CD40/CD40L and Related Signaling Pathways in Cardiovascular Health and Disease—The Pros and Cons for Cardioprotection




Abstract
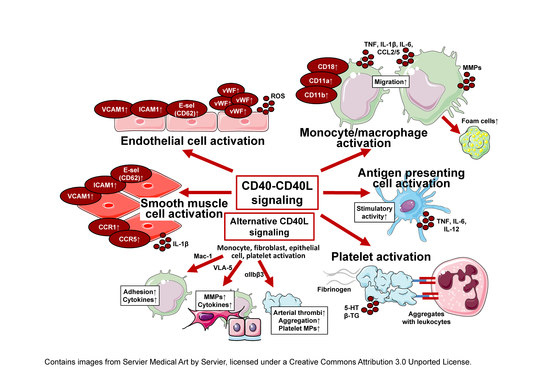
:
1. Introduction
2. Targeting Inflammation in Patients Decreases Cardiovascular Risk
3. Mechanistic Background of CD40–CD40L Signaling
4. CD40–CD40L Dyad in Cardiovascular Diseases
4.1. Dyad in Atherosclerosis, Evidence from Experimental Models
4.2. Dyad in Myocardial Infarction, Heart Failure
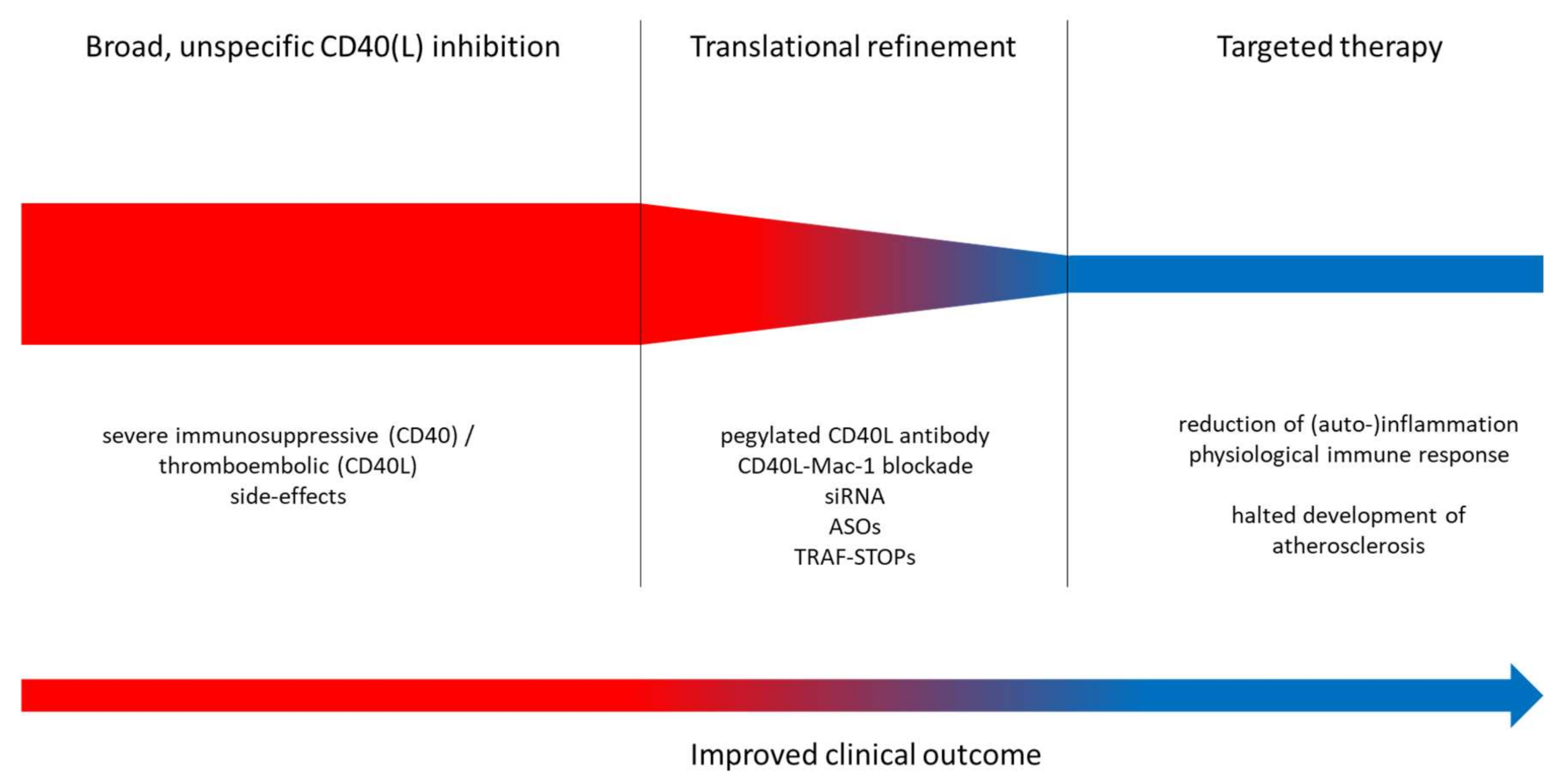
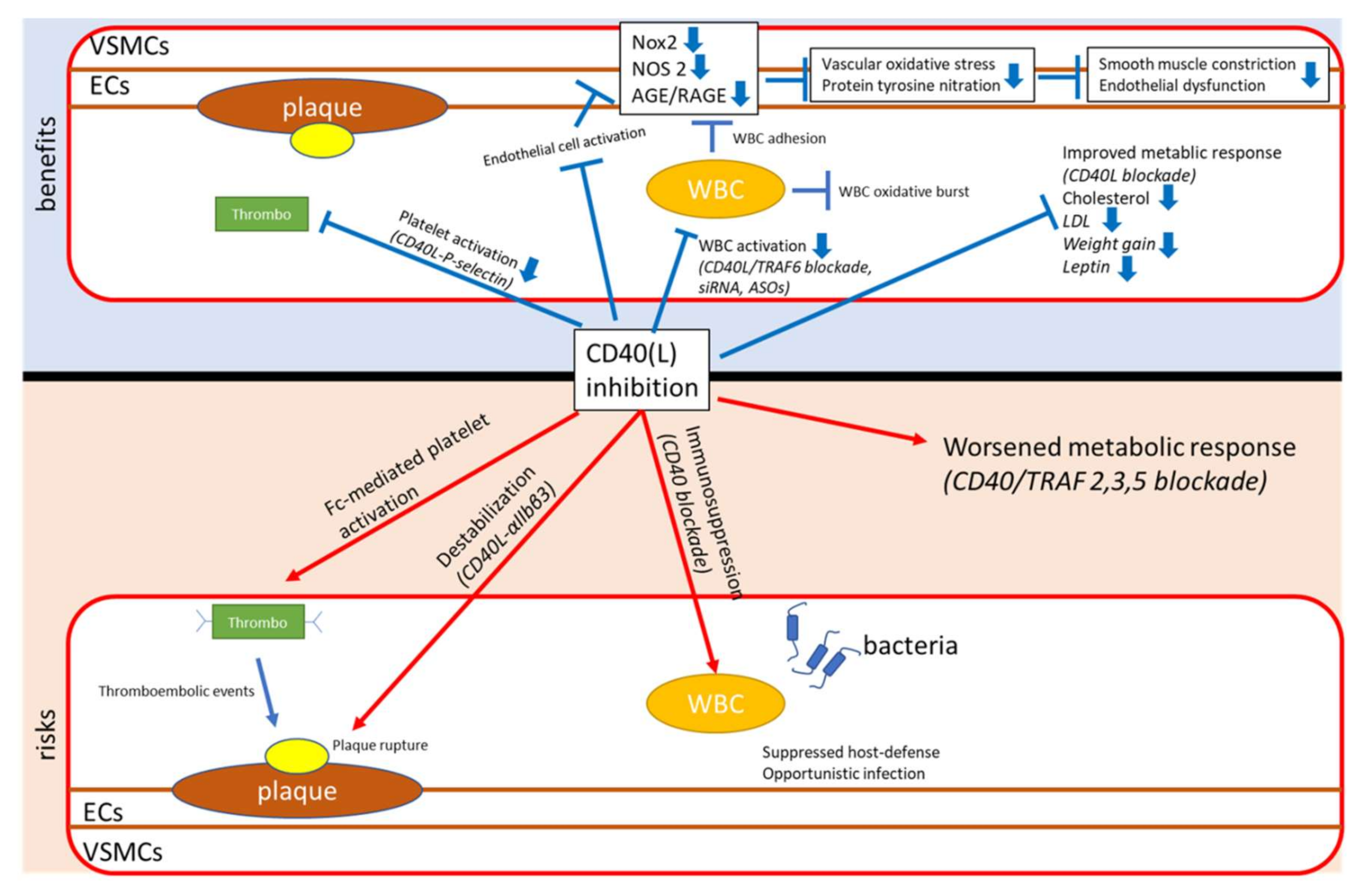
5. Traditional Approaches
5.1. General CD40(L) Blockade
5.1.1. CD40−/− Animals and a General CD40 Blockade
5.1.2. CD40L−/− Animals and a General CD40L Blockade
5.2. Novel Approaches Targeting the CD40–CD40L Dyad
5.2.1. TRAF-STOPS
5.2.2. Silencing RNA
5.2.3. Antisense Oligonucleotides
5.2.4. Specific Blockade of the CD40L-Mac-1 Interaction
6. Clinical Trials
6.1. Blocking CD40L
6.2. Blocking CD40
7. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Aday, A.W.; Ridker, P.M. Targeting Residual Inflammatory Risk: A Shifting Paradigm for Atherosclerotic Disease. Front. Cardiovasc. Med. 2019, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef]
- Libby, P.; Nahrendorf, M.; Swirski, F.K. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum”. J. Am. Coll. Cardiol. 2016, 67, 1091–1103. [Google Scholar] [CrossRef]
- de Winther, M.P.J.; Lutgens, E. The Link between Hematopoiesis and Atherosclerosis. N. Engl. J. Med. 2019, 380, 1869–1871. [Google Scholar] [CrossRef]
- Peikert, A.; Kaier, K.; Merz, J.; Manhart, L.; Schafer, I.; Hilgendorf, I.; Hehn, P.; Wolf, D.; Willecke, F.; Sheng, X.; et al. Residual inflammatory risk in coronary heart disease: Incidence of elevated high-sensitive CRP in a real-world cohort. Clin. Res. Cardiol. 2020, 109, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Conen, D.; Ridker, P.M. Clinical significance of high-sensitivity C-reactive protein in cardiovascular disease. Biomark. Med. 2007, 1, 229–241. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E.; Pravastatin or Atorvastatin, E.; Infection Therapy-Thrombolysis in Myocardial Infarction, I. C-reactive protein levels and outcomes after statin therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef]
- Klarin, D.; Zhu, Q.M.; Emdin, C.A.; Chaffin, M.; Horner, S.; McMillan, B.J.; Leed, A.; Weale, M.E.; Spencer, C.C.A.; Aguet, F.; et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat. Genet. 2017, 49, 1392–1397. [Google Scholar] [CrossRef]
- Howson, J.M.M.; Zhao, W.; Barnes, D.R.; Ho, W.K.; Young, R.; Paul, D.S.; Waite, L.L.; Freitag, D.F.; Fauman, E.B.; Salfati, E.L.; et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat. Genet. 2017, 49, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Soltesz, P.; Kerekes, G.; Der, H.; Szucs, G.; Szanto, S.; Kiss, E.; Bodolay, E.; Zeher, M.; Timar, O.; Szodoray, P.; et al. Comparative assessment of vascular function in autoimmune rheumatic diseases: Considerations of prevention and treatment. Autoimmun. Rev. 2011, 10, 416–425. [Google Scholar] [CrossRef]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spano, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012. [Google Scholar] [CrossRef] [PubMed]
- Vena, G.A.; Vestita, M.; Cassano, N. Psoriasis and cardiovascular disease. Dermatol. Ther. 2010, 23, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Hak, A.E.; Karlson, E.W.; Feskanich, D.; Stampfer, M.J.; Costenbader, K.H. Systemic lupus erythematosus and the risk of cardiovascular disease: Results from the nurses’ health study. Arthritis Rheum. 2009, 61, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaptoge, S.; Seshasai, S.R.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; Rumley, A.; Lowe, G.D.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases--the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M. A Test in Context: High-Sensitivity C-Reactive Protein. J. Am. Coll. Cardiol. 2016, 67, 712–723. [Google Scholar] [CrossRef]
