Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment



{kind=link}
Abstract
:1. Introduction
2. Neurotransmitter/Neuronal Modulation of Glioma Progression
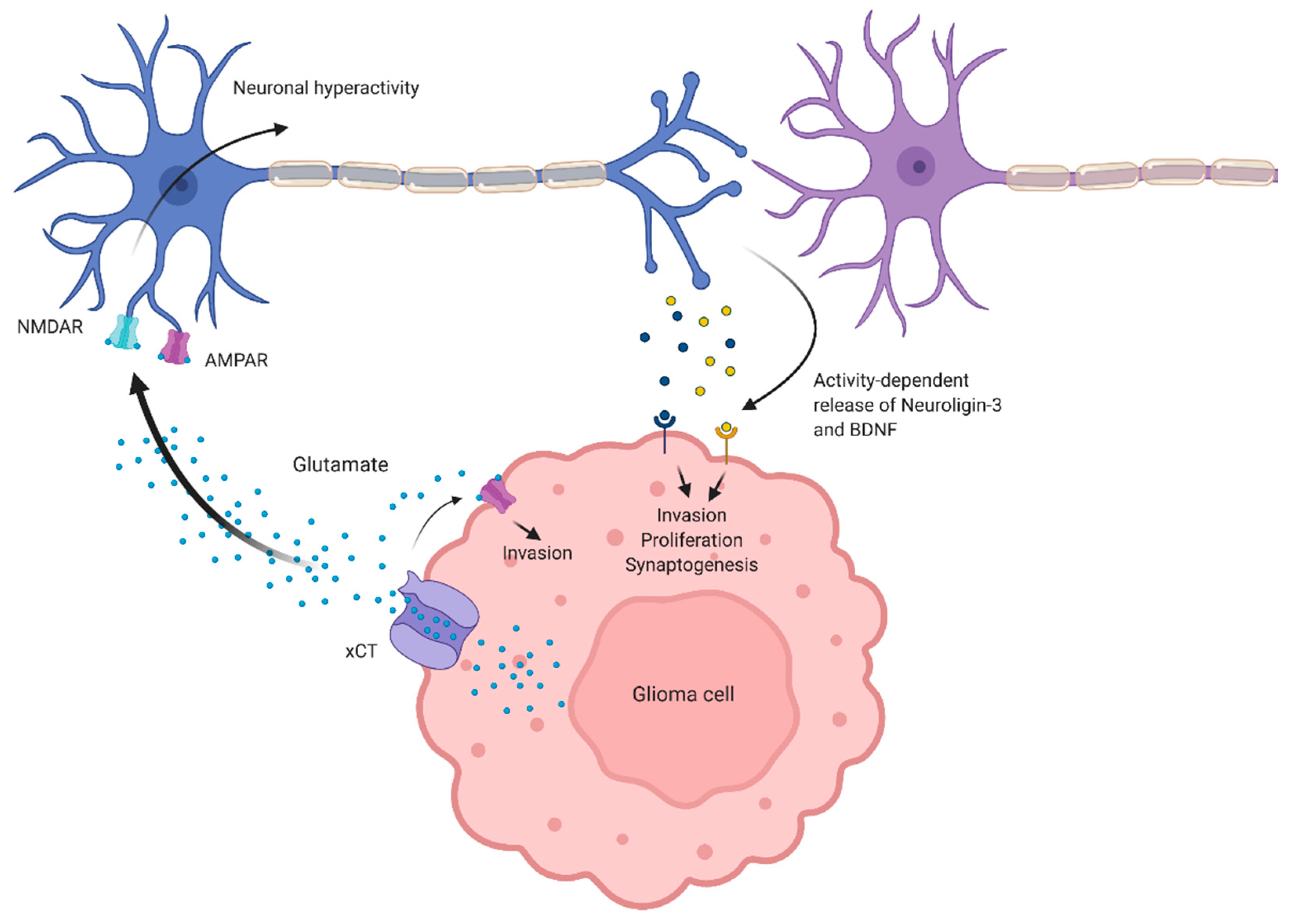
2.1. Pathophysiological Hallmarks of Aberrant Glutamate Secretion
2.2. GABAergic Signaling in the Glioma Microenvironment
2.3. Aberrant Communication between Glioma Cells and Peritumoral Neurons
3. Microglia: Potential Role as Synaptic Sculptors?
3.1. Context-Dependent Actions on Synapse Formation and Pruning
3.2. Pilot Studies to Discern the Role of Synapse Formation and Maintenance
4. Sex-Specific Microglial Modulation of Glioma Progression
5. Microglia and Macrophages: Partners in Crime or Divergent Players in Glioma?
6. Translational Considerations
6.1. Pharmacological Targeting of Aberrant Glutamatergic Action in Glioma
6.2. Targeting Myeloid Cells during Glioma Treatment
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| CSF1R | Colony Stimulating Factor 1 receptor |
| CNS | Central Nervous System |
| EGFR | Epidermal growth factor receptor |
| NMDAR | N-methyl-D-aspartate receptor |
| GABA | γ-Aminobutyric acid |
| TTX | Tetrodotoxin |
| NKCC1 | Na-K-Cl cotransporter |
| KCC2 | Chloride potassium symporter 5 |
| BDNF | Brain-derived neurotrophic factor |
| NBQX | 2,3-dioxo-6-nitro-7-sulfamoyl-benzo[f]quinoxaline |
| NASPM | 1-napthyl acetyl spermine |
| WT | Wild-type |
| DN | Dominant-Negative |
| IDH | Isocitrate dehydrogenase |
| CNQX | 6-cyano-7-nitroquinoxaline-2,3-dione |
| LPS | Lipopolysaccharide |
| CCL2 | of C-C motif chemokine ligand 2 |
| TGF-B1 | Transforming growth factor – beta 1 |
| JAM-A | Junctional adhesion molecule A |
| NGS | neuro-glioma synapses |
| GB | Glioblastoma |
| OPC | Oligodendrocyte precursor cell |
| TM | Tumor microtubes |
References
- Omuro, A. Glioblastoma and Other Malignant Gliomas. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, T.S.; Grant, R.; Gilbert, M.R.; Lee, J.W.; Norden, A.D. Epilepsy in glioma patients: Mechanisms, management, and impact of anticonvulsant therapy. Neuro-Oncology 2015, 18, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iuchi, T.; Hasegawa, Y.; Kawasaki, K.; Sakaida, T. Epilepsy in patients with gliomas: Incidence and control of seizures. J. Clin. Neurosci. 2015, 22, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Buckingham, S.C.; Sontheimer, H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 2012, 53, 1360–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nat. Cell Biol. 2017, 549, 533–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mega, A.; Nilsen, M.H.; Leiss, L.W.; Tobin, N.P.; Miletic, H.; Sleire, L.; Strell, C.; Nelander, S.; Krona, C.; Hägerstrand, D.; et al. Astrocytes enhance glioblastoma growth. Glia 2020, 68, 316–327. [Google Scholar] [CrossRef]
- Hide, T.; Komohara, Y.; Miyasato, Y.; Nakamura, H.; Makino, K.; Takeya, M.; Kuratsu, J.