Diabetes Mellitus Is a Chronic Disease that Can Benefit from Therapy with Induced Pluripotent Stem Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
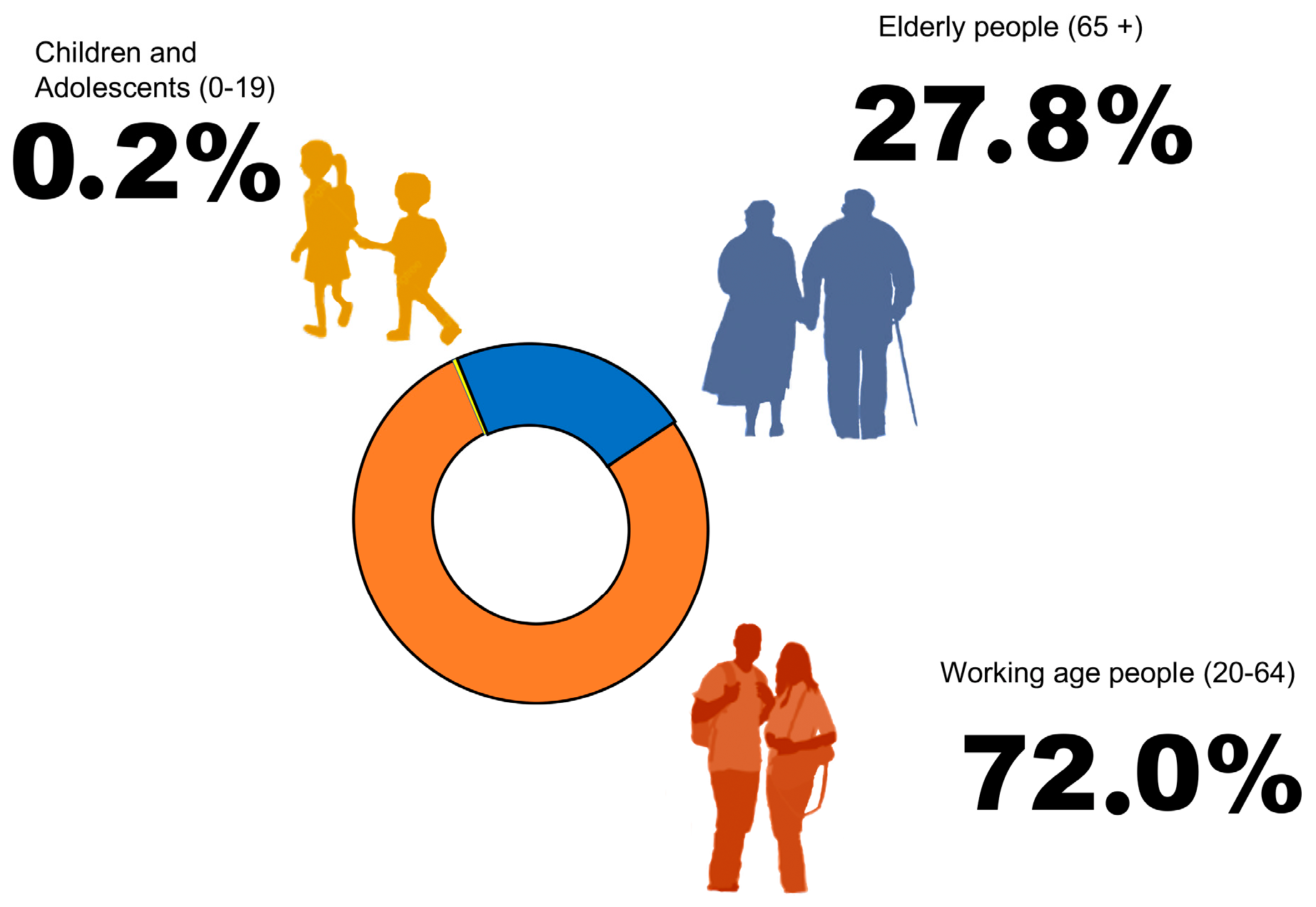
2. DM and Public Health
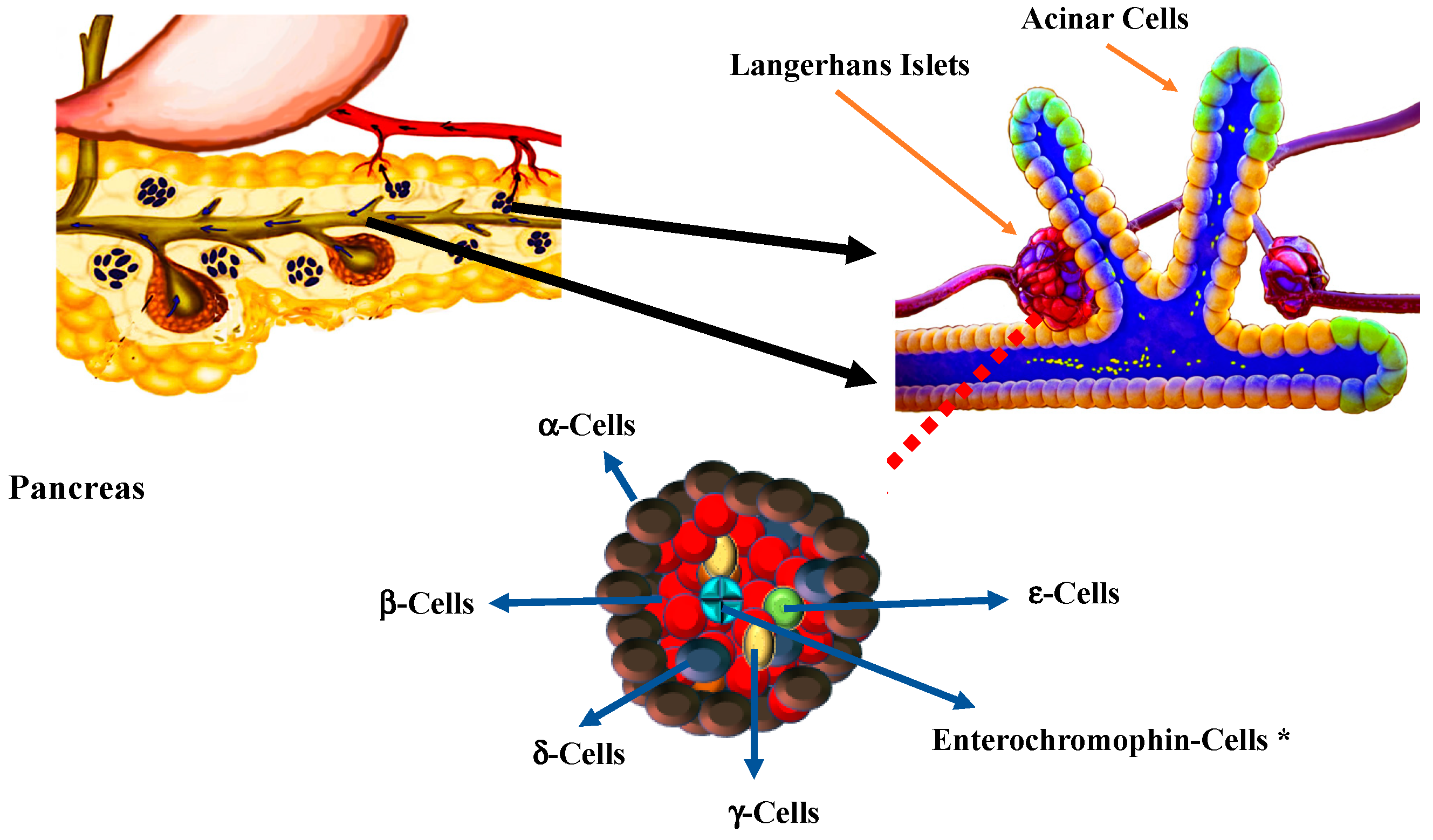
3. Pancreas and β-Cell Development
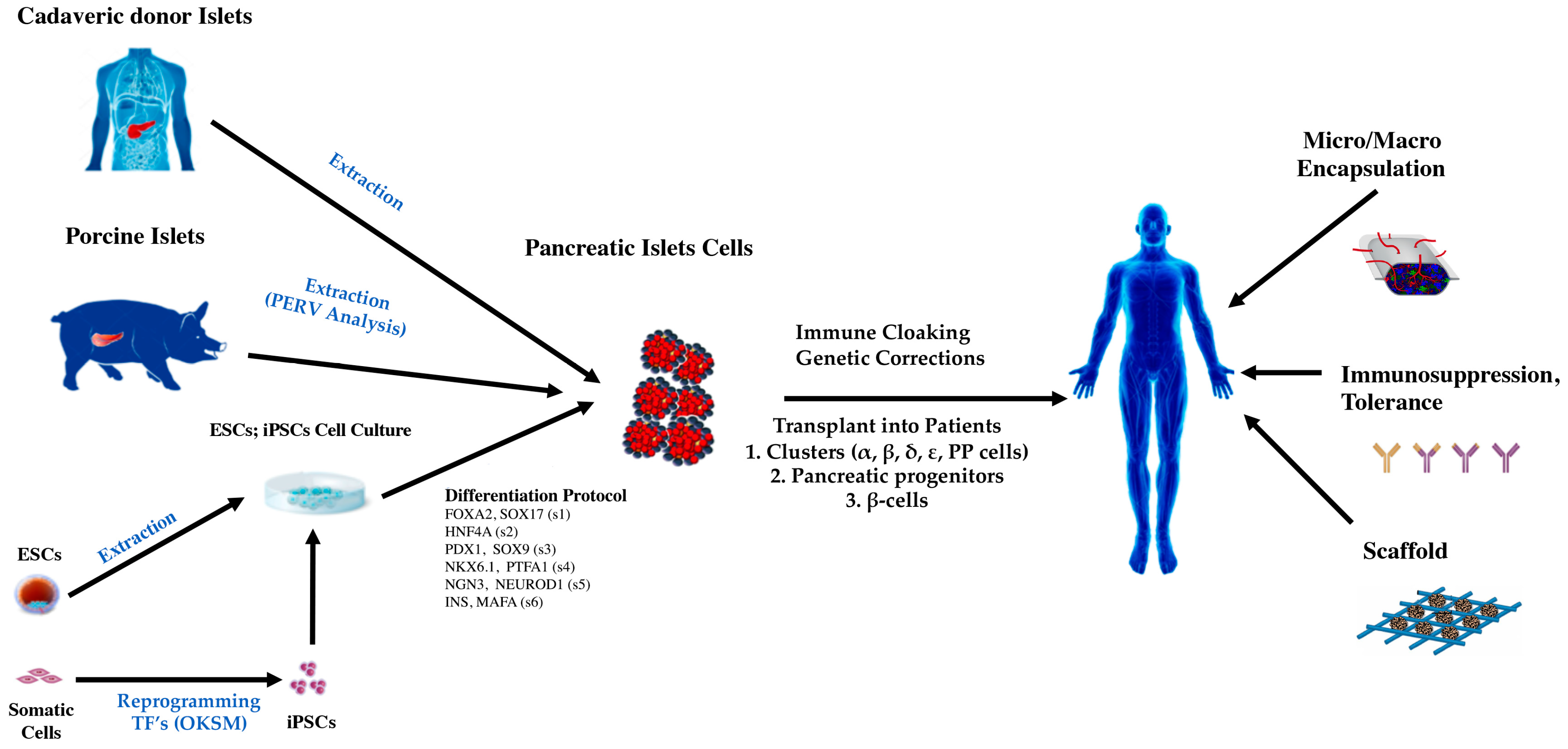
4. Current Treatments of Regenerative Medicine for DM
5. Human Pluripotent Cells and DM
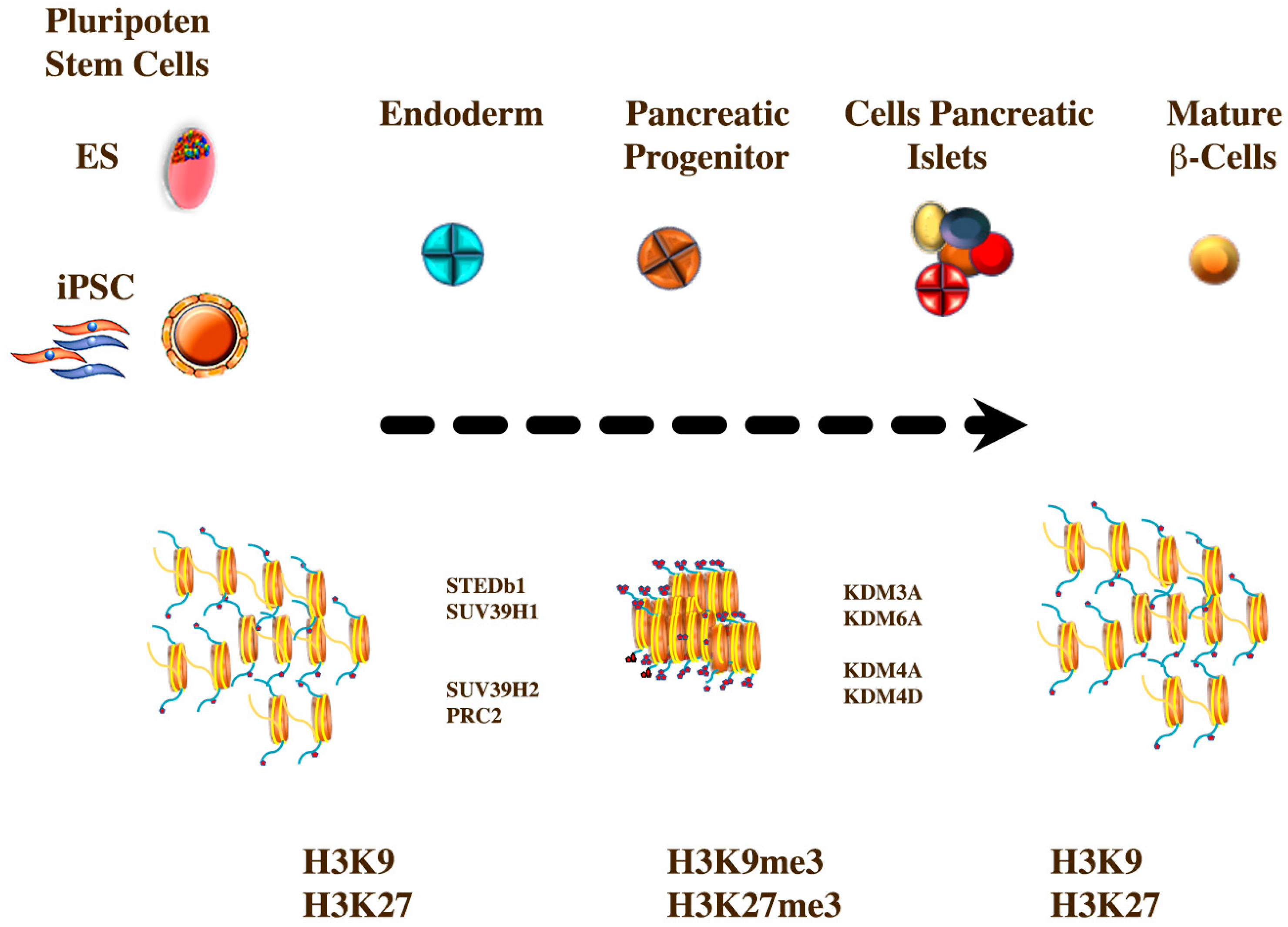
6. Epigenetics and Mechanisms of Chromatin Modification
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Skyler, J.S.; Riddle, M.C.; Ferrannini, E. Diabetes Research and Care Through the Ages. Diabetes Care 2017, 40, 1302–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes, A. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42 (Suppl. 1), S13–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovatchev, B.P.; Brown, S.A.; Beck, R.W. Multicenter Trial of Closed-Loop Control in Type 1 Diabetes Reply. N. Engl. J. Med. 2020, 382, 578. [Google Scholar] [PubMed]
- Bally, L.; Thabit, H.; Hartnell, S.; Andereggen, E.; Ruan, Y.; Wilinska, M.E.; Evans, M.L.; Wertli, M.M.; Coll, A.P.; Stettler, C.; et al. Closed-Loop Insulin Delivery for Glycemic Control in Noncritical Care. N. Engl. J. Med. 2018, 379, 547–556. [Google Scholar] [CrossRef]
- Heinemann, L.; Freckmann, G.; Ehrmann, D.; Faber-Heinemann, G.; Guerra, S.; Waldenmaier, D.; Hermanns, N. Real-time continuous glucose monitoring in adults with type 1 diabetes and impaired hypoglycaemia awareness or severe hypoglycaemia treated with multiple daily insulin injections (HypoDE): A multicentre, randomised controlled trial. Lancet 2018, 391, 1367–1377. [Google Scholar] [CrossRef]
- Cernea, S.; Raz, I. Insulin Therapy: Future Perspectives. Am. J. Ther. 2020, 27, e121–e132. [Google Scholar] [CrossRef]
- Ladisch, M.R.; Kohlmann, K.L. Recombinant human insulin. Biotechnol. Prog. 1992, 8, 469–478. [Google Scholar] [CrossRef]
- Danne, T.; Tamborlane, W.V.; Malievsky, O.A.; Franco, D.R.; Kawamura, T.; Demissie, M.; Niemoeller, E.; Goyeau, H.; Wardecki, M.; Battelino, T. Efficacy and Safety of Insulin Glargine 300 Units/mL (Gla-300) Versus Insulin Glargine 100 Units/mL (Gla-100) in Children and Adolescents (6–17 years) With Type 1 Diabetes: Results of the EDITION JUNIOR Randomized Controlled Trial. Diabetes Care 2020, 43, 1512–1519. [Google Scholar] [CrossRef]
- Danne, T.; Heinemann, L.; Bolinder, J. New Insulins, Biosimilars, and Insulin Therapy. Diabetes Technol. Ther. 2020, 22, S32–S46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovc, K.; Piona, C.; Yesiltepe Mutlu, G.; Bratina, N.; Jenko Bizjan, B.; Lepej, D.; Nimri, R.; Atlas, E.; Muller, I.; Kordonouri, O.; et al. Faster Compared With Standard Insulin Aspart During Day-and-Night Fully Closed-Loop Insulin Therapy in Type 1 Diabetes: A Double-Blind Randomized Crossover Trial. Diabetes Care 2020, 43, 29–36. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Inzucchi, S.; Abdul-Ghani, M.; Nissen, S.E. Pioglitazone: The forgotten, cost-effective cardioprotective drug for type 2 Diabetes. Diab. Vasc. Dis. Res. 2019, 16, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [Green Version]
- Lizcano, F.; Vargas, D. Diverse coactivator recruitment through differential PPARgamma nuclear receptor agonism. Genet. Mol. Biol. 2013, 36, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, E.J.; Newman, J.D. Review of cardiovascular outcomes trials of sodium-glucose cotransporter-2 inhibitors and glucagon-like peptide-1 receptor agonists. Curr. Opin. Cardiol. 2019, 34, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F. DPP-4 inhibitors, heart failure and type 2 diabetes: All eyes on safety. Cardiovasc. Diagn. Ther. 2015, 5, 471–478. [Google Scholar]
- Kristensen, S.L.; Rorth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Kober, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Handelsman, Y.; Mathieu, C.; Del Prato, S.; Johnsson, E.; Kurlyandskaya, R.; Iqbal, N.; Garcia-Sanchez, R.; Rosenstock, J. Sustained 52-week efficacy and safety of triple therapy with dapagliflozin plus saxagliptin versus dual therapy with sitagliptin added to metformin in patients with uncontrolled type 2 Diabetes. Diabetes Obes. Metab. 2019, 21, 883–892. [Google Scholar] [CrossRef]
- Ghosh, R.K.; Ghosh, G.C.; Gupta, M.; Bandyopadhyay, D.; Akhtar, T.; Deedwania, P.; Lavie, C.J.; Fonarow, G.C.; Aneja, A. Sodium Glucose Co-transporter 2 Inhibitors and Heart Failure. Am. J. Cardiol. 2019, 124, 1790–1796. [Google Scholar] [CrossRef]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus: Atherosclerotic Cardiovascular Disease and Heart Failure in Type 2 Diabetes Mellitus—Mechanisms, Management, and Clinical Considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S. Mitigating Cardiovascular Risk in Type 2 Diabetes With Antidiabetes Drugs: A Review of Principal Cardiovascular Outcome Results of EMPA-REG OUTCOME, LEADER, and SUSTAIN-6 Trials. Diabetes Care 2017, 40, 821–831. [Google Scholar] [CrossRef] [Green Version]
- McGuire, D.K.; Marx, N.; Johansen, O.E.; Inzucchi, S.E.; Rosenstock, J.; George, J.T. FDA guidance on antihyperglyacemic therapies for type 2 diabetes: One decade later. Diabetes Obes. Metab. 2019, 21, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Viscoli, C.M.; Young, L.H.; Kernan, W.N. Diabetes prevention and cardiovascular complications. Diabetologia 2019, 62, 2161–2162. [Google Scholar] [CrossRef]
- Suchy, F.; Yamaguchi, T.; Nakauchi, H. iPSC-Derived Organs In Vivo: Challenges and Promise. Cell Stem Cell 2018, 22, 21–24. [Google Scholar] [CrossRef]
- Stirban, A.O.; Tschoepe, D. Cardiovascular complications in diabetes: Targets and interventions. Diabetes Care 2008, 31 (Suppl. 2), S215–S221. [Google Scholar] [CrossRef] [Green Version]
- Isobe, K.; Cheng, Z.; Nishio, N.; Suganya, T.; Tanaka, Y.; Ito, S. iPSCs, aging and age-related diseases. N. Biotechnol. 2014, 31, 411–421. [Google Scholar] [CrossRef]