- Emerging Risk Factors, C.; Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Barron, E.; Lara, J.; White, M.; Mathers, J.C. Blood-borne biomarkers of mortality risk: Systematic review of cohort studies. PLoS ONE 2015, 10, e0127550. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Group, C.T. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T.; Group, C.P.I. Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: A phase IIb randomized, placebo-controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tassell, B.W.; Toldo, S.; Mezzaroma, E.; Abbate, A. Targeting interleukin-1 in heart disease. Circulation 2013, 128, 1910–1923. [Google Scholar] [CrossRef] [Green Version]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2012, 51 (Suppl. S5), v3–v11. [Google Scholar] [CrossRef] [Green Version]
- Pasceri, V.; Yeh, E.T. A tale of two diseases: Atherosclerosis and rheumatoid arthritis. Circulation 1999, 100, 2124–2126. [Google Scholar] [CrossRef] [Green Version]
- Di Cesare, A.; Di Meglio, P.; Nestle, F.O. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J. Investig. Derm. 2009, 129, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Papp, K.A.; Leonardi, C.; Menter, A.; Ortonne, J.P.; Krueger, J.G.; Kricorian, G.; Aras, G.; Li, J.; Russell, C.B.; Thompson, E.H.; et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 2012, 366, 1181–1189. [Google Scholar] [CrossRef]
- Crispin, J.C.; Tsokos, G.C. IL-17 in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010, 943254. [Google Scholar] [CrossRef] [Green Version]
- Opstal, T.S.J.; Hoogeveen, R.M.; Fiolet, A.T.L.; Silvis, M.J.M.; The, S.H.K.; Bax, W.A.; de Kleijn, D.P.V.; Mosterd, A.; Stroes, E.S.G.; Cornel, J.H. Colchicine Attenuates Inflammation Beyond the Inflammasome in Chronic Coronary Artery Disease: A LoDoCo2 Proteomic Substudy. Circulation 2020. [Google Scholar] [CrossRef]
- Soehnlein, O.; Schmeisser, A.; Cicha, I.; Reiss, C.; Ulbrich, H.; Lindbom, L.; Daniel, W.G.; Garlichs, C.D. ACE inhibition lowers angiotensin-II-induced monocyte adhesion to HUVEC by reduction of p65 translocation and AT 1 expression. J. Vasc. Res. 2005, 42, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Caspritz, G.; Alpermann, H.G.; Schleyerbach, R. Influence of the new angiotensin converting enzyme inhibitor ramipril on several models of acute inflammation and the adjuvant arthritis in the rat. Arzneim. Forsch. 1986, 36, 1605–1608. [Google Scholar]
- Suzuki, J.; Iwai, M.; Mogi, M.; Oshita, A.; Yoshii, T.; Higaki, J.; Horiuchi, M. Eplerenone with valsartan effectively reduces atherosclerotic lesion by attenuation of oxidative stress and inflammation. Arter. Thromb. Vasc. Biol. 2006, 26, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Iwai, M.; Nakagami, H.; Li, Z.; Chen, R.; Suzuki, J.; Akishita, M.; de Gasparo, M.; Horiuchi, M. Roles of angiotensin II type 2 receptor stimulation associated with selective angiotensin II type 1 receptor blockade with valsartan in the improvement of inflammation-induced vascular injury. Circulation 2001, 104, 2716–2721. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Patel, T.N.; Shishehbor, M.H.; Bhatt, D.L. A review of high-dose statin therapy: Targeting cholesterol and inflammation in atherosclerosis. Eur. Heart J. 2007, 28, 664–672. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Eikelboom, J.W.; Connolly, S.J.; Bosch, J.; Dagenais, G.R.; Hart, R.G.; Shestakovska, O.; Diaz, R.; Alings, M.; Lonn, E.M.; Anand, S.S.; et al. Rivaroxaban with or without Aspirin in Stable Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 1319–1330. [Google Scholar] [CrossRef]