-I.; Mukasa, A.; Yano, S. Oligodendrocyte Progenitor Cells and Macrophages/Microglia Produce Glioma Stem Cell Niches at the Tumor Border. EBioMedicine 2018, 30, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Zhai, H.; Heppner, F.L.; Tsirka, S.E. Microglia/macrophages promote glioma progression. Glia 2011, 59, 472–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyauchi, J.T.; Caponegro, M.D.; Chen, D.; Choi, M.K.; Li, M.; Tsirka, S.E. Deletion of Neuropilin 1 from Microglia or Bone Marrow-Derived Macrophages Slows Glioma Progression. Cancer Res. 2017, 78, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, J.T.; Chen, D.; Choi, M.; Nissen, J.C.; Shroyer, K.R.; Djordevic, S.; Zachary, I.C.; Selwood, D.; Tsirka, S.E. Ablation of Neuropilin 1 from glioma-associated microglia and macrophages slows tumor progression. Oncotarget 2016, 7, 9801–9814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, N.; Du, X.; He, Y.; Chen, S.; Shao, Q.; Ma, C.; Huang, B.; Chen, A.; Zhao, P.; et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med. Oncol. 2015, 32, 43. [Google Scholar] [CrossRef] [PubMed]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-Oncology 2015, 18, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nat. Cell Biol. 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nat. Cell Biol. 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nat. Cell Biol. 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkhof, M.; Dielemans, J.C.M.; van Breemen, M.S.; Zwinkels, H.; Walchenbach, R.; Taphoorn, M.J.; Vecht, C.J. Effect of valproic acid on seizure control and on survival in patients with glioblastoma multiforme. Neuro-Oncology 2013, 15, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkhof, M.; Vecht, C.J. Seizure characteristics and prognostic factors of gliomas. Epilepsia 2013, 54, 12–17. [Google Scholar] [CrossRef] [PubMed]
- van Breemen, M.S.M.; Wilms, E.B.; Vecht, C.J. Epilepsy in patients with brain tumours: Epidemiology, mechanisms, and management. Lancet Neurol. 2007, 6, 421–430. [Google Scholar] [CrossRef]
- Ye, Z.C.; Sontheimer, H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999, 59, 4383–4391. [Google Scholar] [PubMed]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef]
- Robert, S.M.; Buckingham, S.C.; Campbell, S.L.; Robel, S.; Holt, K.T.; Ogunrinu-Babarinde, T.; Warren, P.P.; White, D.M.; Reid, M.A.; Eschbacher, J.M.; et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 2015, 7, 289ra86. [Google Scholar] [CrossRef] [Green Version]
- Savaskan, N.; Seufert, S.; Hauke, J.; Tränkle, C.; Eyüpoglu, I.Y.; Hahnen, E. Dissection of mitogenic and neurodegenerative actions of cystine and glutamate in malignant gliomas. Oncogene 2010, 30, 43–53. [Google Scholar] [CrossRef]
- Takano, T.; Lin, J.H.-C.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef]
- Tsuchihashi, K.; Okazaki, S.; Ohmura, M.; Ishikawa, M.; Sampetrean, O.; Onishi, N.; Wakimoto, H.; Yoshikawa, M.; Seishima, R.; Iwasaki, Y.; et al. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(—). Cancer Res. 2016, 76, 2954–2963. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.-C.; Rothstein, J.D.; Sontheimer, H. Compromised Glutamate Transport in Human Glioma Cells: Reduction–Mislocalization of Sodium-Dependent Glutamate Transporters and Enhanced Activity of Cystine–Glutamate Exchange. J. Neurosci. 1999, 19, 10767–10777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, H.; Carpenter, K.L.H.; Price, S.J.; Hutchinson, P. In Vivo assessment of high-grade glioma biochemistry using microdialysis: A study of energy-related molecules, growth factors and cytokines. J. Neuro-Oncol. 2009, 97, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Lu, L.; de Groot, J. AMPA receptors promote perivascular glioma invasion via beta1 integrin-dependent adhesion to the extracellular matrix. Neuro Oncol. 