Influence of Phosphodiesterase Inhibition on CRE- and EGR1-Dependent Transcription in a Mouse Hippocampal Cell Line


Abstract
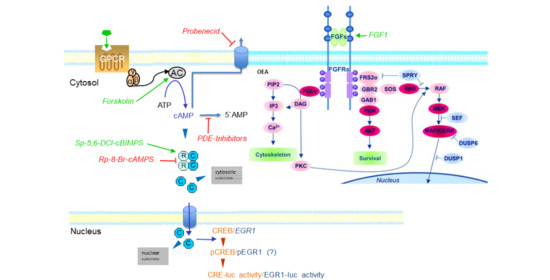
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Experimental Data
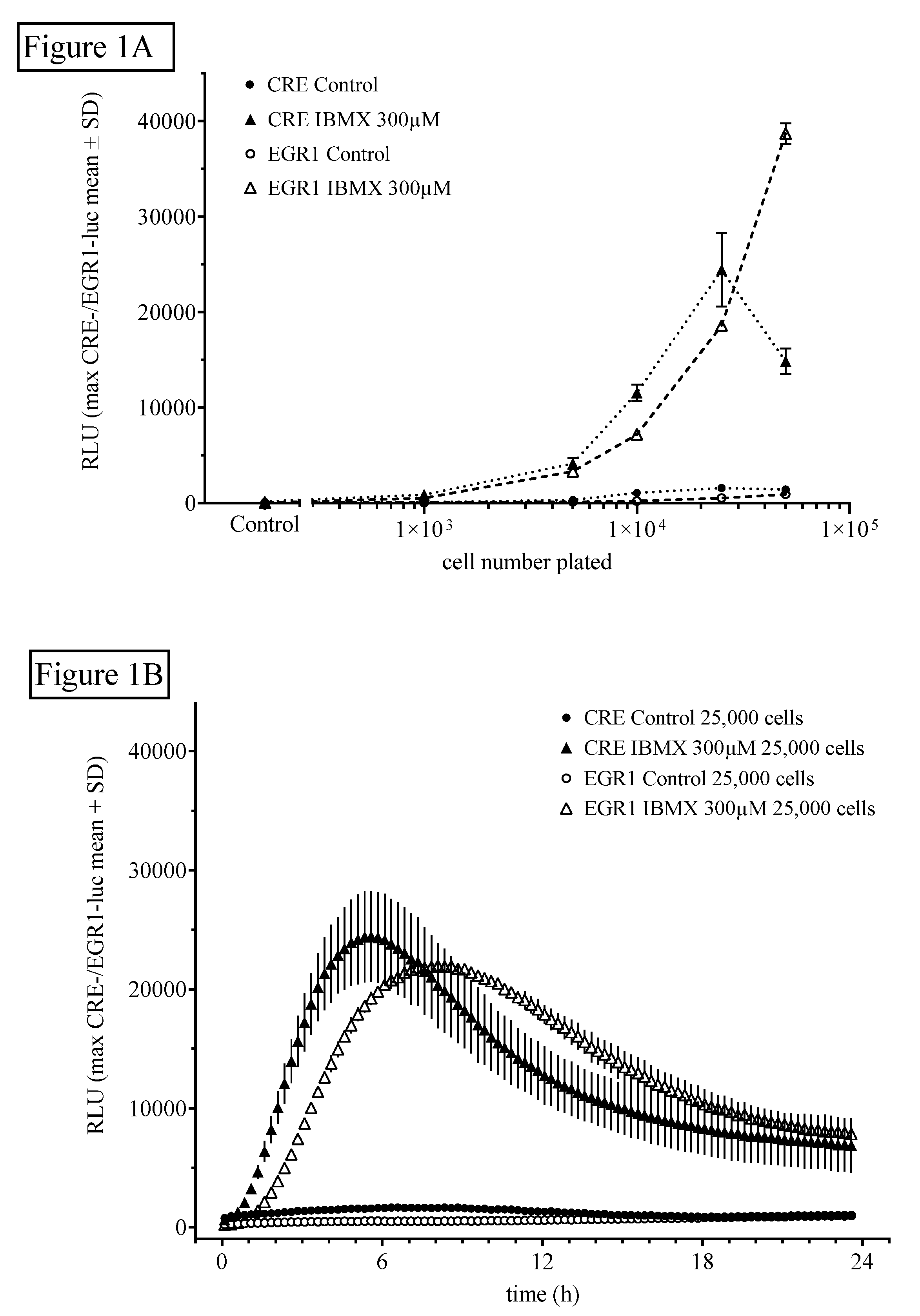
2.1.1. Influence of Cell Number and Concentration of Luciferin on CRE- and EGR1-Dependent Luciferase Activity
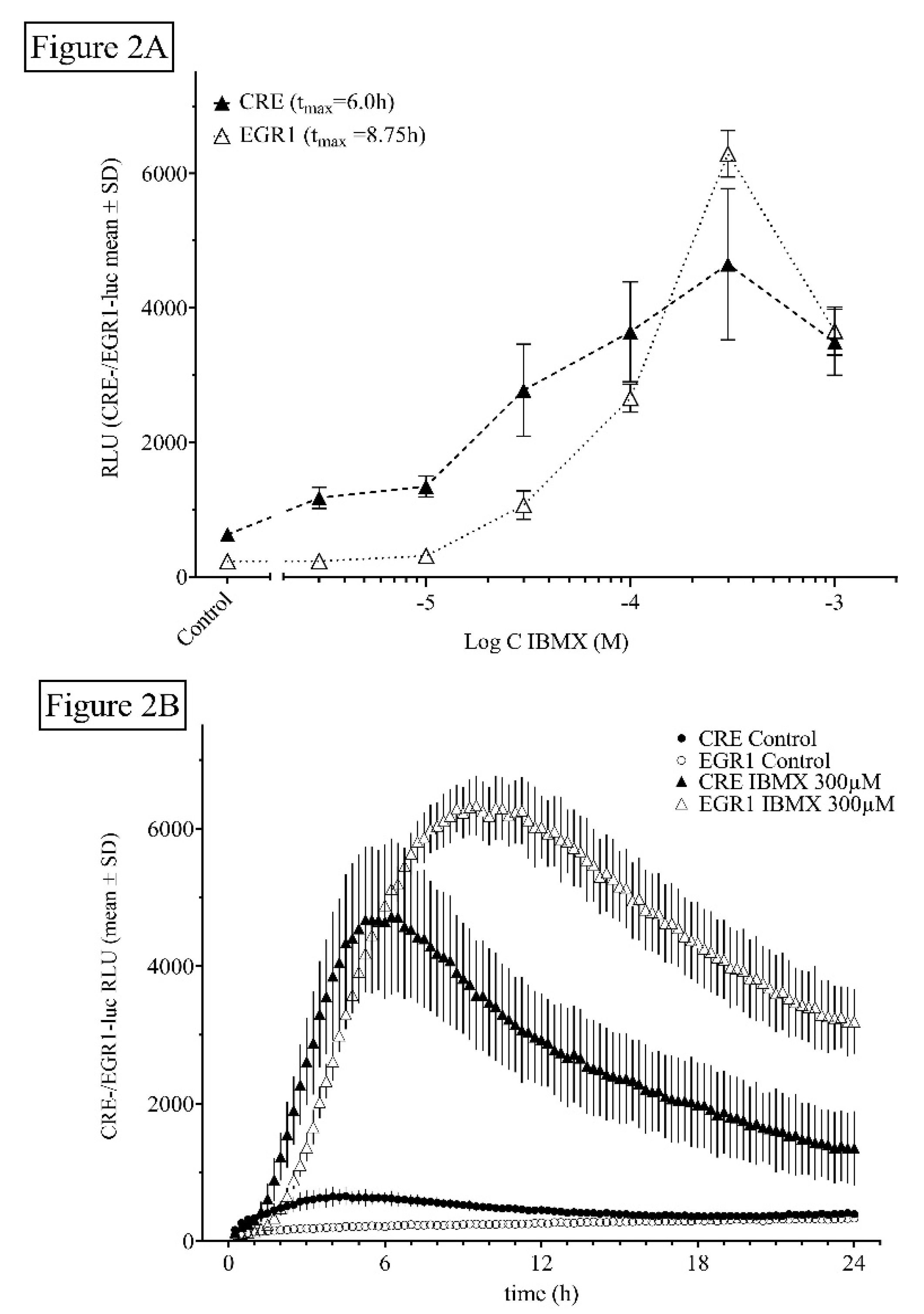
2.1.2. Influence of Time and Dose of PDE Inhibitors and Other Agents on Luciferin-Dependent CRE- and EGR1-Luciferase Activity
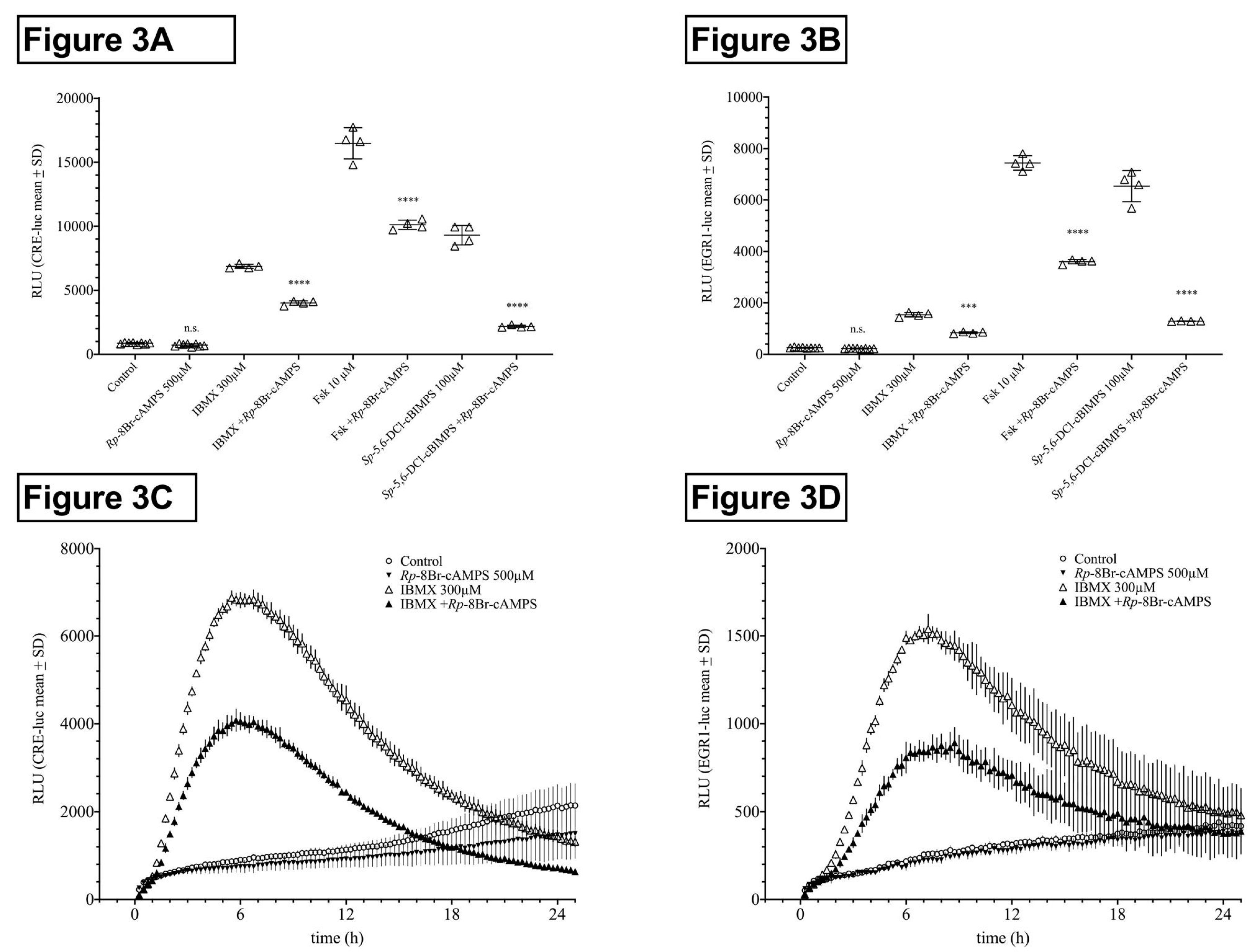
2.1.3. Influence of Protein Kinase A (PKA) Inhibition on HT22CRE- and EGR1-Luciferase Activity Elevated by PDE Inhibitors
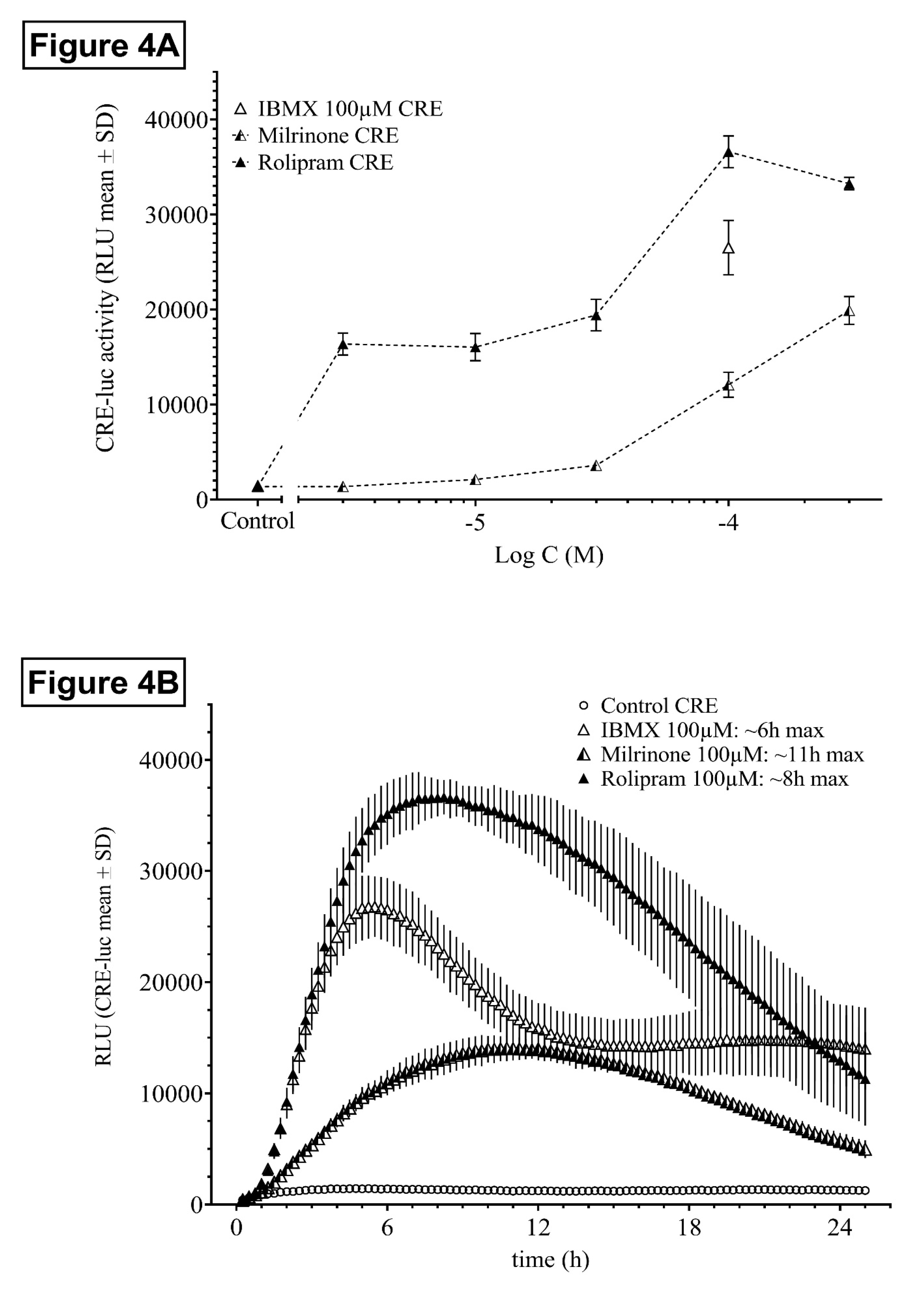
2.1.4. Influence of the PDE3- and PDE4-Specific Inhibitors Milrinone and Rolipram
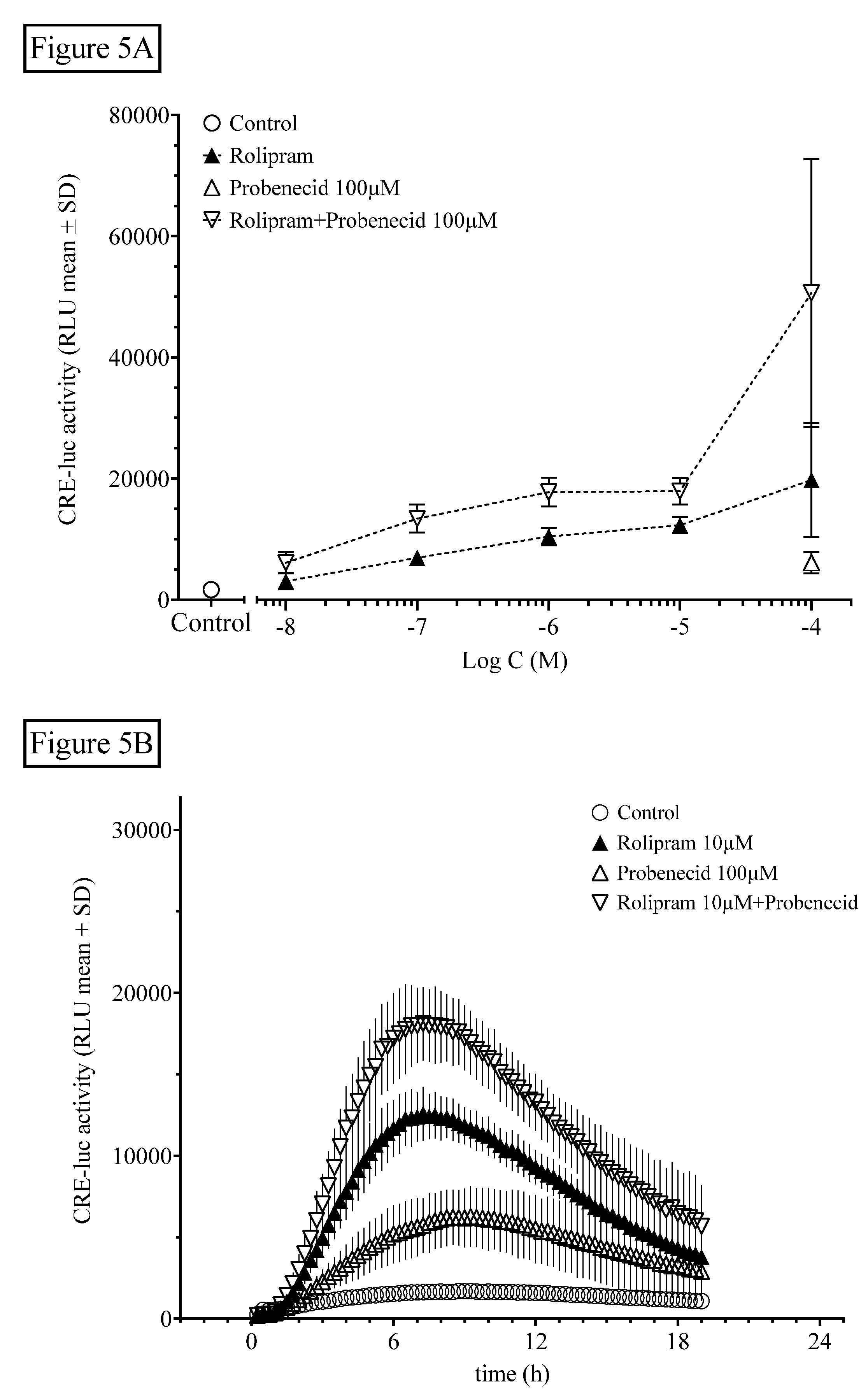
2.1.5. The Influence of Organic Anion Exchangers/Transporters (OAEs)
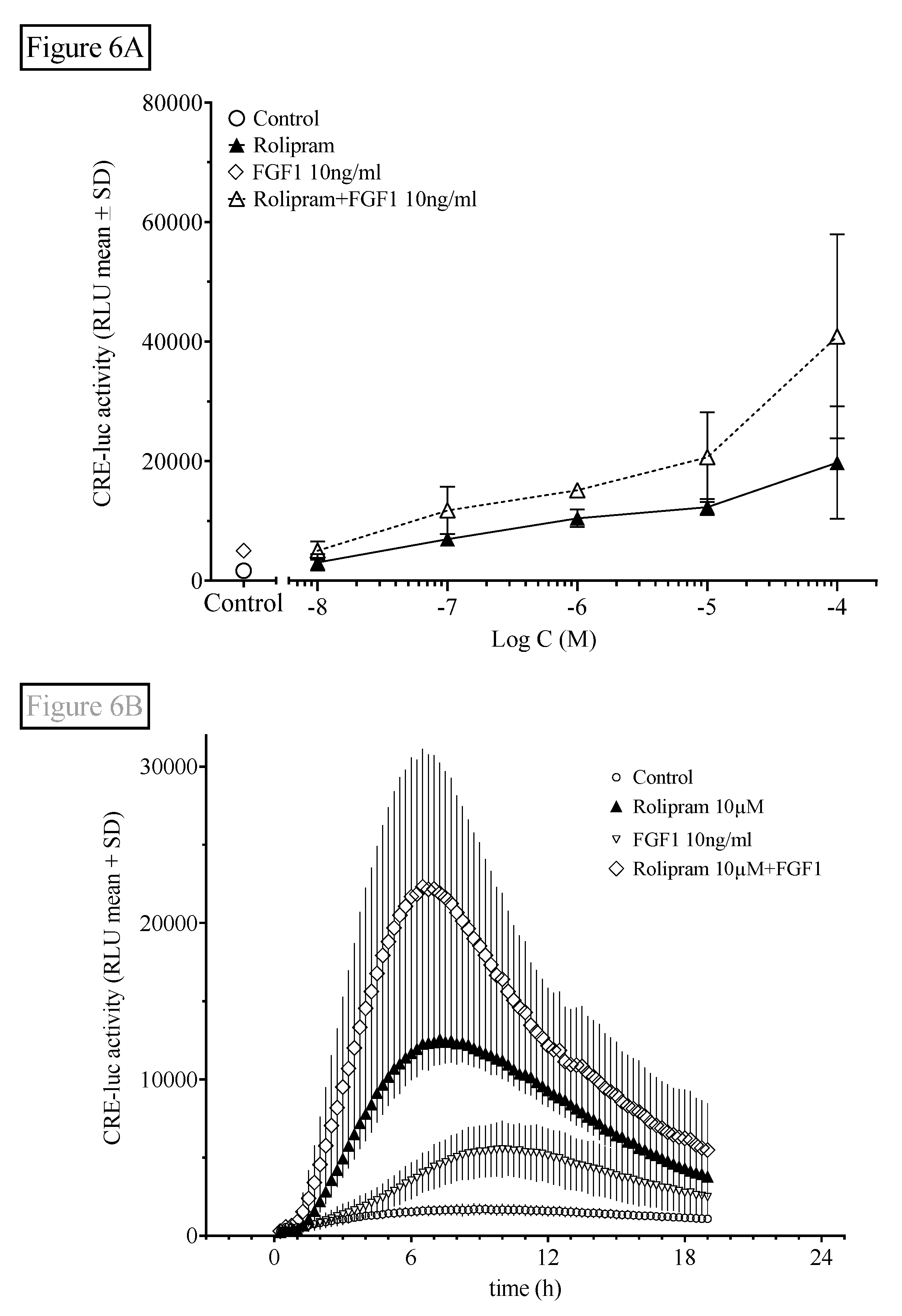
2.1.6. Interaction of Rolipram with the FGF Receptor/Protein Tyrosine Kinase Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Determination of Luminescence Activity
4.3. Mitochondrial Lactate Dehydrogenase Activity (Water Soluble Tetrazolium (WST-1) Assay)
4.4. Chemicals
4.5. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Adenylate cyclase/adenylyl cyclase |