- Buller, H.R.; Bethune, C.; Bhanot, S.; Gailani, D.; Monia, B.P.; Raskob, G.E.; Segers, A.; Verhamme, P.; Weitz, J.I.; Investigators, F.-A.T. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N. Engl. J. Med. 2015, 372, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N. New insights into the mechanisms of venous thrombosis. J. Clin. Investig. 2012, 122, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Michel, N.A.; Zirlik, A.; Wolf, D. CD40L and Its Receptors in Atherothrombosis-An Update. Front. Cardiovasc. Med. 2017, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Paulie, S.; Koho, H.; Ben-Aissa, H.; Hansson, Y.; Lundblad, M.L.; Perlmann, P. Monoclonal antibodies to antigens associated with transitional cell carcinoma of the human urinary bladder. II. Identification of the cellular target structures by immunoprecipitation and SDS-PAGE analysis. Cancer Immunol. Immunother. 1984, 17, 173–179. [Google Scholar] [CrossRef]
- Yazdani, R.; Fekrvand, S.; Shahkarami, S.; Azizi, G.; Moazzami, B.; Abolhassani, H.; Aghamohammadi, A. The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin. Immunol. 2019, 198, 19–30. [Google Scholar] [CrossRef]
- de la Morena, M.T. Clinical Phenotypes of Hyper-IgM Syndromes. J. Allergy Clin. Immunol. Pract. 2016, 4, 1023–1036. [Google Scholar] [CrossRef]
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug Deliv. Rev. 2019, 141, 92–103. [Google Scholar] [CrossRef]
- Zhang, J.; Varas, F.; Stadtfeld, M.; Heck, S.; Faust, N.; Graf, T. CD41-YFP mice allow in vivo labeling of megakaryocytic cells and reveal a subset of platelets hyperreactive to thrombin stimulation. Exp. Hematol. 2007, 35, 490–499. [Google Scholar] [CrossRef]
- Henn, V.; Slupsky, J.R.; Grafe, M.; Anagnostopoulos, I.; Forster, R.; Muller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef]
- Erez, O.; Romero, R.; Hoppensteadt, D.; Fareed, J.; Chaiworapongsa, T.; Kusanovic, J.P.; Mazaki-Tovi, S.; Gotsch, F.; Than, N.G.; Vaisbuch, E.; et al. Premature labor: A state of platelet activation? J. Perinat. Med. 2008, 36, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Antoniades, C.; Bakogiannis, C.; Tousoulis, D.; Antonopoulos, A.S.; Stefanadis, C. The CD40/CD40 ligand system: Linking inflammation with atherothrombosis. J. Am. Coll. Cardiol. 2009, 54, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Mach, F.; Schonbeck, U.; Libby, P. CD40 signaling in vascular cells: A key role in atherosclerosis? Atherosclerosis 1998, 137, S89–S95. [Google Scholar] [CrossRef]
- Lievens, D.; Eijgelaar, W.J.; Biessen, E.A.; Daemen, M.J.; Lutgens, E. The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb. Haemost. 2009, 102, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Durie, F.H.; Fava, R.A.; Foy, T.M.; Aruffo, A.; Ledbetter, J.A.; Noelle, R.J. Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science 1993, 261, 1328–1330. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Elwood, E.T.; Alexander, D.Z.; Ritchie, S.C.; Hendrix, R.; Tucker-Burden, C.; Cho, H.R.; Aruffo, A.; Hollenbaugh, D.; Linsley, P.S.; et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature 1996, 381, 434–438. [Google Scholar] [CrossRef]
- Homann, D.; Jahreis, A.; Wolfe, T.; Hughes, A.; Coon, B.; van Stipdonk, M.J.; Prilliman, K.R.; Schoenberger, S.P.; von Herrath, M.G. CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity 2002, 16, 403–415. [Google Scholar] [CrossRef] [Green Version]