- Zhou, Q.; Melton, D.A. Pancreas regeneration. Nature 2018, 557, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.; Ramzy, A.; Kieffer, T.J. Regenerative medicine and cell-based approaches to restore pancreatic function. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 612–628. [Google Scholar] [CrossRef]
- Nakagawa, M.; Yamanaka, S. Reprogramming of somatic cells to pluripotency. Adv. Exp. Med. Biol. 2010, 695, 215–224. [Google Scholar] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.J.; Lopez, A.D. Measuring the global burden of disease. N. Engl. J. Med. 2013, 369, 448–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerging Risk Factors, C.; Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef] [Green Version]
- Bommer, C.; Sagalova, V.; Heesemann, E.; Manne-Goehler, J.; Atun, R.; Barnighausen, T.; Davies, J.; Vollmer, S. Global Economic Burden of Diabetes in Adults: Projections From 2015 to 2030. Diabetes Care 2018, 41, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Sharp, S.A.; Rich, S.S.; Wood, A.R.; Jones, S.E.; Beaumont, R.N.; Harrison, J.W.; Schneider, D.A.; Locke, J.M.; Tyrrell, J.; Weedon, M.N.; et al. Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis. Diabetes Care 2019, 42, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.C.; Wallin, A.; Hakansson, N.; Stackelberg, O.; Back, M.; Wolk, A. Type 1 and type 2 diabetes mellitus and incidence of seven cardiovascular diseases. Int. J. Cardiol. 2018, 262, 66–70. [Google Scholar] [CrossRef] [Green Version]
- Boyko, E. IDF plans dynamic 2019 congress. Diabetes Res. Clin. Pract. 2019, 150, 342–343. [Google Scholar] [CrossRef]
- Borgnakke, W.S. IDF Diabetes Atlas: Diabetes and oral health—A two-way relationship of clinical importance. Diabetes Res. Clin. Pract. 2019, 157, 107839. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.L.; Halim, S.; Gurudas, S.; Sivaprasad, S.; Owens, D.R. IDF Diabetes Atlas: A review of studies utilising retinal photography on the global prevalence of diabetes related retinopathy between 2015 and 2018. Diabetes Res. Clin. Pract. 2019, 157, 107840. [Google Scholar] [CrossRef]
- Cheng, Y.J.; Kanaya, A.M.; Araneta, M.R.G.; Saydah, S.H.; Kahn, H.S.; Gregg, E.W.; Fujimoto, W.Y.; Imperatore, G. Prevalence of Diabetes by Race and Ethnicity in the United States, 2011–2016. JAMA 2019, 322, 2389–2398. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Economic Burden of Cardiovascular Disease in Type 2 Diabetes: A Systematic Review. Value Health 2018, 21, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Kharroubi, A.T.; Darwish, H.M. Diabetes mellitus: The epidemic of the century. World J. Diabetes 2015, 6, 850–867. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.; Sas, K.M.; Abcouwer, S.F.; Feldman, E.L.; Gardner, T.W.; Pennathur, S.; Fort, P.E. New insights into the mechanisms of diabetic complications: Role of lipids and lipid metabolism. Diabetologia 2019, 62, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, L.; Menon, V.; Kashyap, S. Cardiovascular and Renal Outcomes of Newer Anti-Diabetic Medications in High-Risk Patients. Curr. Cardiol. Rep. 2018, 20, 65. [Google Scholar] [CrossRef]
- Koivula, R.W.; Forgie, I.M.; Kurbasic, A.; Vinuela, A.; Heggie, A.; Giordano, G.N.; Hansen, T.H.; Hudson, M.; Koopman, A.D.M.; Rutters, F.; et al. Discovery of biomarkers for glycaemic deterioration before and after the onset of type 2 diabetes: Descriptive characteristics of the epidemiological studies within the IMI DIRECT Consortium. Diabetologia 2019, 62, 1601–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beulens, J.; Rutters, F.; Ryden, L.; Schnell, O.; Mellbin, L.; Hart, H.E.; Vos, R.C. Risk and management of pre-Diabetes. Eur. J. Prev. Cardiol. 2019, 26 (Suppl. 2), 47–54. [Google Scholar] [CrossRef] [Green Version]
- Rawshani, A.; Rawshani, A.; Franzen, S.; Sattar, N.; Eliasson, B.; Svensson, A.M.; Zethelius, B.; Miftaraj, M.; McGuire, D.K.; Rosengren, A.; et al. Risk Factors, Mortality, and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2018, 379, 633–644. [Google Scholar] [CrossRef]
- Federation, I.D. Idf diabetes atlas. In IDF Diabetes, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; pp. 50–61. [Google Scholar]
- Collaboration, N.C.D.R.F. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Danaei, G.; Finucane, M.M.; Lu, Y.; Singh, G.M.; Cowan, M.J.; Paciorek, C.J.; Lin, J.K.; Farzadfar, F.; Khang, Y.H.; Stevens, G.A.; et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011, 378, 31–40. [Google Scholar] [CrossRef]
- Zhu, Y.; Sidell, M.A.; Arterburn, D.; Daley, M.F.; Desai, J.; Fitzpatrick, S.L.; Horberg, M.A.; Koebnick, C.; McCormick, E.; Oshiro, C.; et al. Racial/Ethnic Disparities in the Prevalence of Diabetes and Prediabetes by BMI: Patient Outcomes Research To Advance Learning (PORTAL) Multisite Cohort of Adults in the U.S. Diabetes Care 2019, 42, 2211–2219. [Google Scholar] [CrossRef]
- Chudasama, Y.V.; Zaccardi, F.; Gillies, C.L.; Dhalwani, N.N.; Yates, T.; Rowlands, A.V.; Davies, M.J.; Khunti, K. Leisure-time physical activity and life expectancy in people with cardiometabolic multimorbidity and depression. J. Intern. Med. 2020, 287, 87–99. [Google Scholar] [CrossRef]
- Hartmann-Boyce, J.; Morris, E.; Goyder, C.; Kinton, J.; Perring, J.; Nunan, D.; Mahtani, K.; Buse, J.B.; Del Prato, S.; Ji, L.; et al. Diabetes and COVID-19: Risks, Management, and Learnings From Other National Disasters. Diabetes Care 2020, 43, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Grenard, J.L.; Munjas, B.A.; Adams, J.L.; Suttorp, M.; Maglione, M.; McGlynn, E.A.; Gellad, W.F. Depression and medication adherence in the treatment of chronic diseases in the United States: A meta-analysis. J. Gen. Intern. Med. 2011, 26, 1175–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstein, H.C. Diabetes: Dysglycaemia as a cause of cardiovascular outcomes. Nat. Rev. Endocrinol. 2015, 11, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Vargas, D.; Lopez, C.; Acero, E.; Benitez, E.; Wintaco, A.; Camacho, J.; Carreno, M.; Umana, J.; Jimenez, D.; Diaz, S.; et al. Thermogenic capacity of human periaortic adipose tissue is transformed by body weight. PLoS ONE 2018, 13, e0194269. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Diaz, J.; Herrington, W.; Lopez-Cervantes, M.; Gnatiuc, L.; Ramirez, R.; Hill, M.; Baigent, C.; McCarthy, M.I.; Lewington, S.; Collins, R.; et al. Diabetes and Cause-Specific Mortality in Mexico City. N. Engl. J. Med. 2016, 375, 1961–1971. [Google Scholar] [CrossRef]