| ATP | Adenosine 5’-triphosphate |
| cAMP | 3’,5’-adenosine monophosphate |
| CNG | Cyclic nucleotide-gated ion channel |
| CRE | Cyclic nucleotide-regulated element (TGACGTCA) |
| CREB | Cyclic nucleotide-regulated element-binding protein |
| DMEM | Dulbecco’s minimal essential medium |
| EGR1 | Early growth response protein 1 (synonyms: zif268, NGFI-A, Krox-24, TIS8, ZENK) |
| EPAC | Exchange factor directly activated by cAMP |
| FBS | Fetal bovine serum |
| FGF1 | Fibroblast growth factor 1/ acidic fibroblast growth factor |
| OEA | Organic anion exchanger/transporter |
| PDE | 3’,5’-cyclic nucleotide phosphodiesterase |
| PKA | 3’,5’-monophosphate-dependent protein kinase |
| RLU | Relative luminescence unit |
References
- Seifert, R.; Lushington, G.H.; Mou, T.-C.; Gille, A.; Sprang, S.R. Inhibitors of membranous adenylyl cyclases. Trends Pharmacol. Sci. 2012, 33, 64–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dremier, S.; Kopperud, R.; Doskeland, S.O.; Dumont, J.E.; Maenhaut, C. Search for new cyclic AMP-binding proteins. FEBS Lett. 2003, 546, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Michalakis, S.; Kleppisch, T.; Polta, S.A.; Wotjak, C.T.; Koch, S.; Rammes, G.; Matt, L.; Becirovic, E.; Biel, M. Altered synaptic plasticity and behavioral abnormalities in CNGA3-deficient mice. Genes Brain Behav. 