- Seales, E.C.; Shaikh, F.M.; Woodard-Grice, A.V.; Aggarwal, P.; McBrayer, A.C.; Hennessy, K.M.; Bellis, S.L. A protein kinase C/Ras/ERK signaling pathway activates myeloid fibronectin receptors by altering beta1 integrin sialylation. J. Biol. Chem. 2005, 280, 37610–37615. [Google Scholar] [CrossRef] [Green Version]
- Zirlik, A.; Maier, C.; Gerdes, N.; MacFarlane, L.; Soosairajah, J.; Bavendiek, U.; Ahrens, I.; Ernst, S.; Bassler, N.; Patko, Z.; et al. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation 2007, 115, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- André, P.; Prasad, K.S.S.; Denis, C.V.; He, M.; Papalia, J.M.; Hynes, R.O.; Phillips, D.R.; Wagner, D.D. CD40L stabilizes arterial thrombi by a beta3 integrin--dependent mechanism. Nat. Med. 2002, 8, 247–252. [Google Scholar] [CrossRef]
- Prasad, K.S.; Andre, P.; He, M.; Bao, M.; Manganello, J.; Phillips, D.R. Soluble CD40 ligand induces beta3 integrin tyrosine phosphorylation and triggers platelet activation by outside-in signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 12367–12371. [Google Scholar] [CrossRef] [Green Version]
- Li, D.K.; Wang, W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol. Lett. 2020, 20, 176. [Google Scholar] [CrossRef]
- Bosmans, L.A.; Bosch, L.; Kusters, P.J.H.; Lutgens, E.; Seijkens, T.T.P. The CD40-CD40L Dyad as Immunotherapeutic Target in Cardiovascular Disease. J. Cardiovasc. Transl. Res. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoni, A.; Alabiso, O.; Galetto, A.S.; Baldanzi, G. Integrins in T Cell Physiology. Int. J. Mol. Sci. 2018, 19, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korniluk, A.; Kemona, H.; Dymicka-Piekarska, V. Multifunctional CD40L: Pro- and anti-neoplastic activity. Tumour Biol. 2014, 35, 9447–9457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muto, S.; Vetvicka, V.; Ross, G.D. CR3 (CD11b/CD18) expressed by cytotoxic T cells and natural killer cells is upregulated in a manner similar to neutrophil CR3 following stimulation with various activating agents. J. Clin. Immunol. 1993, 13, 175–184. [Google Scholar] [CrossRef]
- Engel, D.; Seijkens, T.; Poggi, M.; Sanati, M.; Thevissen, L.; Beckers, L.; Wijnands, E.; Lievens, D.; Lutgens, E. The immunobiology of CD154-CD40-TRAF interactions in atherosclerosis. Semin. Immunol. 2009, 21, 308–312. [Google Scholar] [CrossRef]
- Arron, J.R.; Pewzner-Jung, Y.; Walsh, M.C.; Kobayashi, T.; Choi, Y. Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J. Exp. Med. 2002, 196, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, S.R.; Angelini, F.; Bacharier, L.B.; Blom, A.M.; Mizoguchi, E.; Fujiwara, H.; Plebani, A.; Notarangelo, L.D.; Dahlback, B.; Tsitsikov, E.; et al. C4b-binding protein (C4BP) activates B cells through the CD40 receptor. Immunity 2003, 18, 837–848. [Google Scholar] [CrossRef]
- Wang, Y.; Kelly, C.G.; Karttunen, J.T.; Whittall, T.; Lehner, P.J.; Duncan, L.; MacAry, P.; Younson, J.S.; Singh, M.; Oehlmann, W.; et al. CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC-chemokines. Immunity 2001, 15, 971–983. [Google Scholar] [CrossRef] [Green Version]
