Axon Injury-Induced Autophagy Activation Is Impaired in a C. elegans Model of Tauopathy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Axon Injury-Triggered Autophagy Induction Is Abolished in a Transgenic Tauopathy Model
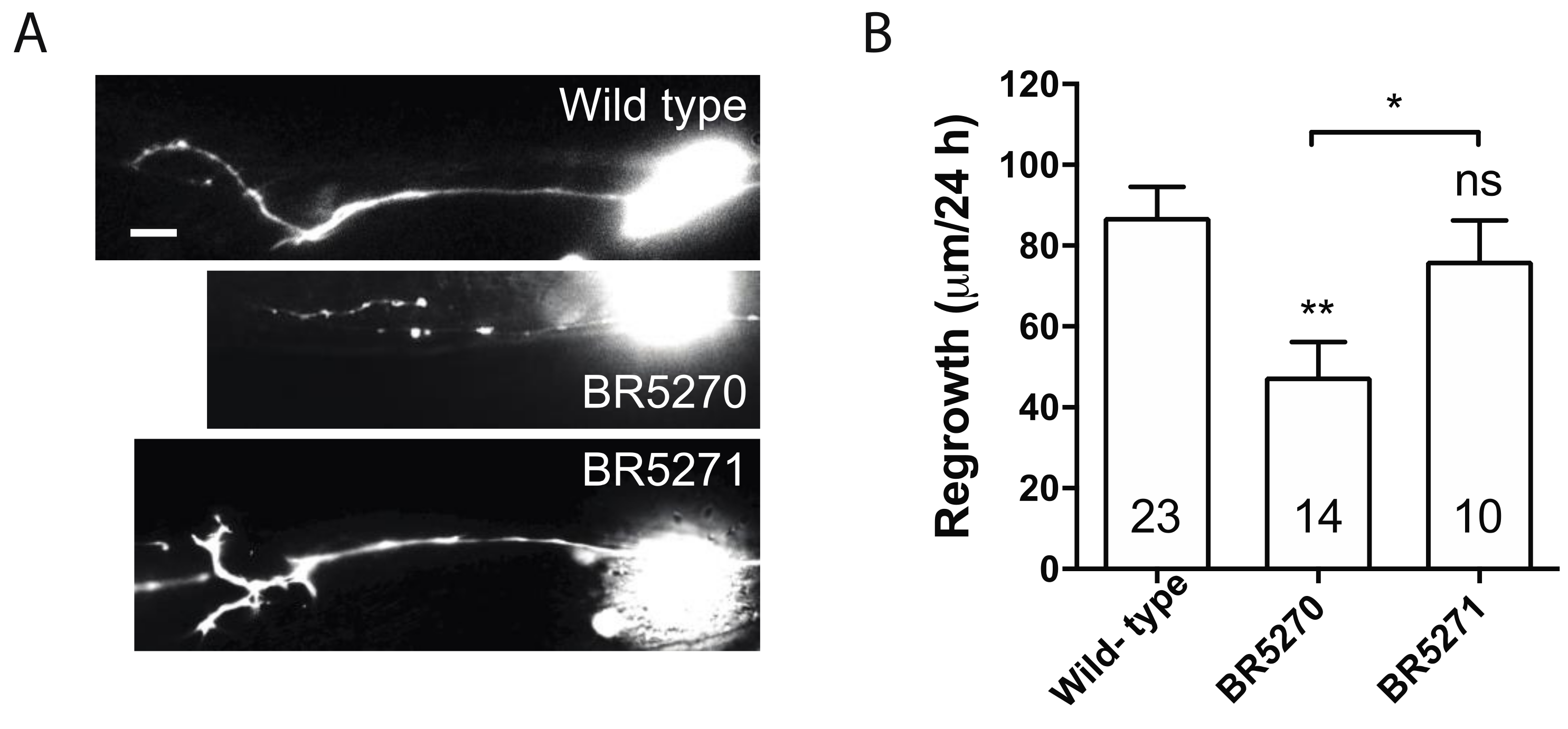
2.2. Transgenic Expression of Pro-Aggregant Tau Leads to Reduced Axon Regeneration after Injury
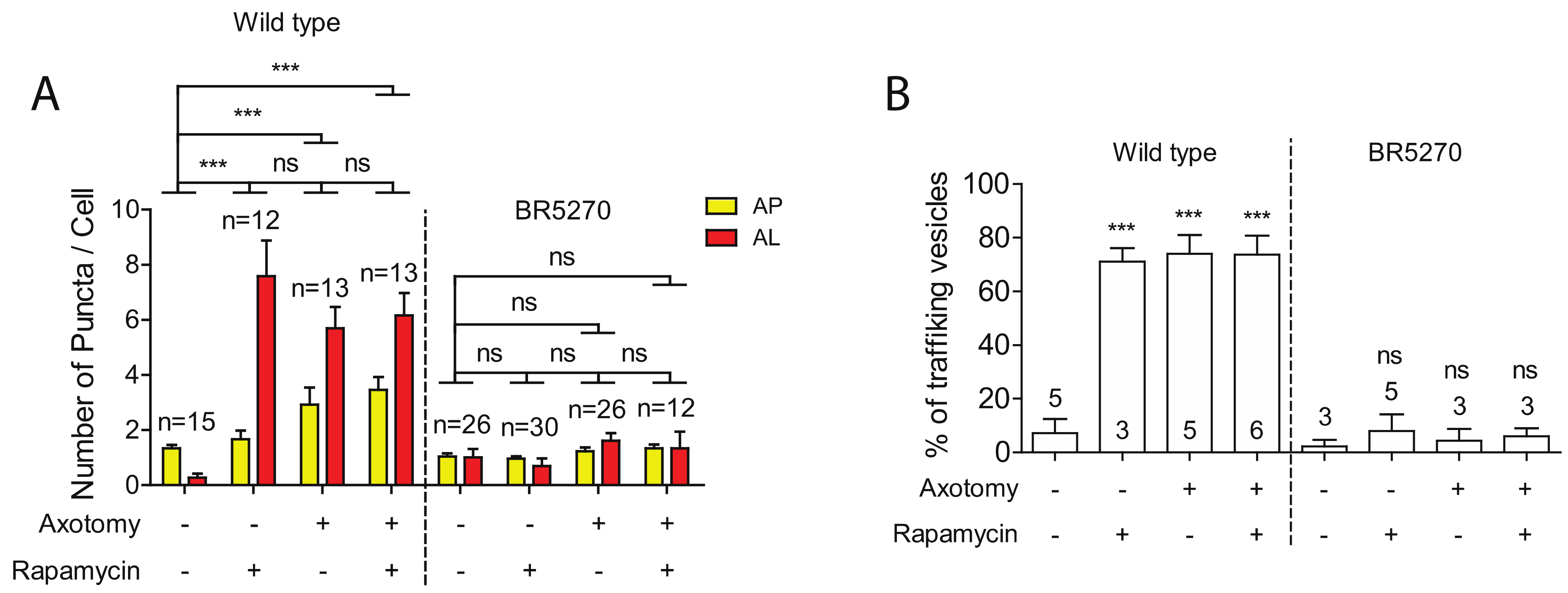
2.3. Injury Induces Autophagic Vesicles Trafficking in Wild-Type Neurons but Not in Neurons Expressing Pro-Aggregant Tau
2.4. Autophagy Activator Rapamycin Failed to Rescue Autophagy Defects in the Tauopathy Model
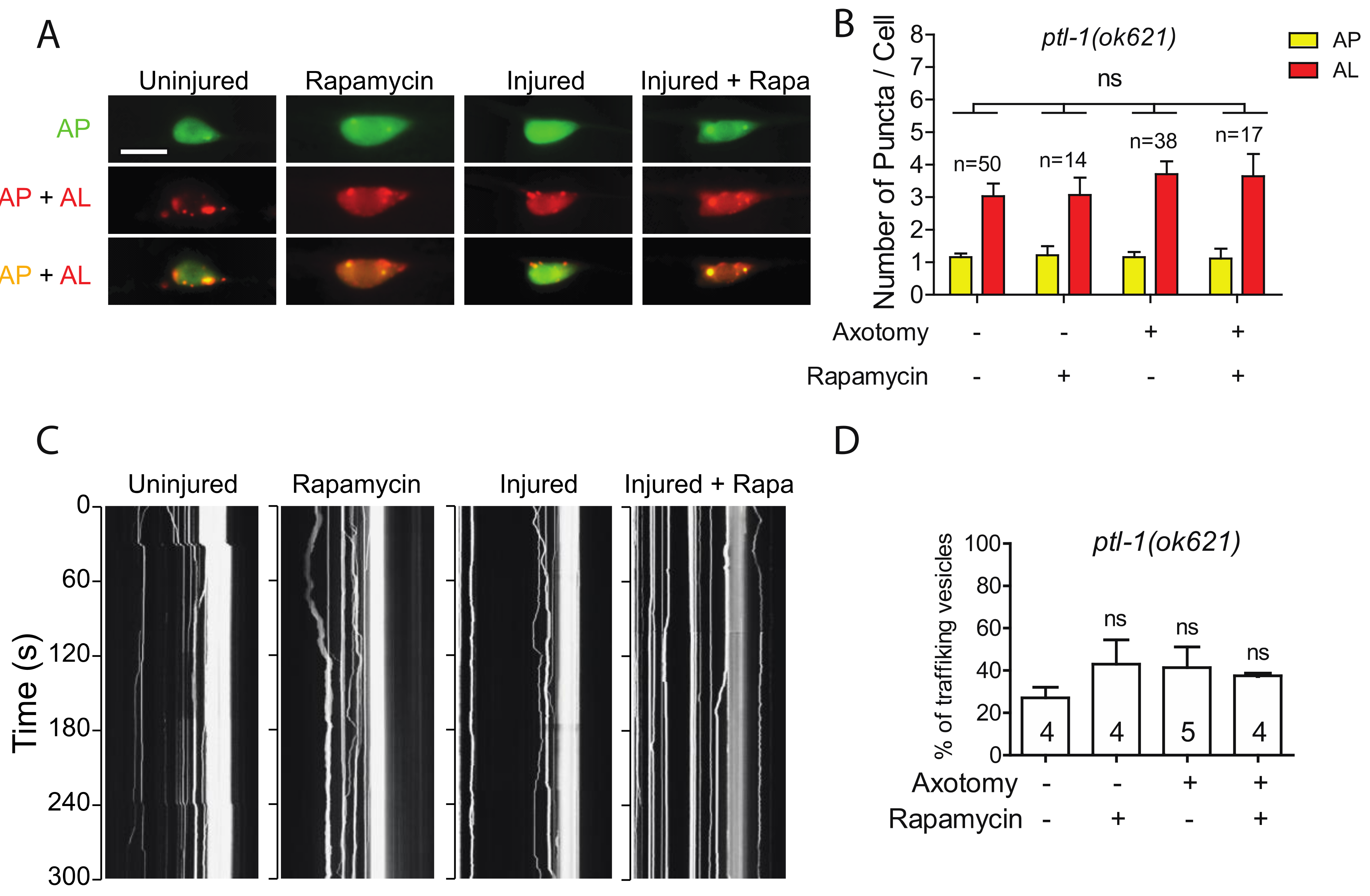
2.5. Loss of PTL-1 Blocks Injury-Induced Autophagy Activation
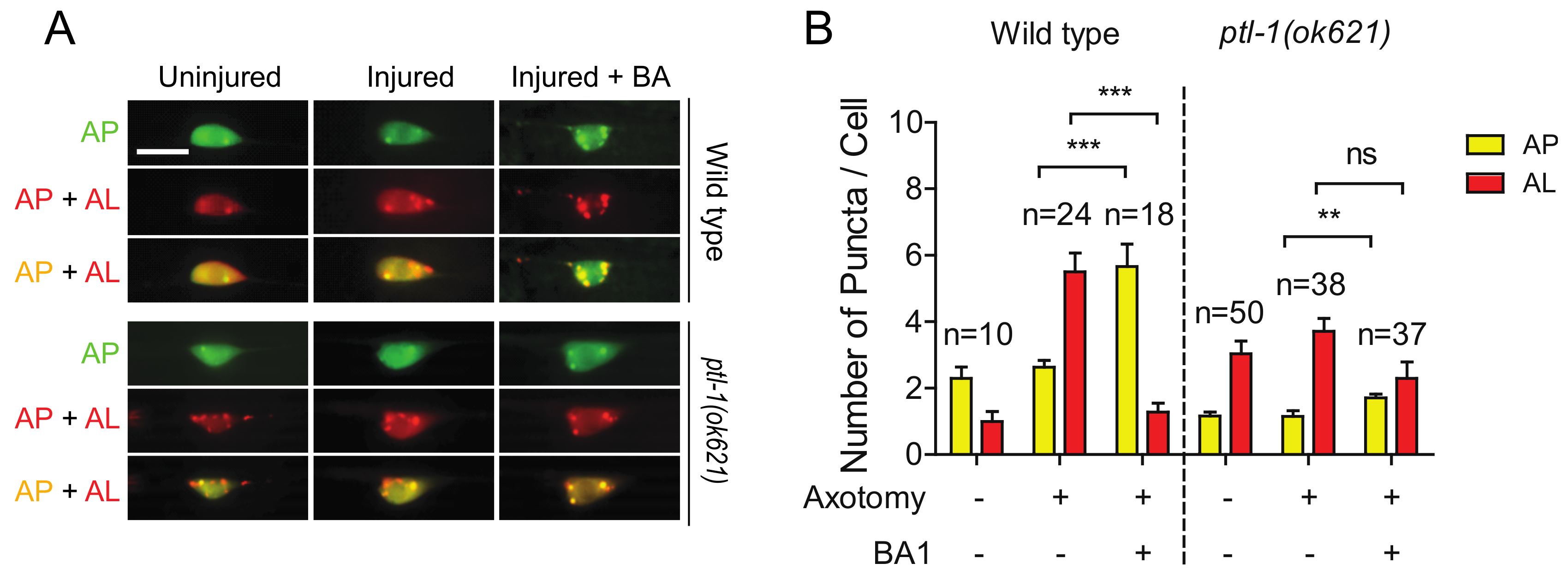
2.6. The High Basal Level of Autolysosomes in Ptl-1 Mutant Is Resistant to Autophagy Inhibitor BA1
3. Discussion
4. Materials and Methods
4.1. C. elegans Genetics
4.2. Transgene Construction
4.3. Laser Axotomy
4.4. Rapamycin and Bafilomycin A1 treatment
4.5. Quantification of Autophagic Vesicles
4.6. Live imaging of Autophagy Dynamics
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AL | Autolysosome |
| AP | Autophagosome |
| BA1 | Bafilomycin A1 |
| CNS | Central Nervous System |
| LGG-1 | LC3, GABARAP and GATE-16 family-1 |
| MT | Microtubule |
| PLM | Posterior Lateral Mechanosensory |
| PNS | Peripheral Nervous System |
| PTL-1 | Protein with Tau-like repeats-1 |
References