2011, 10, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Epac proteins: Multi-purpose cAMP targets. Trends Pharmacol. Sci. 2006, 31, 680–686. [Google Scholar] [CrossRef]
- Døskeland, S.O.; Maronde, E.; Gjertsen, B.T. The genetic subtypes of cAMP-dependent protein kinase--functionally different or redundant? Biochim. Biophys. Acta 1993, 1178, 249–258. [Google Scholar] [CrossRef]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Ribaudo, G.; Ongaro, A.; Zagotto, G.; Memo, M.; Gianoncelli, A. Therapeutic Potential of Phosphodiesterase Inhibitors against Neurodegeneration: The Perspective of the Medicinal Chemist. ACS Chem. Neurosci. 2020, 11, 1726–1739. [Google Scholar] [CrossRef]
- Kandel, E.R.; Dudai, Y.; Mayford, M.R. The Molecular and Systems Biology of Memory. Cell 2014, 157, 163–186. [Google Scholar] [CrossRef] [Green Version]
- Rawashdeh, O.; Parsons, R.; Maronde, E. Clocking in Time to Gate Memory Processes: The Circadian Clock Is Part of the Ins and Outs of Memory. Neural Plast. 2018, 2018, 6238989. [Google Scholar] [CrossRef] [Green Version]
- Alberini, C.M.; Kandel, E.R. The regulation of transcription in memory consolidation. Cold Spring Harb. Perspect. Biol. 2015, 7, a021741. [Google Scholar] [CrossRef] [Green Version]