- Andre, P.; Nannizzi-Alaimo, L.; Prasad, S.K.; Phillips, D.R. Platelet-derived CD40L: The switch-hitting player of cardiovascular disease. Circulation 2002, 106, 896–899. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, N.; Seijkens, T.; Lievens, D.; Kuijpers, M.J.; Winkels, H.; Projahn, D.; Hartwig, H.; Beckers, L.; Megens, R.T.; Boon, L.; et al. Platelet CD40 Exacerbates Atherosclerosis by Transcellular Activation of Endothelial Cells and Leukocytes. Arter. Thromb. Vasc. Biol. 2016, 36, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Popa, M.; Tahir, S.; Elrod, J.; Kim, S.H.; Leuschner, F.; Kessler, T.; Bugert, P.; Pohl, U.; Wagner, A.H.; Hecker, M. Role of CD40 and ADAMTS13 in von Willebrand factor-mediated endothelial cell-platelet-monocyte interaction. Proc. Natl. Acad. Sci. USA 2018, 115, E5556–E5565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotowicz, K.; Dixon, G.L.; Klein, N.J.; Peters, M.J.; Callard, R.E. Biological function of CD40 on human endothelial cells: Costimulation with CD40 ligand and interleukin-4 selectively induces expression of vascular cell adhesion molecule-1 and P-selectin resulting in preferential adhesion of lymphocytes. Immunology 2000, 100, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Fu, H.; Ren, L.; Wang, H.; Guo, W. Soluble CD40 ligand promotes macrophage foam cell formation in the etiology of atherosclerosis. Cardiology 2015, 131, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Donners, M.M.; Beckers, L.; Lievens, D.; Munnix, I.; Heemskerk, J.; Janssen, B.J.; Wijnands, E.; Cleutjens, J.; Zernecke, A.; Weber, C.; et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood 2008, 111, 4596–4604. [Google Scholar] [CrossRef] [Green Version]
- Lutgens, E.; Lievens, D.; Beckers, L.; Wijnands, E.; Soehnlein, O.; Zernecke, A.; Seijkens, T.; Engel, D.; Cleutjens, J.; Keller, A.M.; et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J. Exp. Med. 2010, 207, 391–404. [Google Scholar] [CrossRef]
- Chand Dakal, T.; Dhabhai, B.; Agarwal, D.; Gupta, R.; Nagda, G.; Meena, A.R.; Dhakar, R.; Menon, A.; Mathur, R.; Mona, A.; et al. Mechanistic basis of co-stimulatory CD40-CD40L ligation mediated regulation of immune responses in cancer and autoimmune disorders. Immunobiology 2020, 225, 151899. [Google Scholar] [CrossRef]
- Hausding, M.; Jurk, K.; Daub, S.; Kroller-Schon, S.; Stein, J.; Schwenk, M.; Oelze, M.; Mikhed, Y.; Kerahrodi, J.G.; Kossmann, S.; et al. CD40L contributes to angiotensin II-induced pro-thrombotic state, vascular inflammation, oxidative stress and endothelial dysfunction. Basic Res. Cardiol. 2013, 108, 386. [Google Scholar] [CrossRef]
- Steven, S.; Dib, M.; Hausding, M.; Kashani, F.; Oelze, M.; Kroller-Schon, S.; Hanf, A.; Daub, S.; Roohani, S.; Gramlich, Y.; et al. CD40L controls obesity-associated vascular inflammation, oxidative stress, and endothelial dysfunction in high fat diet-treated and db/db mice. Cardiovasc. Res. 2018, 114, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Sultan, C.S.; Weitnauer, M.; Turinsky, M.; Kessler, T.; Brune, M.; Gleissner, C.A.; Leuschner, F.; Wagner, A.H.; Hecker, M. Functional association of a CD40 gene single-nucleotide polymorphism with the pathogenesis of coronary heart disease. Cardiovasc. Res. 2020, 116, 1214–1225. [Google Scholar] [CrossRef]
- Heeschen, C.; Dimmeler, S.; Hamm, C.W.; van den Brand, M.J.; Boersma, E.; Zeiher, A.M.; Simoons, M.L.; Investigators, C.S. Soluble CD40 ligand in acute coronary syndromes. N. Engl. J. Med. 2003, 348, 1104–1111. [Google Scholar] [CrossRef] [Green Version]
- Pusuroglu, H.; Akgul, O.; Erturk, M.; Uyarel, H.; Bulut, U.; Akkaya, E.; Buturak, A.; Surgit, O.; Fuat, A.; Cetin, M.; et al. Predictive value of elevated soluble CD40 ligand in patients undergoing primary angioplasty for ST-segment elevation myocardial infarction. Coron. Artery Dis. 2014, 25, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Aukrust, P.; Yndestad, A.; Otterdal, K.; Froland, S.S.; Dickstein, K.; Kjekshus, J.; Gullestad, L.; Damas, J.K. Soluble CD40 ligand in acute and chronic heart failure. Eur. Heart J. 2005, 26, 1101–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabe, T.; Naka, T.; Yoshida, K.; Tanaka, T.; Fujiwara, H.; Suematsu, S.; Yoshida, N.; Kishimoto, T.; Kikutani, H. The immune responses in CD40-deficient mice: Impaired immunoglobulin class switching and germinal center formation. Immunity 1994, 1, 167–178. [Google Scholar] [CrossRef]