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in Age-Associated Neurodegeneration. Cells 2018, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; Gessani, A.; Fini, N.; Fasano, A.; Caponnetto, C.; Chio, A.; Dalla Bella, E.; Lunetta, C.; et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine 2018, 97, e11119. [Google Scholar] [CrossRef]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.L.; Jahrling, J.B.; Zhang, W.; DeRosa, N.; Bakshi, V.; Romero, P.; Galvan, V.; Richardson, A. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2017, 37, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS ONE 2013, 8, e62459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siman, R.; Cocca, R.; Dong, Y. The mTOR Inhibitor Rapamycin Mitigates Perforant Pathway Neurodegeneration and Synapse Loss in a Mouse Model of Early-Stage Alzheimer-Type Tauopathy. PLoS ONE 2015, 10, e0142340. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Choi, K.J.; Cho, S.J.; Yun, S.M.; Jeon, J.P.; Koh, Y.H.; Song, J.; Johnson, G.V.; Jo, C. Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors. Sci. Rep. 2016, 6, 24933. [Google Scholar] [CrossRef]
- Kruger, U.; Wang, Y.; Kumar, S.; Mandelkow, E.M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem. Soc. Trans. 2012, 40, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Lim, F.; Hernandez, F.; Lucas, J.J.; Gomez-Ramos, P.; Moran, M.A.; Avila, J. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol. Cell. Neurosci. 2001, 18, 702–714. [Google Scholar] [CrossRef]