- Kandel, E.R. The molecular biology of memory storage: A dialogue between genes and synapses. Science 2001, 294, 1030–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Yan, C. An update of cyclic nucleotide phosphodiesterase as a target for cardiac diseases. Expert Opin. Drug Discov. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vecsey, C.G.; Baillie, G.S.; Jaganath, D.; Havekes, R.; Daniels, A.; Wimmer, M.; Huang, T.; Brown, K.M.; Li, X.-Y.; Descalzi, G.; et al. Sleep deprivation impairs cAMP signalling in the hippocampus. Nature 2009, 461, 1122–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maronde, E. Trehalose Activates CRE-Dependent Transcriptional Signaling in HT22 Mouse Hippocampal Neuronal Cells: A Central Role for PKA without cAMP Elevation. Front. Mol. Neurosci. 2018, 11, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudernova, I.; Foldynova-Trantirkova, S.; Ghannamova, B.E.; Fafilek, B.; Varecha, M.; Balek, L.; Hruba, E.; Jonatova, L.; Jelinkova, I.; Kunova Bosakova, M.; et al. One reporter for in-cell activity profiling of majority of protein kinase oncogenes. eLife 2017, 6, 443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seamon, K.B.; Daly, J.W. Forskolin: Its biological and chemical properties. Adv. Cycl. Nucleotide Protein Phosphorylation Res. 1986, 20, 1–150. [Google Scholar]