- Reaven, P.D.; Emanuele, N.V.; Wiitala, W.L.; Bahn, G.D.; Reda, D.J.; McCarren, M.; Duckworth, W.C.; Hayward, R.A.; Investigators, V. Intensive Glucose Control in Patients with Type 2 Diabetes—15-Year Follow-up. N. Engl. J. Med. 2019, 380, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Rawshani, A.; Sattar, N.; Franzen, S.; Rawshani, A.; Hattersley, A.T.; Svensson, A.M.; Eliasson, B.; Gudbjornsdottir, S. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: A nationwide, register-based cohort study. Lancet 2018, 392, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. (Lond) 2015, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy Chowdhury, S.; Thomas, R.L.; Dunseath, G.J.; Peter, R.; Rees, D.A.; North, R.V.; Luzio, S.D.; Owens, D.R. Diabetic Retinopathy in Newly Diagnosed Subjects With Type 2 Diabetes Mellitus: Contribution of beta-Cell Function. J. Clin. Endocrinol. Metab. 2016, 101, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, A.I.; Stevens, R.J.; Manley, S.E.; Bilous, R.W.; Cull, C.A.; Holman, R.R.; Ukpds, G. Development and progression of nephropathy in type 2 diabetes: The United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 2003, 63, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-Induced Reactive Oxygen Species: Mechanism of Their Generation and Role in Renal Injury. J. Diabetes Res. 2017, 2017, 8379327. [Google Scholar] [CrossRef]
- Komenda, P.; Ferguson, T.W.; Macdonald, K.; Rigatto, C.; Koolage, C.; Sood, M.M.; Tangri, N. Cost-effectiveness of primary screening for CKD: A systematic review. Am. J. Kidney Dis. 2014, 63, 789–797. [Google Scholar] [CrossRef]
- Steinke, J.M. The natural progression of kidney injury in young type 1 diabetic patients. Curr. Diab. Rep. 2009, 9, 473–479. [Google Scholar] [CrossRef]
- Vincent, A.M.; Feldman, E.L. New insights into the mechanisms of diabetic neuropathy. Rev. Endocr. Metab. Disord. 2004, 5, 227–236. [Google Scholar] [CrossRef]
- Callaghan, B.C.; Cheng, H.T.; Stables, C.L.; Smith, A.L.; Feldman, E.L. Diabetic neuropathy: Clinical manifestations and current treatments. Lancet Neurol. 2012, 11, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.M.; Russell, J.W.; Low, P.; Feldman, E.L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 2004, 25, 612–628. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S.; Mizukami, H.; Sugimoto, K. Mechanism of diabetic neuropathy: Where are we now and where to go? J. Diabetes Investig. 2011, 2, 18–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers. 2019, 5, 41. [Google Scholar] [CrossRef]
- Rawshani, A.; Rawshani, A.; Franzen, S.; Eliasson, B.; Svensson, A.M.; Miftaraj, M.; McGuire, D.K.; Sattar, N.; Rosengren, A.; Gudbjornsdottir, S. Range of Risk Factor Levels: Control, Mortality, and Cardiovascular Outcomes in Type 1 Diabetes Mellitus. Circulation 2017, 135, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Roep, B.O.; Posgai, A.; Wheeler, D.C.S.; Peakman, M. The challenge of modulating beta-cell autoimmunity in type 1 Diabetes. Lancet Diabetes Endocrinol. 2019, 7, 52–64. [Google Scholar] [CrossRef]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Type 1 Diabetes Genetics, C. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 Diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Onengut-Gumuscu, S.; Chen, W.M.; Burren, O.; Cooper, N.J.; Quinlan, A.R.; Mychaleckyj, J.C.; Farber, E.; Bonnie, J.K.; Szpak, M.; Schofield, E.; et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 2015, 47, 381–386. [Google Scholar] [CrossRef]
- Hunter, C.A. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 2005, 5, 521–531. [Google Scholar] [CrossRef]
- Meka, R.R.; Venkatesha, S.H.; Dudics, S.; Acharya, B.; Moudgil, K.D. IL-27-induced modulation of autoimmunity and its therapeutic potential. Autoimmun. Rev. 2015, 14, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Amaro, R.; Cortes, J.R.; Sanchez-Madrid, F.; Martin, P. Is CD69 an effective brake to control inflammatory diseases? Trends Mol. Med. 2013, 19, 625–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Mirza, A.H.; Brorsson, C.A.; Floyel, T.; Storling, J.; Mortensen, H.B.; Pociot, F.; Hvidoere International Study Group. The genetic and regulatory architecture of ERBB3-type 1 diabetes susceptibility locus. Mol. Cell Endocrinol. 2016, 419, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Floyel, T.; Brorsson, C.; Nielsen, L.B.; Miani, M.; Bang-Berthelsen, C.H.; Friedrichsen, M.; Overgaard, A.J.; Berchtold, L.A.; Wiberg, A.; Poulsen, P.; et al. CTSH regulates beta-cell function and disease progression in newly diagnosed type 1 diabetes patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10305–10310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, T.; Segre, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 Diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar]
- Pandey, A.; Patel, K.V.; Bahnson, J.L.; Gaussoin, S.A.; Martin, C.K.; Balasubramanyam, A.; Johnson, K.C.; McGuire, D.K.; Bertoni, A.G.; Kitzman, D.; et al. Association of Intensive Lifestyle Intervention, Fitness, and Body Mass Index With Risk of Heart Failure in Overweight or Obese Adults With Type 2 Diabetes Mellitus: An Analysis From the Look AHEAD Trial. Circulation 2020, 141, 1295–1306. [Google Scholar] [CrossRef]
- Rawshani, A.; Rawshani, A.; Franzen, S.; Eliasson, B.; Svensson, A.M.; Miftaraj, M.; McGuire, D.K.; Sattar, N.; Rosengren, A.; Gudbjornsdottir, S. Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes. N. Engl. J. Med. 2017, 376, 1407–1418. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, M.H.; Sawyer, J.M.; Afelik, S.; Jensen, J.; Leach, S.D. Exocrine ontogenies: On the development of pancreatic acinar, ductal and centroacinar cells. Semin. Cell Dev. Biol. 2012, 23, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Citro, A.; Ott, H.C. Can We Re-Engineer the Endocrine Pancreas? Curr. Diab. Rep. 2018, 18, 122. [Google Scholar] [CrossRef]