- Wang, Y.; Song, M.; Song, F. Neuronal autophagy and axon degeneration. Cell Mol. Life Sci. 2018, 75, 2389–2406. [Google Scholar] [CrossRef]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maday, S. Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res. 2016, 1649, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, J.; Miura, E.; Mizushima, N.; Watanabe, M.; Yuzaki, M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007, 3, 591–596. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Jin, Y. Intrinsic Control of Axon Regeneration. Neuron 2016, 90, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Itoi, E. The role of autophagy in spinal cord injury. Autophagy 2009, 5, 390–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Muela, N.; Boya, P. Axonal damage, autophagy and neuronal survival. Autophagy 2012, 8, 286–288. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, M.M.; Wu, J.; Faden, A.I.; Sarkar, C. Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma. Antioxid. Redox Sign. 2015, 23, 565–577. [Google Scholar] [CrossRef]
- He, M.; Ding, Y.; Chu, C.; Tang, J.; Xiao, Q.; Luo, Z.G. Autophagy induction stabilizes microtubules and promotes axon regeneration after spinal cord injury. Proc. Natl. Acad. Sci. USA 2016, 113, 11324–11329. [Google Scholar] [CrossRef] [Green Version]
- Saraswat Ohri, S.; Bankston, A.N.; Mullins, S.A.; Liu, Y.; Andres, K.R.; Beare, J.E.; Howard, R.M.; Burke, D.A.; Riegler, A.S.; Smith, A.E.; et al. Blocking Autophagy in Oligodendrocytes Limits Functional Recovery after Spinal Cord Injury. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 5900–5912. [Google Scholar] [CrossRef]