2009, 11, 260–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vuurden, D.G.; Yazdani, M.; Bosma, I.; Broekhuizen, A.J.F.; Postma, T.J.; Heimans, J.J.; van der Valk, P.; Aronica, E.; Tannous, B.A.; Wurdinger, T.; et al. Attenuated AMPA Receptor Expression Allows Glioblastoma Cell Survival in Glutamate-Rich Environment. PLoS ONE 2009, 4, e5953. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nat. Cell Biol. 2008, 455, 1061–1068. [CrossRef]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.-G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.L.; Robel, S.; Cuddapah, V.A.; Robert, S.; Buckingham, S.C.; Kahle, K.T.; Sontheimer, H. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 2015, 63, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Conti, L.; Palma, E.; Roseti, C.; Lauro, C.; Cipriani, R.; de Groot, M.; Aronica, E.; Limatola, C. Anomalous levels of Cl− transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia 2011, 52, 1635–1644. [Google Scholar] [CrossRef]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Labussiere, M.; Cresto, N.; Dieme, M.-J.; Baulac, M.; Duyckaerts, C.; et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014, 6, 244ra89. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Deeb, T.Z.; Walker, J.A.; Davies, P.A.; Moss, S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor–mediated currents. Nat. Neurosci. 2011, 14, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Blanchart, A.; Fernando, R.; Häring, M.; Assaife-Lopes, N.; Romanov, R.A.; Andäng, M.; Harkany, T.; Ernfors, P. Endogenous GABAA receptor activity suppresses glioma growth. Oncogene 2016, 36, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.M.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal Activity Promotes Oligodendrogenesis and Adaptive Myelination in the Mammalian Brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Lin, C.-C.J.; Hatcher, A.; Lozzi, B.; Kong, K.; Huang-Hobbs, E.; Cheng, Y.-T.; Beechar, V.B.; Zhu, W.; Zhang, Y.; et al. PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nat. Cell Biol. 2020, 578, 166–171. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; D’Ambrosio, R.; Janigro, D. Heterogeneity of Astrocyte Resting Membrane Potentials and Intercellular Coupling Revealed by Whole-Cell and Gramicidin-Perforated Patch Recordings from Cultured Neocortical and Hippocampal Slice Astrocytes. J. Neurosci. 1997, 17, 6850–6863. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, M.; Hartley, M.; Heinemann, S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science 1991, 252, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Sommer, B.; Köhler, M.; Sprengel, R.; Seeburg, P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Maas, S.; Patt, S.; Schrey, M.; Rich, A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. USA 2001, 98, 14687–14692. [Google Scholar] [CrossRef] [Green Version]
- Oakes, E.; Anderson, A.; Cohen-Gadol, A.; Hundley, H.A. Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to Glutamate Receptor Subunit B Pre-mRNA Inhibits RNA Editing in Glioblastoma. J. Biol. Chem. 2017, 292, 4326–4335. [Google Scholar] [CrossRef] [Green Version]