- Peloso, A.; Citro, A.; Zoro, T.; Cobianchi, L.; Kahler-Quesada, A.; Bianchi, C.M.; Andres, A.; Berishvili, E.; Piemonti, L.; Berney, T.; et al. Regenerative Medicine and Diabetes: Targeting the Extracellular Matrix Beyond the Stem Cell Approach and Encapsulation Technology. Front. Endocrinol. (Lausanne) 2018, 9, 445. [Google Scholar] [CrossRef] [Green Version]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.; Harb, G.; Poh, Y.C.; Sintov, E.; Gurtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef]
- O’Rahilly, R.; Muller, F. Developmental stages in human embryos: Revised and new measurements. Cells Tissues Organs 2010, 192, 73–84. [Google Scholar] [CrossRef]
- Jennings, R.E.; Berry, A.A.; Kirkwood-Wilson, R.; Roberts, N.A.; Hearn, T.; Salisbury, R.J.; Blaylock, J.; Piper Hanley, K.; Hanley, N.A. Development of the human pancreas from foregut to endocrine commitment. Diabetes 2013, 62, 3514–3522. [Google Scholar] [CrossRef] [Green Version]
- Rankin, S.A.; McCracken, K.W.; Luedeke, D.M.; Han, L.; Wells, J.M.; Shannon, J.M.; Zorn, A.M. Timing is everything: Reiterative Wnt, BMP and RA signaling regulate developmental competence during endoderm organogenesis. Dev. Biol. 2018, 434, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.; Papadopoulou, S.; Edlund, H. Fgf10 maintains notch activation, stimulates proliferation, and blocks differentiation of pancreatic epithelial cells. Dev. Dyn. 2003, 228, 185–193. [Google Scholar] [CrossRef]
- Jorgensen, M.C.; Ahnfelt-Ronne, J.; Hald, J.; Madsen, O.D.; Serup, P.; Hecksher-Sorensen, J. An illustrated review of early pancreas development in the mouse. Endocr. Rev. 2007, 28, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Pan, F.C.; Wright, C. Pancreas organogenesis: From bud to plexus to gland. Dev. Dyn. 2011, 240, 530–565. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, M.; Delgado, I.; Soria, B.; Martin, F.; Rojas, A. GATA4 and GATA6 control mouse pancreas organogenesis. J. Clin. Investig. 2012, 122, 3504–3515. [Google Scholar] [CrossRef] [Green Version]
- Wandzioch, E.; Zaret, K.S. Dynamic signaling network for the specification of embryonic pancreas and liver progenitors. Science 2009, 324, 1707–1710. [Google Scholar] [CrossRef] [Green Version]
- Jennings, R.E.; Berry, A.A.; Gerrard, D.T.; Wearne, S.J.; Strutt, J.; Withey, S.; Chhatriwala, M.; Piper Hanley, K.; Vallier, L.; Bobola, N.; et al. Laser Capture and Deep Sequencing Reveals the Transcriptomic Programmes Regulating the Onset of Pancreas and Liver Differentiation in Human Embryos. Stem Cell Rep. 2017, 9, 1387–1394. [Google Scholar] [CrossRef] [Green Version]
- Jennings, R.E.; Berry, A.A.; Strutt, J.P.; Gerrard, D.T.; Hanley, N.A. Human pancreas development. Development 2015, 142, 3126–3137. [Google Scholar] [CrossRef] [Green Version]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar]
- Salisbury, R.J.; Blaylock, J.; Berry, A.A.; Jennings, R.E.; De Krijger, R.; Piper Hanley, K.; Hanley, N.A. The window period of NEUROGENIN3 during human gestation. Islets 2014, 6, e954436. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Cho, H.; Rickert, R.W.; Li, Q.V.; Pulecio, J.; Leslie, C.S.; Huangfu, D. FOXA2 Is Required for Enhancer Priming during Pancreatic Differentiation. Cell Rep. 2019, 28, 382–393.e7. [Google Scholar] [CrossRef]
- Taylor, B.L.; Liu, F.F.; Sander, M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013, 4, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, A.E.; Taylor, B.L.; Benthuysen, J.R.; Liu, J.; Thorel, F.; Yuan, W.; Jiao, Y.; Kaestner, K.H.; Herrera, P.L.; Magnuson, M.A.; et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PLoS Genet. 2013, 9, e1003274. [Google Scholar] [CrossRef] [Green Version]
- Villamayor, L.; Rodriguez-Seguel, E.; Araujo, R.; Carrasco, M.; Bru-Tari, E.; Mellado-Gil, J.M.; Gauthier, B.R.; Martinelli, P.; Quesada, I.; Soria, B.; et al. GATA6 Controls Insulin Biosynthesis and Secretion in Adult beta-Cells. Diabetes 2018, 67, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhoose, A.M.; Samaras, S.; Artner, I.; Henderson, E.; Hang, Y.; Stein, R. MafA and MafB regulate Pdx1 transcription through the Area II control region in pancreatic beta cells. J. Biol. Chem. 2008, 283, 22612–22619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, N.; LeLay, J.; Vatamaniuk, M.Z.; Rieck, S.; Friedman, J.R.; Kaestner, K.H. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes. Dev. 2008, 22, 3435–3448. [Google Scholar] [CrossRef] [Green Version]
- Harrison, K.A.; Thaler, J.; Pfaff, S.L.; Gu, H.; Kehrl, J.H. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat. Genet. 1999, 23, 71–75. [Google Scholar] [CrossRef]
- Sharma, S.; Leonard, J.; Lee, S.; Chapman, H.D.; Leiter, E.H.; Montminy, M.R. Pancreatic islet expression of the homeobox factor STF-1 relies on an E-box motif that binds USF. J. Biol. Chem. 1996, 271, 2294–2299. [Google Scholar] [CrossRef] [Green Version]
- Melloul, D.; Marshak, S.; Cerasi, E. Regulation of pdx-1 gene expression. Diabetes 2002, 51 (Suppl. 3), S320–S325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, S.; Roy, N.; Russ, H.A.; Leonhardt, L.; French, E.K.; Roy, R.; Bengtsson, H.; Scott, D.K.; Stewart, A.F.; Hebrok, M. Replication confers beta cell immaturity. Nat. Commun. 2018, 9, 485. [Google Scholar] [CrossRef]
- Helman, A.; Avrahami, D.; Klochendler, A.; Glaser, B.; Kaestner, K.H.; Ben-Porath, I.; Dor, Y. Effects of ageing and senescence on pancreatic beta-cell function. Diabetes Obes. Metab. 2016, 18 (Suppl. 1), 58–62. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516. [Google Scholar] [CrossRef]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 Diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.J. The Current Drug Treatment Landscape for Diabetes and Perspectives for the Future. Clin. Pharmacol. Ther. 2015, 98, 170–184. [Google Scholar] [CrossRef]