- Ko, S.H.; Apple, E.C.; Liu, Z.; Chen, L. Age-dependent autophagy induction after injury promotes axon regeneration by limiting NOTCH. Autophagy 2020, 16, 2052–2068. [Google Scholar] [CrossRef]
- Fatouros, C.; Pir, G.J.; Biernat, J.; Koushika, S.P.; Mandelkow, E.; Mandelkow, E.M.; Schmidt, E.; Baumeister, R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 2012, 21, 3587–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, A.; Fraldi, A. Protein Aggregation and Dysfunction of Autophagy-Lysosomal Pathway: A Vicious Cycle in Lysosomal Storage Diseases. Front. Mol. Neurosci. 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Biernat, J.; Pickhardt, M.; Mandelkow, E.; Mandelkow, E.M. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. USA 2007, 104, 10252–10257. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Martinez-Vicente, M.; Kruger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.T.; Kumsta, C.; Hellman, A.B.; Adams, L.M.; Hansen, M. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. eLife 2017, 6, e18459. [Google Scholar] [CrossRef]
- Hubert, T.; Wu, Z.; Chisholm, A.D.; Jin, Y. S6 kinase inhibits intrinsic axon regeneration capacity via AMP kinase in Caenorhabditis elegans. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Ghosh-Roy, A.; Wu, Z.; Goncharov, A.; Jin, Y.; Chisholm, A.D. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 3175–3183. [Google Scholar] [CrossRef] [Green Version]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Monastyrska, I.; Rieter, E.; Klionsky, D.J.; Reggiori, F. Multiple roles of the cytoskeleton in autophagy. Biol. Rev. Camb. Philos. Soc. 2009, 84, 431–448. [Google Scholar] [CrossRef] [Green Version]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Baur, C.P.; Ahringer, J.; Jakes, R.; Hasegawa, M.; Spillantini, M.G.; Smith, M.J.; Hill, F. PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J. Cell Sci. 1996, 109, 2661–2672. [Google Scholar] [PubMed]
- McDermott, J.B.; Aamodt, S.; Aamodt, E. ptl-1, a Caenorhabditis elegans gene whose products are homologous to the tau microtubule-associated proteins. Biochemistry 1996, 35, 9415–9423. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.; Jasionowski, T.; Tuttle, D.L.; Lenk, S.E.; Dunn, W.A., Jr. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J. Cell. Physiol. 1992, 152, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Kochl, R.; Hu, X.W.; Chan, E.Y.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef]
- Reunanen, H.; Marttinen, M.; Hirsimaki, P. Effects of griseofulvin and nocodazole on the accumulation of autophagic vacuoles in Ehrlich ascites tumor cells. Exp. Mol. Pathol. 1988, 48, 97–102. [Google Scholar] [CrossRef]
- Mackeh, R.; Perdiz, D.; Lorin, S.; Codogno, P.; Pous, C. Autophagy and microtubules—New story, old players. J. Cell Sci. 2013, 126, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Ben-Zvi, A.; Miller, E.A.; Morimoto, R.I. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. USA 2009, 106, 14914–14919. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Geeraert, C.; Ratier, A.; Pfisterer, S.G.; Perdiz, D.; Cantaloube, I.; Rouault, A.; Pattingre, S.; Proikas-Cezanne, T.; Codogno, P.; Pous, C. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 2010, 285, 24184–24194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ghosh-Roy, A.; Yanik, M.F.; Zhang, J.Z.; Jin, Y.; Chisholm, A.D. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc. Natl. Acad. Sci. USA 2007, 104, 15132–15137. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, S.-H.; Gonzalez, G.; Liu, Z.; Chen, L. Axon Injury-Induced Autophagy Activation Is Impaired in a C. elegans Model of Tauopathy. Int. J. Mol. Sci. 2020, 21, 8559. https://doi.org/10.3390/ijms21228559
Ko S-H, Gonzalez G, Liu Z, Chen L. Axon Injury-Induced Autophagy Activation Is Impaired in a C. elegans Model of Tauopathy. International Journal of Molecular Sciences. 2020; 21(22):8559. https://doi.org/10.3390/ijms21228559
Chicago/Turabian StyleKo, Su-Hyuk, Gilberto Gonzalez, Zhijie Liu, and Lizhen Chen. 2020. "Axon Injury-Induced Autophagy Activation Is Impaired in a C. elegans Model of Tauopathy" International Journal of Molecular Sciences 21, no. 22: 8559. https://doi.org/10.3390/ijms21228559
APA StyleKo, S. -H., Gonzalez, G., Liu, Z., & Chen, L. (2020). Axon Injury-Induced Autophagy Activation Is Impaired in a C. elegans Model of Tauopathy. International Journal of Molecular Sciences, 21(22), 8559. https://doi.org/10.3390/ijms21228559