- Monier, A.; Adle-Biassette, H.; Delezoide, A.-L.; Evrard, P.; Gressens, P.; Verney, C. Entry and Distribution of Microglial Cells in Human Embryonic and Fetal Cerebral Cortex. J. Neuropathol. Exp. Neurol. 2007, 66, 372–382. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.-D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liston, C.; Cichon, J.M.; Jeanneteau, F.; Jia, Z.; Chao, M.V.; Gan, W.-B. Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat. Neurosci. 2013, 16, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.W.H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, A.W.I.Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.-H.; Park, E.; You, B.; Jung, Y.; Park, A.-R.; Park, S.G.; Lee, J.-R. Neuronal Synapse Formation Induced by Microglia and Interleukin 10. PLoS ONE 2013, 8, e81218. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Xin, Y.-R.; Jiang, J.-X.; Hu, Y.; Pan, J.-P.; Mi, X.-N.; Gao, Q.; Xiao, F.; Zhang, W.; Luo, H.-M. The Immune System Drives Synapse Loss during Lipopolysaccharide-Induced Learning and Memory Impairment in Mice. Front. Aging Neurosci. 2019, 11, 279. [Google Scholar] [CrossRef]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef]
- Sarkar, S.; Döring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, X.; Dunn, J.F. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Ann. Neurosci. 2013, 20, 154. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Jopson, T.; Paladini, M.-S.; Liu, S.; West, B.; Gupta, N.; Rosi, S. Colony-stimulating factor 1 receptor blockade prevents fractionated whole-brain irradiation-induced memory deficits. J. Neuroinflamm. 2016, 13, 215. [Google Scholar] [CrossRef] [Green Version]
- Acharya, M.M.; Green, K.N.; Allen, B.D.; Najafi, A.R.; Syage, A.; Minasyan, H.; Le, M.T.; Kawashita, T.; Giedzinski, E.; Parihar, V.K.; et al. Elimination of microglia improves cognitive function following cranial irradiation. Sci. Rep. 2016, 6, 31545. [Google Scholar] [CrossRef]
- Feng, X.; Liu, S.; Chen, D.; Rosi, S.; Gupta, N. Rescue of cognitive function following fractionated brain irradiation in a novel preclinical glioma model. eLife 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Kokkosis, A.G.; Tsirka, S.E. Neuroimmune mechanisms and sex/gender-dependent effects in the pathophysiology of mental disorders. J. Pharmacol. Exp. Ther. 2020, 375, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, J.L.; Burns, C.M.B.; Wellman, C.L. Differential effects of stress on microglial cell activation in male and female medial prefrontal cortex. Brain Behav. Immun. 2016, 52, 88–97. [Google Scholar] [CrossRef]
- Sarvari, M.; Hrabovszky, E.; Kalló, I.; Solymosi, N.; Tóth, K.; Likó, I.; Liposits, Z. Estrogens regulate neuroinflammatory genes via estrogen receptors alpha and beta in the frontal cortex of middle-aged female rats. J. Neuroinflamm. 2011, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turaga, S.M.; Silver, D.J.; Bayik, D.; Paouri, E.; Peng, S.; Lauko, A.; Alban, T.J.; Borjini, N.; Stanko, S.; Naik, U.P.; et al. JAM-A functions as a female microglial tumor suppressor in glioblastoma. Neuro-Oncology 2020, 761445. [Google Scholar] [CrossRef]
- Gittleman, H.; Ostrom, Q.T.; Stetson, L.C.; Waite, K.; Hodges, T.R.; Wright, C.H.; Wright, J.; Rubin, J.B.; E Berens, M.; Lathia, J.; et al. Sex is an important prognostic factor for glioblastoma but not for nonglioblastoma. Neuro-Oncol. Pract. 2019, 6, 451–462. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Rubin, J.B.; Lathia, J.D.; Berens, M.E.; Barnholtz-Sloan, J.S. Females have the survival advantage in glioblastoma. Neuro-Oncology 2018, 20, 576–577. [Google Scholar] [CrossRef]