- Wilke, T.; Mueller, S.; Groth, A.; Fuchs, A.; Seitz, L.; Kienhofer, J.; Maywald, U.; Lundershausen, R.; Wehling, M. Treatment-dependent and treatment-independent risk factors associated with the risk of diabetes-related events: A retrospective analysis based on 229,042 patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2015, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.A.; Creager, M.A. Vascular Complications of Diabetes. Circ. Res. 2016, 118, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Latres, E.; Finan, D.A.; Greenstein, J.L.; Kowalski, A.; Kieffer, T.J. Navigating Two Roads to Glucose Normalization in Diabetes: Automated Insulin Delivery Devices and Cell Therapy. Cell Metab. 2019, 29, 545–563. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Gamble, A.; Pepper, A.R.; Bruni, A.; Shapiro, A.M.J. The journey of islet cell transplantation and future development. Islets 2018, 10, 80–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, N.J.; Powers, A.C. Use of human islets to understand islet biology and diabetes: Progress, challenges and suggestions. Diabetologia 2019, 62, 212–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickels, M.R.; Robertson, R.P. Pancreatic Islet Transplantation in Humans: Recent Progress and Future Directions. Endocr. Rev. 2019, 40, 631–668. [Google Scholar] [CrossRef] [Green Version]
- Vantyghem, M.C.; de Koning, E.J.P.; Pattou, F.; Rickels, M.R. Advances in beta-cell replacement therapy for the treatment of type 1 Diabetes. Lancet 2019, 394, 1274–1285. [Google Scholar] [CrossRef]
- Gamble, A.; Pawlick, R.; Pepper, A.R.; Bruni, A.; Adesida, A.; Senior, P.A.; Korbutt, G.S.; Shapiro, A.M.J. Improved islet recovery and efficacy through co-culture and co-transplantation of islets with human adipose-derived mesenchymal stem cells. PLoS ONE 2018, 13, e0206449. [Google Scholar] [CrossRef]
- Johnson, I.S. Human insulin from recombinant DNA technology. Science 1983, 219, 632–637. [Google Scholar] [CrossRef]
- MacLeod, K.M.; Gold, A.E.; Frier, B.M. Frequency, severity and symptomatology of hypoglycaemia: A comparative trial of human and porcine insulins in type 1 diabetic patients. Diabet. Med. 1995, 12, 134–141. [Google Scholar] [CrossRef]
- Griesemer, A.; Yamada, K.; Sykes, M. Xenotransplantation: Immunological hurdles and progress toward tolerance. Immunol. Rev. 2014, 258, 241–258. [Google Scholar] [CrossRef] [Green Version]
- Hering, B.J.; Clarke, W.R.; Bridges, N.D.; Eggerman, T.L.; Alejandro, R.; Bellin, M.D.; Chaloner, K.; Czarniecki, C.W.; Goldstein, J.S.; Hunsicker, L.G.; et al. Phase 3 Trial of Transplantation of Human Islets in Type 1 Diabetes Complicated by Severe Hypoglycemia. Diabetes Care 2016, 39, 1230–1240. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Platero-Luengo, A.; Sakurai, M.; Sugawara, A.; Gil, M.A.; Yamauchi, T.; Suzuki, K.; Bogliotti, Y.S.; Cuello, C.; Morales Valencia, M.; et al. Interspecies Chimerism with Mammalian Pluripotent Stem Cells. Cell 2017, 168, 473–486.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Sato, H.; Kato-Itoh, M.; Goto, T.; Hara, H.; Sanbo, M.; Mizuno, N.; Kobayashi, T.; Yanagida, A.; Umino, A.; et al. Interspecies organogenesis generates autologous functional islets. Nature 2017, 542, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Kemter, E.; Klymiuk, N.; Reichart, B. Genetically modified pigs as donors of cells, tissues, and organs for xenotransplantation. Anim. Front. 2019, 9, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klymiuk, N.; van Buerck, L.; Bahr, A.; Offers, M.; Kessler, B.; Wuensch, A.; Kurome, M.; Thormann, M.; Lochner, K.; Nagashima, H.; et al. Xenografted islet cell clusters from INSLEA29Y transgenic pigs rescue diabetes and prevent immune rejection in humanized mice. Diabetes 2012, 61, 1527–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samy, K.P.; Martin, B.M.; Turgeon, N.A.; Kirk, A.D. Islet cell xenotransplantation: A serious look toward the clinic. Xenotransplantation 2014, 21, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.K.; Matsumoto, S.; Abalovich, A.; Itoh, T.; Mourad, N.I.; Gianello, P.R.; Wolf, E.; Cozzi, E. Progress in Clinical Encapsulated Islet Xenotransplantation. Transplantation 2016, 100, 2301–2308. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teta, M.; Long, S.Y.; Wartschow, L.M.; Rankin, M.M.; Kushner, J.A. Very slow turnover of beta-cells in aged adult mice. Diabetes 2005, 54, 2557–2567. [Google Scholar] [CrossRef] [Green Version]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Fiaschi-Taesch, N.M.; Vasavada, R.C.; Scott, D.K.; Garcia-Ocana, A.; Stewart, A.F. Diabetes mellitus--advances and challenges in human beta-cell proliferation. Nat. Rev. Endocrinol. 2015, 11, 201–212. [Google Scholar] [CrossRef]
- Saunders, D.; Powers, A.C. Replicative capacity of beta-cells and type 1 Diabetes. J. Autoimmun. 2016, 71, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Gregg, B.E.; Moore, P.C.; Demozay, D.; Hall, B.A.; Li, M.; Husain, A.; Wright, A.J.; Atkinson, M.A.; Rhodes, C.J. Formation of a human beta-cell population within pancreatic islets is set early in life. J. Clin. Endocrinol. Metab. 2012, 97, 3197–3206. [Google Scholar] [CrossRef]
- Wang, P.; Alvarez-Perez, J.C.; Felsenfeld, D.P.; Liu, H.; Sivendran, S.; Bender, A.; Kumar, A.; Sanchez, R.; Scott, D.K.; Garcia-Ocana, A.; et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat. Med. 2015, 21, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dancik, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A Stimulates Human beta-Cell Proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef] [Green Version]
- Thorel, F.; Nepote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chera, S.; Baronnier, D.; Ghila, L.; Cigliola, V.; Jensen, J.N.; Gu, G.; Furuyama, K.; Thorel, F.; Gribble, F.M.; Reimann, F.; et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature 2014, 514, 503–507. [Google Scholar] [CrossRef]