- Maronde, E.; Korf, H.W.; Niemann, P.; Genieser, H.G. Direct comparison of the potency of three novel cAMP analogs to induce CREB-phophorylation in rat pinealocytes. J. Pineal Res. 2001, 31, 183–185. [Google Scholar] [CrossRef]
- Maronde, E.; Motzkus, D. Oscillation of human period 1 (hPER1) reporter gene activity in human neuroblastoma cells in vivo. Chronobiol. Int. 2003, 20, 671–681. [Google Scholar] [CrossRef]
- Fricke, K.; Heitland, A.; Maronde, E. Cooperative activation of lipolysis by protein kinase A and protein kinase C pathways in 3T3-L1 adipocytes. Endocrinology 2004, 145, 4940–4947. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, M.; Butt, E.; Nolte, C.; Fischer, L.; Halbrügge, M.; Beltman, J.; Jahnsen, T.; Genieser, H.G.; Jastorff, B.; Walter, U. Characterization of Sp-5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole- 3′,5′-monophosphorothioate (Sp-5,6-DCl-cBiMPS) as a potent and specific activator of cyclic-AMP-dependent protein kinase in cell extracts and intact cells. Biochem. J. 1991, 279 Pt 2, 521–527. [Google Scholar] [CrossRef]
- Schwede, F.; Maronde, E.; Genieser, H.; Jastorff, B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther. 2000, 87, 199–226. [Google Scholar] [CrossRef]