- Ingersoll, S.B.; Langer, F.; Walker, J.M.; Meyer, T.; Robson, T.; Amaya, M.; Desai, H.; Francis, J.L.; Amirkhosravi, A. Deficiencies in the CD40 and CD154 receptor-ligand system reduce experimental lung metastasis. Clin. Exp. Metastasis 2009, 26, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Perper, S.J.; Westmoreland, S.V.; Karman, J.; Twomey, R.; Seagal, J.; Wang, R.; McRae, B.L.; Clarke, S.H. Treatment with a CD40 Antagonist Antibody Reverses Severe Proteinuria and Loss of Saliva Production and Restores Glomerular Morphology in Murine Systemic Lupus Erythematosus. J. Immunol. 2019, 203, 58–75. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Andrews, D.; Colvin, R.B.; Sachs, D.H.; Cosimi, A.B. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat. Med. 2000, 6, 114. [Google Scholar] [CrossRef]
- Zarzycka, B.; Seijkens, T.; Nabuurs, S.B.; Ritschel, T.; Grommes, J.; Soehnlein, O.; Schrijver, R.; van Tiel, C.M.; Hackeng, T.M.; Weber, C.; et al. Discovery of small molecule CD40-TRAF6 inhibitors. J. Chem. Inf. Model. 2015, 55, 294–307. [Google Scholar] [CrossRef]
- Chatzigeorgiou, A.; Seijkens, T.; Zarzycka, B.; Engel, D.; Poggi, M.; van den Berg, S.; Soehnlein, O.; Winkels, H.; Beckers, L. Blocking CD40-TRAF6 signaling is a therapeutic target in obesity-associated insulin resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 2686–2691. [Google Scholar] [CrossRef] [Green Version]
- Bosch, L.; de Haan, J.; Seijkens, T.; van Tiel, C.; Brans, M.; Pasterkamp, G.; Lutgens, E.; de Jager, S. Small molecule-mediated inhibition of CD40-TRAF6 reduces adverse cardiac remodelling in pressure overload induced heart failure. Int. J. Cardiol. 2019, 279, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Seijkens, T.T.P.; van Tiel, C.M.; Kusters, P.J.H.; Atzler, D.; Soehnlein, O.; Zarzycka, B.; Aarts, S.; Lameijer, M.; Gijbels, M.J.; Beckers, L.; et al. Targeting CD40-Induced TRAF6 Signaling in Macrophages Reduces Atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 527–542. [Google Scholar] [CrossRef]
- Lameijer, M.; Binderup, T.; van Leent, M.M.T.; Senders, M.L.; Fay, F.; Malkus, J.; Sanchez-Gaytan, B.L.; Teunissen, A.J.P.; Karakatsanis, N.; Robson, P.; et al. Efficacy and safety assessment of a TRAF6-targeted nanoimmunotherapy in atherosclerotic mice and non-human primates. Nat. Biomed. Eng. 2018, 2, 279–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, M.W.; Moore, K.J. MicroRNA Regulation of Atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreou, I.; Sun, X.; Stone, P.H.; Edelman, E.R.; Feinberg, M.W. miRNAs in atherosclerotic plaque initiation, progression, and rupture. Trends Mol. Med. 2015, 21, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Hueso, M.; De Ramon, L.; Navarro, E.; Ripoll, E.; Cruzado, J.M.; Grinyo, J.M.; Torras, J. Datasets for the validation of the “in vivo” siRNA-silencing of CD40 and for the detection of new markers of atherosclerosis progression in ApoE-deficient mice. Data Brief. 2016, 9, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hueso, M.; De Ramon, L.; Navarro, E.; Ripoll, E.; Cruzado, J.M.; Grinyo, J.M.; Torras, J. Silencing of CD40 in vivo reduces progression of experimental atherogenesis through an NF-kappaB/miR-125b axis and reveals new potential mediators in the pathogenesis of atherosclerosis. Atherosclerosis 2016, 255, 80–89. [Google Scholar] [CrossRef]