- Kopp, J.L.; Grompe, M.; Sander, M. Stem cells versus plasticity in liver and pancreas regeneration. Nat. Cell Biol. 2016, 18, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Guo, P.; Shiota, C.; Zhang, T.; Coudriet, G.M.; Fischbach, S.; Prasadan, K.; Fusco, J.; Ramachandran, S.; Witkowski, P.; et al. Endogenous Reprogramming of Alpha Cells into Beta Cells, Induced by Viral Gene Therapy, Reverses Autoimmune Diabetes. Cell Stem Cell 2018, 22, 78–90 e4. [Google Scholar] [CrossRef] [Green Version]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Courtney, M.; Gjernes, E.; Druelle, N.; Ravaud, C.; Vieira, A.; Ben-Othman, N.; Pfeifer, A.; Avolio, F.; Leuckx, G.; Lacas-Gervais, S.; et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet. 2013, 9, e1003934. [Google Scholar] [CrossRef]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Kitamura, T.; DePinho, R.A.; Accili, D. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nat. Genet. 2012, 44, 406–412. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Cavelti-Weder, C.; Zhang, Y.; Clement, K.; Donovan, S.; Gonzalez, G.; Zhu, J.; Stemann, M.; Xu, K.; Hashimoto, T.; et al. Long-term persistence and development of induced pancreatic beta cells generated by lineage conversion of acinar cells. Nat. Biotechnol. 2014, 32, 1223–1230. [Google Scholar] [CrossRef]
- Lemper, M.; Leuckx, G.; Heremans, Y.; German, M.S.; Heimberg, H.; Bouwens, L.; Baeyens, L. Reprogramming of human pancreatic exocrine cells to beta-like cells. Cell Death Differ. 2015, 22, 1117–1130. [Google Scholar] [CrossRef] [Green Version]
- Meivar-Levy, I.; Ferber, S. Liver to Pancreas Transdifferentiation. Curr. Diab. Rep. 2019, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Galivo, F.; Benedetti, E.; Wang, Y.; Pelz, C.; Schug, J.; Kaestner, K.H.; Grompe, M. Reprogramming human gallbladder cells into insulin-producing beta-like cells. PLoS ONE 2017, 12, e0181812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Fischbach, G.D.; Fischbach, R.L. Stem cells: Science, policy, and ethics. J. Clin. Investig. 2004, 114, 1364–1370. [Google Scholar] [CrossRef]
- Mertes, H.; Pennings, G.; Van Steirteghem, A. An ethical analysis of alternative methods to obtain pluripotent stem cells without destroying embryos. Hum. Reprod. 2006, 21, 2749–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Los Angeles, A.; Ferrari, F.; Xi, R.; Fujiwara, Y.; Benvenisty, N.; Deng, H.; Hochedlinger, K.; Jaenisch, R.; Lee, S.; Leitch, H.G.; et al. Hallmarks of pluripotency. Nature 2015, 525, 469–478. [Google Scholar] [CrossRef]
- Bramswig, N.C.; Everett, L.J.; Schug, J.; Dorrell, C.; Liu, C.; Luo, Y.; Streeter, P.R.; Naji, A.; Grompe, M.; Kaestner, K.H. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J. Clin. Investig. 2013, 123, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.D. The quest to make fully functional human pancreatic beta cells from embryonic stem cells: Climbing a mountain in the clouds. Diabetologia 2016, 59, 2047–2057. [Google Scholar] [CrossRef]
- Kieffer, T.J. Closing in on Mass Production of Mature Human Beta Cells. Cell Stem Cell 2016, 18, 699–702. [Google Scholar] [CrossRef] [Green Version]
- Saxena, P.; Heng, B.C.; Bai, P.; Folcher, M.; Zulewski, H.; Fussenegger, M. A programmable synthetic lineage-control network that differentiates human IPSCs into glucose-sensitive insulin-secreting beta-like cells. Nat. Commun. 2016, 7, 11247. [Google Scholar] [CrossRef] [Green Version]
- Ameri, J.; Borup, R.; Prawiro, C.; Ramond, C.; Schachter, K.A.; Scharfmann, R.; Semb, H. Efficient Generation of Glucose-Responsive Beta Cells from Isolated GP2(+) Human Pancreatic Progenitors. Cell Rep. 2017, 19, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Kelly, O.G.; Chan, M.Y.; Martinson, L.A.; Kadoya, K.; Ostertag, T.M.; Ross, K.G.; Richardson, M.; Carpenter, M.K.; D’Amour, K.A.; Kroon, E.; et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol. 2011, 29, 750–756. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [Green Version]
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541. [Google Scholar] [CrossRef]
- Korytnikov, R.; Nostro, M.C. Generation of polyhormonal and multipotent pancreatic progenitor lineages from human pluripotent stem cells. Methods 2016, 101, 56–64. [Google Scholar] [CrossRef]
- Shahjalal, H.M.; Shiraki, N.; Sakano, D.; Kikawa, K.; Ogaki, S.; Baba, H.; Kume, K.; Kume, S. Generation of insulin-producing beta-like cells from human iPS cells in a defined and completely xeno-free culture system. J. Mol. Cell Biol. 2014, 6, 394–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452. [Google Scholar] [CrossRef] [PubMed]
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.C.; Young, H.Y.; Agulnick, A.D.; Babin, M.J.; Baetge, E.E.; Bang, A.G.; Bhoumik, A.; Cepa, I.; Cesario, R.M.; Haakmeester, C.; et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS ONE 2012, 7, e37004. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Au, M.; Lu, K.; Eshpeter, A.; Korbutt, G.; Fisk, G.; Majumdar, A.S. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells 2007, 25, 1940–1953. [Google Scholar] [CrossRef]
- Nostro, M.C.; Sarangi, F.; Ogawa, S.; Holtzinger, A.; Corneo, B.; Li, X.; Micallef, S.J.; Park, I.H.; Basford, C.; Wheeler, M.B.; et al. Stage-specific signaling through TGFbeta family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development 2011, 138, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nostro, M.C.; Sarangi, F.; Yang, C.; Holland, A.; Elefanty, A.G.; Stanley, E.G.; Greiner, D.L.; Keller, G. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015, 4, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Bruin, J.E.; Erener, S.; Vela, J.; Hu, X.; Johnson, J.D.; Kurata, H.T.; Lynn, F.C.; Piret, J.M.; Asadi, A.; Rezania, A.; et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014, 12, 194–208. [Google Scholar] [CrossRef] [Green Version]
- Lee Chong, T.; Ahearn, E.L.; Cimmino, L. Reprogramming the Epigenome With Vitamin C. Front. Cell Dev. Biol. 2019, 7, 128. [Google Scholar] [CrossRef]