- Gjertsen, B.T.; Mellgren, G.; Otten, A.; Maronde, E.; Genieser, H.G.; Jastorff, B.; Vintermyr, O.K.; McKnight, G.S.; Døskeland, S.O. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J. Biol. Chem. 1995, 270, 20599–20607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmiski, J.B.; MacVicar, B.A. Cyclic Nucleotide-Gated Channels Contribute to the Cholinergic Plateau Potential in Hippocampal CA1 Pyramidal Neurons. J. Neurosci. 2001, 21, 8707–8714. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enserink, J.M.; Christensen, A.E.; Rooij, J.; de van Triest, M.; Schwede, F.; Genieser, H.G.; Døskeland, S.O.; Blank, J.L.; Bos, J.L. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 2002, 4, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.; Butcher, R.W. The quantitative relationship between intracellular concentration and egress of cyclic AMP from cultured cells. Mol. Pharmacol. 1981, 19, 38–43. [Google Scholar]
- Robbins, N.; Koch, S.E.; Tranter, M.; Rubinstein, J. The history and future of probenecid. Cardiovasc. Toxicol. 2012, 12, 1–9. [Google Scholar] [CrossRef]
- Burckhardt, B.C.; Burckhardt, G. Transport of organic anions across the basolateral membrane of proximal tubule cells. Rev. Physiol. Biochem. Pharmacol. 2003, 146, 95–158. [Google Scholar] [CrossRef]