- Toonen, J.A.; Solga, A.C.; Ma, Y.; Gutmann, D.H. Estrogen activation of microglia underlies the sexually dimorphic differences in Nf1 optic glioma–induced retinal pathology. J. Exp. Med. 2016, 214, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Bayik, D.; Zhou, Y.; Park, C.; Hong, C.; Vail, D.; Silver, D.J.; Lauko, A.; Roversi, G.; Watson, D.C.; Lo, A.; et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discov. 2020, 10, 1210–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, C.C.; Gordon, P.M.K.; Liu, K.; Yang, R.; Sarkar, S.; Mirzaei, R.; Ahmad, S.T.; Hughes, M.L.; Yong, V.W.; Kelly, J.J. Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 2019, 10, 3129–3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660. [Google Scholar] [CrossRef]
- Caponegro, M.D. A Microglial Subset at the Tumor-Stroma Interface of Glioma. bioRxiv 2020, 76. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Glass, R.; Synowitz, M.; van Rooijen, N.; Kettenmann, H. Microglia Stimulate the Invasiveness of Glioma Cells by Increasing the Activity of Metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kalin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Burguillos, M.A.; Osman, A.M.; Frijhoff, J.; Carrillo-Jiménez, A.; Kanatani, S.; Augsten, M.; Saidi, D.; Rodhe, J.; Kavanagh, E.; et al. Glioma-induced inhibition of caspase-3 in microglia promotes a tumor-supportive phenotype. Nat. Immunol. 2016, 17, 1282–1290. [Google Scholar] [CrossRef]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; MacSwords, J. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Yong, V. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ke, S.Q.; Huang, Z.; A Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xu, Y.; Sun, J.; Chen, W.; Zhao, L.; Ma, C.; Wang, Q.; Sun, J.; Huang, B.; Zhang, Y.; et al. M2-like tumor-associated macrophages drive vasculogenic mimicry through amplification of IL-6 expression in glioma cells. Oncotarget 2016, 8, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro-Oncology 2017, 19, 1435–1446. [Google Scholar] [CrossRef] [Green Version]
- Alcoreza, O.; Tewari, B.P.; Bouslog, A.; Savoia, A.; Sontheimer, H.; Campbell, S.L. Sulfasalazine decreases mouse cortical hyperexcitability. Epilepsia 2019, 60, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Savaskan, N.; Heckel, A.; Hahnen, E.; Engelhorn, T.; Doerfler, A.; Ganslandt, O.; Nimsky, C.; Buchfelder, M.; Eyüpoglu, I.Y. Small interfering RNA–mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008, 14, 629–632. [Google Scholar] [CrossRef]
- Hatcher, A.; Yu, K.; Meyer, J.; Aiba, I.; Deneen, B.; Noebels, J.L. Pathogenesis of peritumoral hyperexcitability in an immunocompetent CRISPR-based glioblastoma model. J. Clin. Investig. 2020, 130, 2286–2300. [Google Scholar] [CrossRef]
- Ignarro, R.S.; Facchini, G.; Vieira, A.S.; de Melo, D.R.; Lopes-Cendes, I.; Castilho, R.F.; Rogerio, F. Sulfasalazine intensifies temozolomide cytotoxicity in human glioblastoma cells. Mol. Cell. Biochem. 2016, 418, 167–178. [Google Scholar] [CrossRef]