- Young, J.I.; Zuchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef] [Green Version]
- Bruin, J.E.; Saber, N.; O’Dwyer, S.; Fox, J.K.; Mojibian, M.; Arora, P.; Rezania, A.; Kieffer, T.J. Hypothyroidism Impairs Human Stem Cell-Derived Pancreatic Progenitor Cell Maturation in Mice. Diabetes 2016, 65, 1297–1309. [Google Scholar] [CrossRef] [Green Version]
- Kotliar, D.; Veres, A.; Nagy, M.A.; Tabrizi, S.; Hodis, E.; Melton, D.A.; Sabeti, P.C. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Helman, A.; Melton, D.A. A Stem Cell Approach to Cure Type 1 Diabetes. Cold Spring Harb. Perspect. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; Wei, Z.; Lin, C.S.; Fang, S.; Ahmadian, M.; Kida, Y.; Tseng, T.; Dai, Y.; Yu, R.T.; Liddle, C.; et al. ERRgamma Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive beta Cells. Cell Metab. 2016, 23, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.C.; Alves, T.C.; Helman, A.; Chen, J.C.; Kenty, J.H.; Cardone, R.L.; Liu, D.R.; Kibbey, R.G.; Melton, D.A. Glucose Response by Stem Cell-Derived beta Cells In Vitro Is Inhibited by a Bottleneck in Glycolysis. Cell Rep. 2020, 31, 107623. [Google Scholar] [CrossRef]
- Vegas, A.J.; Veiseh, O.; Gurtler, M.; Millman, J.R.; Pagliuca, F.W.; Bader, A.R.; Doloff, J.C.; Li, J.; Chen, M.; Olejnik, K.; et al. Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nat. Med. 2016, 22, 306–311. [Google Scholar] [CrossRef]
- Stock, A.A.; Manzoli, V.; De Toni, T.; Abreu, M.M.; Poh, Y.C.; Ye, L.; Roose, A.; Pagliuca, F.W.; Thanos, C.; Ricordi, C.; et al. Conformal Coating of Stem Cell-Derived Islets for beta Cell Replacement in Type 1 Diabetes. Stem Cell Rep. 2020, 14, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Okano, H.; Sipp, D. New trends in cellular therapy. Development 2020, 147. [Google Scholar] [CrossRef]
- Millman, J.R.; Xie, C.; Van Dervort, A.; Gurtler, M.; Pagliuca, F.W.; Melton, D.A. Generation of stem cell-derived beta-cells from patients with type 1 Diabetes. Nat. Commun. 2016, 7, 11463. [Google Scholar] [CrossRef] [Green Version]
- Bhansali, S.; Dutta, P.; Kumar, V.; Yadav, M.K.; Jain, A.; Mudaliar, S.; Bhansali, S.; Sharma, R.R.; Jha, V.; Marwaha, N.; et al. Efficacy of Autologous Bone Marrow-Derived Mesenchymal Stem Cell and Mononuclear Cell Transplantation in Type 2 Diabetes Mellitus: A Randomized, Placebo-Controlled Comparative Study. Stem Cells Dev. 2017, 26, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, Y.; Gong, H.; Yu, C.; Guo, C.; Wang, F.; Yan, S.; Xu, H. Long term effect and safety of Wharton’s jelly-derived mesenchymal stem cells on type 2 Diabetes. Exp. Ther. Med. 2016, 12, 1857–1866. [Google Scholar] [CrossRef]
- Sood, V.; Bhansali, A.; Mittal, B.R.; Singh, B.; Marwaha, N.; Jain, A.; Khandelwal, N. Autologous bone marrow derived stem cell therapy in patients with type 2 diabetes mellitus—Defining adequate administration methods. World J. Diabetes 2017, 8, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Cerrada, V.; Garcia-Lopez, M.; Moreno-Izquierdo, A.; Villaverde, C.; Zurita, O.; Martin-Merida, M.I.; Arenas, J.; Ayuso, C.; Gallardo, M.E. Derivation of a human DOA iPSC line, IISHDOi006-A, with a mutation in the ACO2 gene: C.1999G>A; p.Glu667Lys. Stem Cell Res. 2019, 40, 101566. [Google Scholar] [CrossRef] [PubMed]
- Garreta, E.; Sanchez, S.; Lajara, J.; Montserrat, N.; Belmonte, J.C.I. Roadblocks in the Path of iPSC to the Clinic. Curr. Transplant. Rep. 2018, 5, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Hwang, G.; Jeong, H.; Yang, H.K.; Kim, H.S.; Hong, H.; Kim, N.J.; Oh, I.H.; Yim, H.W. Efficacies of Stem Cell Therapies for Functional Improvement of the beta Cell in Patients with Diabetes: A Systematic Review of Controlled Clinical Trials. Int. J. Stem Cells 2019, 12, 195–205. [Google Scholar] [CrossRef]
- Legoy, T.A.; Vethe, H.; Abadpour, S.; Strand, B.L.; Scholz, H.; Paulo, J.A.; Raeder, H.; Ghila, L.; Chera, S. Encapsulation boosts islet-cell signature in differentiating human induced pluripotent stem cells via integrin signalling. Sci. Rep. 2020, 10, 414. [Google Scholar] [CrossRef] [Green Version]
- Strand, B.L.; Coron, A.E.; Skjak-Braek, G. Current and Future Perspectives on Alginate Encapsulated Pancreatic Islet. Stem Cells Transl. Med. 2017, 6, 1053–1058. [Google Scholar] [CrossRef]
- Desai, T.; Shea, L.D. Advances in islet encapsulation technologies. Nat. Rev. Drug Discov. 2017, 16, 338–350. [Google Scholar] [CrossRef]
- Clough, D.W.; King, J.L.; Li, F.; Shea, L.D. Integration of Islet/Beta-Cell Transplants with Host Tissue Using Biomaterial Platforms. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- Gebe, J.A.; Preisinger, A.; Gooden, M.D.; D’Amico, L.A.; Vernon, R.B. Local, Controlled Release In Vivo of Vascular Endothelial Growth Factor Within a Subcutaneous Scaffolded Islet Implant Reduces Early Islet Necrosis and Improves Performance of the Graft. Cell Transpl. 2018, 27, 531–541. [Google Scholar] [CrossRef]
- Kondo, Y.; Toyoda, T.; Inagaki, N.; Osafune, K. iPSC technology-based regenerative therapy for Diabetes. J. Diabetes Investig. 2018, 9, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostolou, E.; Stadtfeld, M. Cellular trajectories and molecular mechanisms of iPSC reprogramming. Curr. Opin. Genet. Dev. 2018, 52, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Rao, M.S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2007, 2, 3081–3089. [Google Scholar] [CrossRef]
- Gorecka, J.; Kostiuk, V.; Fereydooni, A.; Gonzalez, L.; Luo, J.; Dash, B.; Isaji, T.; Ono, S.; Liu, S.; Lee, S.R.; et al. The potential and limitations of induced pluripotent stem cells to achieve wound healing. Stem Cell Res. Ther. 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Hochedlinger, K.; Jaenisch, R. Induced Pluripotency and Epigenetic Reprogramming. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Path, G.; Perakakis, N.; Mantzoros, C.S.; Seufert, J. Stem cells in the treatment of diabetes mellitus—Focus on mesenchymal stem cells. Metabolism 2019, 90, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Warren, L.; Lin, C. mRNA-Based Genetic Reprogramming. Mol. Ther. 2019, 27, 729–734. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Tao, Z.; Loo, S.; Su, L.; Chen, X.; Ye, L. Non-viral vector based gene transfection with human induced pluripotent stem cells derived cardiomyocytes. Sci. Rep. 2019, 9, 14404. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Brix, J.; Zhou, Y.; Luo, Y. The Epigenetic Reprogramming Roadmap in Generation of iPSCs from Somatic Cells. J. Genet. Genom. 2015, 42, 661–670. [Google Scholar] [CrossRef]