- Caldwell, G.B.; Howe, A.K.; Nickl, C.K.; Dostmann, W.R.; Ballif, B.A.; Deming, P.B. Direct modulation of the protein kinase a catalytic subunit by growth factor receptor tyrosine kinases. J. Cell Biochem. 2011, 113, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Benz, A.H.; Shajari, M.; Peruzki, N.; Dehghani, F.; Maronde, E. Early growth response-1 induction by fibroblast growth factor-1 via increase of mitogen-activated protein kinase and inhibition of protein kinase B in hippocampal neurons. Br. J. Pharmacol. 2010, 160, 1621–1630. [Google Scholar] [CrossRef] [Green Version]
- Poppe, H.; Rybalkin, S.D.; Rehmann, H.; Hinds, T.R.; Tang, X.-B.; Christensen, A.E.; Schwede, F.; Genieser, H.-G.; Bos, J.L.; Doskeland, S.O.; et al. Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 2008, 5, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Sagara, Y.; Schubert, D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J. Neurosci. 1998, 18, 6662–6671. [Google Scholar] [CrossRef] [PubMed]
- Dargusch, R.; Schubert, D. Specificity of resistance to oxidative stress. J. Neurochem. 2002, 81, 1394–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BOGER, W.P.; STRICKLAND, S.C. Probenecid (benemid); its uses and side-effects in 2502 patients. AMA Arch. Intern. Med. 1955, 95, 83–92. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Santamaría, A. Probenecid: An emerging tool for neuroprotection. CNS Neurol. Disord. Drug Targets 2013, 12, 1050–1065. [Google Scholar] [CrossRef]
