, C.; Lee, M.H.; Pani, B.; et al. Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 2018, 11, eaat7650. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Conditions | [θ]222nm, deg.cm2.dmol−1 | % Helix [a] from [θ]222nm | Helix Length | Av. ΔδHα, ppm | % α-Helix from ΔδHα | Av. ΔδCα, ppm | % α-Helix from ΔδCα | Av. % Helix[c] |
|---|---|---|---|---|---|---|---|---|---|
| CB1391-409 | H2O | −2178.68 | 13 | P394-K402 | −0.09 [b] | 22 [b] | +0.48 [b] | 16 [b] | 19 ± 3 [b] |
| L404-F408 | −0.07 [b] | 18 [b] | +0.33 [b] | 11 [b] | 14 ± 4 [b] | ||||
| TFE | −7931.79 | 28 | P394-K402 | −0.24 | 62 | +2.50 | 81 | 71 ± 9 | |
| L404-F408 | −0.14 | 37 | +1.52 | 49 | 43 ± 6 | ||||
| DPC | −9415.07 | 32 | P394-K402 | −0.25 | 64 | +2.01 | 65 | 64 ± 1 | |
| L404-F408 | −0.23 | 58 | +1.69 | 54 | 56 ± 2 | ||||
| β-arr163-76 | H2O | −1750.71 | 12 | E66-T74 | −0.06 [b] | 16 [b] | +0.56 [b] | 18 [b] | 17 ± 2 [b] |
| TFE | −10229.3 | 34 | E66-T74 | −0.12 | 31 | +1.79 | 58 | 45 ± 13 | |
| DPC | −3626.36 | 17 | R65-T74 | −0.13 | 34 | +1.22 | 39 | 37 ± 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales, P.; Bruix, M.; Jiménez, M.A. Structural Insights into β-arrestin/CB1 Receptor Interaction: NMR and CD Studies on Model Peptides. Int. J. Mol. Sci. 2020, 21, 8111. https://doi.org/10.3390/ijms21218111
Morales P, Bruix M, Jiménez MA. Structural Insights into β-arrestin/CB1 Receptor Interaction: NMR and CD Studies on Model Peptides. International Journal of Molecular Sciences. 2020; 21(21):8111. https://doi.org/10.3390/ijms21218111
Chicago/Turabian StyleMorales, Paula, Marta Bruix, and M. Angeles Jiménez. 2020. "Structural Insights into β-arrestin/CB1 Receptor Interaction: NMR and CD Studies on Model Peptides" International Journal of Molecular Sciences 21, no. 21: 8111. https://doi.org/10.3390/ijms21218111
APA StyleMorales, P., Bruix, M., & Jiménez, M. A. (2020). Structural Insights into β-arrestin/CB1 Receptor Interaction: NMR and CD Studies on Model Peptides. International Journal of Molecular Sciences, 21(21), 8111. https://doi.org/10.3390/ijms21218111