- Hueso, M.; Torras, J.; Carrera, M.; Vidal, A.; Navarro, E.; Grinyo, J. Chronic Kidney Disease is associated with an increase of Intimal Dendritic cells in a comparative autopsy study. J. Inflamm. 2015, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Hueso, M.; Casas, A.; Mallen, A.; de Ramon, L.; Bolanos, N.; Varela, C.; Cruzado, J.M.; Torras, J.; Navarro, E. The double edge of anti-CD40 siRNA therapy: It increases renal microcapillar density but favours the generation of an inflammatory milieu in the kidneys of ApoE (-/-) mice. J. Inflamm. 2019, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Wagner, A.H.; Fankhaenel, S.; Stojanovic, T.; Schweyer, S.; Panzner, S.; Hecker, M. CD40 antisense oligonucleotide inhibition of trinitrobenzene sulphonic acid induced rat colitis. Gut 2005, 54, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Arranz, A.; Reinsch, C.; Papadakis, K.A.; Dieckmann, A.; Rauchhaus, U.; Androulidaki, A.; Zacharioudaki, V.; Margioris, A.N.; Tsatsanis, C.; Panzner, S. Treatment of experimental murine colitis with CD40 antisense oligonucleotides delivered in amphoteric liposomes. J. Control. Release 2013, 165, 163–172. [Google Scholar] [CrossRef]
- Donner, A.J.; Yeh, S.T.; Hung, G.; Graham, M.J.; Crooke, R.M.; Mullick, A.E. CD40 Generation 2.5 Antisense Oligonucleotide Treatment Attenuates Doxorubicin-induced Nephropathy and Kidney Inflammation. Mol. Ther. Nucleic Acids 2015, 4, e265. [Google Scholar] [CrossRef]
- Wolf, D.; Hohmann, J.D.; Wiedemann, A.; Bledzka, K.; Blankenbach, H.; Marchini, T.; Gutte, K.; Zeschky, K.; Bassler, N.; Hoppe, N.; et al. Binding of CD40L to Mac-1’s I-domain involves the EQLKKSKTL motif and mediates leukocyte recruitment and atherosclerosis--but does not affect immunity and thrombosis in mice. Circ. Res. 2011, 109, 1269–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Anto-Michel, N.; Blankenbach, H.; Wiedemann, A.; Buscher, K.; Hohmann, J.D.; Lim, B.; Bauml, M.; Marki, A.; Mauler, M.; et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat. Commun. 2018, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Boumpas, D.T.; Furie, R.; Manzi, S.; Illei, G.G.; Wallace, D.J.; Balow, J.E.; Vaishnaw, A.; Group, B.G.L.N.T. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003, 48, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Robles-Carrillo, L.; Meyer, T.; Hatfield, M.; Desai, H.; Davila, M.; Langer, F.; Amaya, M.; Garber, E.; Francis, J.L.; Hsu, Y.M.; et al. Anti-CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J. Immunol. 2010, 185, 1577–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shock, A.; Burkly, L.; Wakefield, I.; Peters, C.; Garber, E.; Ferrant, J.; Taylor, F.R.; Su, L.; Hsu, Y.M.; Hutto, D.; et al. CDP7657, an anti-CD40L antibody lacking an Fc domain, inhibits CD40L-dependent immune responses without thrombotic complications: An in vivo study. Arthritis Res. Ther. 2015, 17, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, C.; Colman, P.J.; Ranger, A.M.; Burkly, L.C.; Johnston, G.I.; Otoul, C.; Stach, C.; Zamacona, M.; Dorner, T.; Urowitz, M.; et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann. Rheum. Dis. 2017, 76, 1837–1844. [Google Scholar] [CrossRef]
- Tocoian, A.; Buchan, P.; Kirby, H.; Soranson, J.; Zamacona, M.; Walley, R.; Mitchell, N.; Esfandiari, E.; Wagner, F.; Oliver, R. First-in-human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti-CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus 2015, 24, 1045–1056. [Google Scholar] [CrossRef]