- Chong, L.Y.Z.; Satya, K.; Kim, B.; Berkowitz, R. Milrinone Dosing and a Culture of Caution in Clinical Practice. Cardiol. Rev. 2018, 26, 35–42. [Google Scholar] [CrossRef]
- Karppanen, H.; Paakkari, P.; Orma, A.L.; Paakkari, I. Central hypotensive effects of imidazole acetic acid and rolipram (ZK 62 711) in rats proceedings. Agents Actions 1979, 9, 84–86. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maronde, E. Influence of Phosphodiesterase Inhibition on CRE- and EGR1-Dependent Transcription in a Mouse Hippocampal Cell Line. Int. J. Mol. Sci. 2020, 21, 8658. https://doi.org/10.3390/ijms21228658
Maronde E. Influence of Phosphodiesterase Inhibition on CRE- and EGR1-Dependent Transcription in a Mouse Hippocampal Cell Line. International Journal of Molecular Sciences. 2020; 21(22):8658. https://doi.org/10.3390/ijms21228658
Chicago/Turabian StyleMaronde, Erik. 2020. "Influence of Phosphodiesterase Inhibition on CRE- and EGR1-Dependent Transcription in a Mouse Hippocampal Cell Line" International Journal of Molecular Sciences 21, no. 22: 8658. https://doi.org/10.3390/ijms21228658
APA StyleMaronde, E. (2020). Influence of Phosphodiesterase Inhibition on CRE- and EGR1-Dependent Transcription in a Mouse Hippocampal Cell Line. International Journal of Molecular Sciences, 21(22), 8658. https://doi.org/10.3390/ijms21228658