- Takeuchi, S.; Wada, K.; Nagatani, K.; Otani, N.; Osada, H.; Nawashiro, H. Sulfasalazine and temozolomide with radiation therapy for newly diagnosed glioblastoma. Neurol. India 2014, 62, 42–47. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Ye, X.; Chamberlain, M.; Mikkelsen, T.; Batchelor, T.; Desideri, S.; Piantadosi, S.; Fisher, J.; Fine, H.A. Talampanel With Standard Radiation and Temozolomide in Patients With Newly Diagnosed Glioblastoma: A Multicenter Phase II Trial. J. Clin. Oncol. 2009, 27, 4155–4161. [Google Scholar] [CrossRef] [Green Version]
- Izumoto, S.; Miyauchi, M.; Tasaki, T.; Okuda, T.; Nakagawa, N.; Nakano, N.; Kato, A.; Fujita, M. Seizures and Tumor Progression in Glioma Patients with Uncontrollable Epilepsy Treated with Perampanel. Anticancer Res. 2018, 38, 4361–4366. [Google Scholar] [CrossRef] [PubMed]
- Coppola, A.; Zarabla, A.; Maialetti, A.; Villani, V.; Koudriavtseva, T.; Russo, E.; Nozzolillo, A.; Sueri, C.; Belcastro, V.; Balestrini, S.; et al. Perampanel Confirms to Be Effective and Well-Tolerated as an Add-On Treatment in Patients With Brain Tumor-Related Epilepsy (PERADET Study). Front. Neurol. 2020, 11, 592. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.; Devi, N.A.; Fathima, K.H.; Nanjaiah, N.D. Activation of NMDA receptor of glutamate influences MMP-2 activity and proliferation of glioma cells. Neurol. Sci. 2013, 35, 823–829. [Google Scholar] [CrossRef]
- Yoon, W.-S.; Yeom, M.-Y.; Kang, E.-S.; Chung, Y.-A.; Chung, D.-S.; Jeun, S. Memantine Induces NMDAR1-Mediated Autophagic Cell Death in Malignant Glioma Cells. J. Korean Neurosurg. Soc. 2017, 60, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Maraka, S.; Groves, M.D.; Mammoser, A.G.; Melguizo-Gavilanes, I.; Conrad, C.A.; Tremont-Lukats, I.W.; Loghin, M.E.; Brien, B.J.O.; Puduvalli, V.K.; Sulman, E.P.; et al. Phase 1 lead-in to a phase 2 factorial study of temozolomide plus memantine, mefloquine, and metformin as postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer 2018, 125, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Colman, H.; de Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2015, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, H.; Raizer, J.; Walbert, T.; Plotkin, S.; Chamberlain, M.; Wong, E.; Puduvalli, V.; Reardon, D.; Iwamoto, F.; Johnson, B.; et al. ACTR-20. Initial Results of PLX108-08: An Open Label Phase 1B/2 Study of Orally Administred Pexidartinib (PLX3397) in Combination with Radiation Theraphy and Temozolomide in Patients with Newly Diagnpsed Gliobastoma. Neuro-Oncology 2016, 18, vi6. [Google Scholar] [CrossRef] [Green Version]
- Hampson, R.E.; Rogers, G.; Lynch, G.; Deadwyler, S.A. Facilitative Effects of the Ampakine CX516 on Short-Term Memory in Rats: Correlations with Hippocampal Neuronal Activity. J. Neurosci. 1998, 18, 2748–2763. [Google Scholar] [CrossRef] [Green Version]
- Hampson, R.E.; Rogers, G.; Lynch, G.; Deadwyler, S.A. Facilitative Effects of the Ampakine CX516 on Short-Term Memory in Rats: Enhancement of Delayed-Nonmatch-to-Sample Performance. J. Neurosci. 1998, 18, 2740–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingvar, M.; Ambros-Ingerson, J.; Davis, M.; Granger, R.; Kessler, M.; Rogers, G.A.; Schehr, R.S.; Lynch, G. Enhancement by an Ampakine of Memory Encoding in Humans. Exp. Neurol. 1997, 146, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radin, D.P.; Zhong, S.; Purcell, R.; Lippa, A. Acute ampakine treatment ameliorates age-related deficits in long-term potentiation. Biomed. Pharmacother. 2016, 84, 806–809. [Google Scholar] [CrossRef]