- Kalunian, K.C.; Davis, J.C., Jr.; Merrill, J.T.; Totoritis, M.C.; Wofsy, D.; Group, I.-L.S. Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46, 3251–3258. [Google Scholar] [CrossRef]
- Anil Kumar, M.S.; Papp, K.; Tainaka, R.; Valluri, U.; Wang, X.; Zhu, T.; Schwabe, C. Randomized, controlled study of bleselumab (ASKP1240) pharmacokinetics and safety in patients with moderate-to-severe plaque psoriasis. Biopharm. Drug Dispos. 2018, 39, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Albach, F.N.; Wagner, F.; Huser, A.; Igel, J.; Joseph, D.; Hilbert, J.; Schoelch, C.; Padula, S.J.; Steffgen, J. Safety, pharmacokinetics and pharmacodynamics of single rising doses of BI 655064, an antagonistic anti-CD40 antibody in healthy subjects: A potential novel treatment for autoimmune diseases. Eur. J. Clin. Pharm. 2018, 74, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Visvanathan, S.; Daniluk, S.; Ptaszynski, R.; Muller-Ladner, U.; Ramanujam, M.; Rosenstock, B.; Eleftheraki, A.G.; Vinisko, R.; Petrikova, A.; Kellner, H.; et al. Effects of BI 655064, an antagonistic anti-CD40 antibody, on clinical and biomarker variables in patients with active rheumatoid arthritis: A randomised, double-blind, placebo-controlled, phase IIa study. Ann. Rheum. Dis. 2019, 78, 754–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasran, A.; Boon, L.; Wortel, C.H.; Hogezand, R.A.; Schreiber, S.; Goldin, E.; Boer, M.; Geboes, K.; Rutgeerts, P.; Ceuppens, J.L. Safety and tolerability of antagonist anti-human CD40 Mab ch5D12 in patients with moderate to severe Crohn’s disease. Aliment. Pharmacol. Ther. 2005, 22, 111–122. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species. | CVD Entity | Effect | Pathway | Ref |
|---|---|---|---|---|
| mouse | Atherosclerosis | Plaque initiation | CD40L-ULVWF | [71] |
| Leucocyte recruitment/infiltration | CD40L-CD40 (ECs), VCAM-1 | [72] | ||
| Plaque destabilization/rupture | CD40L-Mac1 | [73] | ||
| Myocardial infarction | Thrombus stabilization/platelet activation | CD40L- αIIbβ3 Cd40L-CD40 | [58] | |
| Arterial hypertension | Plaque formation | CD40L- dependent | [77] | |
| High-fat diet | Vascular inflammation | CD40L- dependent | [78] | |
| human | Coronary artery disease | CD40 expression on endothelial cells, increased susceptibility to atherosclerosis | SNP rs1883832 | [79] |
| Acute coronary syndrome | Increased risk for death and recurrent MI | Elevated sCD40L levels in serum | [80] | |
| Myocardial infarction | Increased mortality | Elevated sCD40L levels in serum | [81] | |
| Heart failure | LV dysfunction | CD40L-CD40 | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daub, S.; Lutgens, E.; Münzel, T.; Daiber, A. CD40/CD40L and Related Signaling Pathways in Cardiovascular Health and Disease—The Pros and Cons for Cardioprotection. Int. J. Mol. Sci. 2020, 21, 8533. https://doi.org/10.3390/ijms21228533
Daub S, Lutgens E, Münzel T, Daiber A. CD40/CD40L and Related Signaling Pathways in Cardiovascular Health and Disease—The Pros and Cons for Cardioprotection. International Journal of Molecular Sciences. 2020; 21(22):8533. https://doi.org/10.3390/ijms21228533
Chicago/Turabian StyleDaub, Steffen, Esther Lutgens, Thomas Münzel, and Andreas Daiber. 2020. "CD40/CD40L and Related Signaling Pathways in Cardiovascular Health and Disease—The Pros and Cons for Cardioprotection" International Journal of Molecular Sciences 21, no. 22: 8533. https://doi.org/10.3390/ijms21228533
APA StyleDaub, S., Lutgens, E., Münzel, T., & Daiber, A. (2020). CD40/CD40L and Related Signaling Pathways in Cardiovascular Health and Disease—The Pros and Cons for Cardioprotection. International Journal of Molecular Sciences, 21(22), 8533. https://doi.org/10.3390/ijms21228533