- Samartgis, J.R.; Schachte, L.; Hazi, A.; Crowe, S.F. Piracetam, an AMPAkine drug, facilitates memory consolidation in the day-old chick. Pharmacol. Biochem. Behav. 2012, 103, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Seese, R.R.; Le, A.A.; Wang, K.; Cox, C.D.; Lynch, G.; Gall, C.M. A TrkB agonist and ampakine rescue synaptic plasticity and multiple forms of memory in a mouse model of intellectual disability. Neurobiol. Dis. 2020, 134, 104604. [Google Scholar] [CrossRef]
- Simmons, D.A.; Rex, C.S.; Palmer, L.; Pandyarajan, V.; Fedulov, V.; Gall, C.M.; Lynch, G. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington’s disease knockin mice. Proc. Natl. Acad. Sci. USA 2009, 106, 4906–4911. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Balabhadrapatruni, S.; Masumura, C.; Darlington, C.L.; Smith, P.F. Effects of the putative cognitive-enhancing ampakine, CX717, on attention and object recognition memory. Curr. Alzheimer Res. 2011, 8, 876–882. [Google Scholar] [CrossRef]
- Kuhn, S.A.; van Landeghem, F.K.; Zacharias, R.; Färber, K.; Rappert, A.; Pavlovic, S.; Hoffmann, A.; Nolte, C.; Kettenmann, H. Microglia express GABA B receptors to modulate interleukin release. Mol. Cell. Neurosci. 2004, 25, 312–322. [Google Scholar] [CrossRef]
- Beppu, K.; Kosai, Y.; Kido, M.A.; Akimoto, N.; Mori, Y.; Kojima, Y.; Fujita, K.; Okuno, Y.; Yamakawa, Y.; Ifuku, M.; et al. Expression, subunit composition, and function of AMPA-type glutamate receptors are changed in activated microglia; possible contribution of GluA2 (GluR-B)-deficiency under pathological conditions. Glia 2013, 61, 881–891. [Google Scholar] [CrossRef]
- Hagino, Y.; Kariura, Y.; Manago, Y.; Amano, T.; Wang, B.; Sekiguchi, M.; Nishikawa, K.; Aoki, S.; Wada, K.; Noda, M. Heterogeneity and potentiation of AMPA type of glutamate receptors in rat cultured microglia. Glia 2004, 47, 68–77. [Google Scholar] [CrossRef]
- Noda, M.; Nakanishi, H.; Nabekura, J.; Akaike, N. AMPA–Kainate Subtypes of Glutamate Receptor in Rat Cerebral Microglia. J. Neurosci. 2000, 20, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Leak, R.K.; Hu, X. Neurotransmitter receptors on microglia. Stroke Vasc. Neurol. 2016, 1, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pocock, J.; Kettenmann, H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Nagarajah, R.; Banati, R.; Bennet, M. Glutamate induces directed chemotaxis of microglia. Eur. J. Neurosci. 2009, 29, 1108–1118. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radin, D.P.; Tsirka, S.E. Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment. Int. J. Mol. Sci. 2020, 21, 8476. https://doi.org/10.3390/ijms21228476
Radin DP, Tsirka SE. Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment. International Journal of Molecular Sciences. 2020; 21(22):8476. https://doi.org/10.3390/ijms21228476
Chicago/Turabian StyleRadin, Daniel P., and Stella E. Tsirka. 2020. "Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment" International Journal of Molecular Sciences 21, no. 22: 8476. https://doi.org/10.3390/ijms21228476
APA StyleRadin, D. P., & Tsirka, S. E. (2020). Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment. International Journal of Molecular Sciences, 21(22), 8476. https://doi.org/10.3390/ijms21228476