- Zhao, T.; Zhang, Z.N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Haworth, R.; Sharpe, M. Accept or Reject: The Role of Immune Tolerance in the Development of Stem Cell Therapies and Possible Future Approaches. Toxicol. Pathol. 2020. [Google Scholar] [CrossRef]
- Chakradhar, S. An eye to the future: Researchers debate best path for stem cell-derived therapies. Nat. Med. 2016, 22, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M. Assessing commercial opportunities for autologous and allogeneic cell-based products. Regen. Med. 2012, 7, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Trounson, A.; Boyd, N.R.; Boyd, R.L. Toward a Universal Solution: Editing Compatibility into Pluripotent Stem Cells. Cell Stem Cell 2019, 24, 508–510. [Google Scholar] [CrossRef] [Green Version]
- Pappas, D.J.; Gourraud, P.A.; Le Gall, C.; Laurent, J.; Trounson, A.; DeWitt, N.; Talib, S. Proceedings: Human leukocyte antigen haplo-homozygous induced pluripotent stem cell haplobank modeled after the california population: Evaluating matching in a multiethnic and admixed population. Stem Cells Transl. Med. 2015, 4, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 2019, 24, 566–578 e7. [Google Scholar] [CrossRef] [Green Version]
- Volarevic, V.; Markovic, B.S.; Gazdic, M.; Volarevic, A.; Jovicic, N.; Arsenijevic, N.; Armstrong, L.; Djonov, V.; Lako, M.; Stojkovic, M. Ethical and Safety Issues of Stem Cell-Based Therapy. Int. J. Med. Sci. 2018, 15, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, A.A.; Ross, E.G.; Bolli, R.; Pepine, C.J.; Leeper, N.J.; Yang, P.C. The Promise and Challenge of Induced Pluripotent Stem Cells for Cardiovascular Applications. JACC Basic Transl. Sci. 2016, 1, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.J.; Zhang, G.; Garfield, S.H.; Shi, Y.J.; Chen, K.G.; Robey, P.G.; Leapman, R.D. Variations in Glycogen Synthesis in Human Pluripotent Stem Cells with Altered Pluripotent States. PLoS ONE 2015, 10, e0142554. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lacroix, N.; Li, Q. Histone deacetylase inhibitor valproic acid as a small molecule inducer to direct the differentiation of pluripotent stem cells. Methods Mol. Biol. 2013, 977, 359–363. [Google Scholar]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Chen, J.; Wu, Y.; et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef]
- Chen, T.; Dent, S.Y. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Krentz, N.A.J.; Gloyn, A.L. Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat. Rev. Endocrinol. 2020, 16, 202–212. [Google Scholar] [CrossRef]
- Zimmermann, W.H. Challenges to heart repair with pluripotent stem cell-derived cardiomyocytes. Trends Cardiovasc. Med. 2020. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPR-based functional genomics for neurological disease. Nat. Rev. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nicetto, D.; Donahue, G.; Jain, T.; Peng, T.; Sidoli, S.; Sheng, L.; Montavon, T.; Becker, J.S.; Grindheim, J.M.; Blahnik, K.; et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science 2019, 363, 294–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, D.B.; Bonasio, R.; Kaneko, S.; Li, G.; Li, G.; Margueron, R.; Oda, H.; Sarma, K.; Sims, R.J., 3rd; Son, J.; et al. Chromatin in the nuclear landscape. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 11–22. [Google Scholar] [CrossRef]
- Jacob, Y.; Bergamin, E.; Donoghue, M.T.; Mongeon, V.; LeBlanc, C.; Voigt, P.; Underwood, C.J.; Brunzelle, J.S.; Michaels, S.D.; Reinberg, D.; et al. Selective methylation of histone H3 variant H3.1 regulates heterochromatin replication. Science 2014, 343, 1249–1253. [Google Scholar] [CrossRef] [Green Version]
- Nicetto, D.; Zaret, K.S. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet. Dev. 2019, 55, 1–10. [Google Scholar] [CrossRef]
- Astro, V.; Adamo, A. Epigenetic Control of Endocrine Pancreas Differentiation in vitro: Current Knowledge and Future Perspectives. Front. Cell Dev. Biol. 2018, 6, 141. [Google Scholar] [CrossRef]
- Xie, R.; Everett, L.J.; Lim, H.W.; Patel, N.A.; Schug, J.; Kroon, E.; Kelly, O.G.; Wang, A.; D’Amour, K.A.; Robins, A.J.; et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell 2013, 12, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Wang, X.; Moazed, D. Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature 2018, 558, 615–619. [Google Scholar] [CrossRef]
- Thienpont, B.; Aronsen, J.M.; Robinson, E.L.; Okkenhaug, H.; Loche, E.; Ferrini, A.; Brien, P.; Alkass, K.; Tomasso, A.; Agrawal, A.; et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Investig. 2017, 127, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Papait, R.; Serio, S.; Pagiatakis, C.; Rusconi, F.; Carullo, P.; Mazzola, M.; Salvarani, N.; Miragoli, M.; Condorelli, G. Histone Methyltransferase G9a Is Required for Cardiomyocyte Homeostasis and Hypertrophy. Circulation 2017, 136, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Matsumura, Y.; Fukuda, M.; Kimura, H.; Shinkai, Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 2008, 27, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; McCarthy, R.L.; Sidoli, S.; Donahue, G.; Kaeding, K.E.; He, Z.; Lin, S.; Garcia, B.A.; Zaret, K.S. Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol. Cell 2017, 68, 1023–1037.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, O. On your histone mark, SET, methylate! Epigenetics 2013, 8, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef]
- Wang, C.; Liu, X.; Gao, Y.; Yang, L.; Li, C.; Liu, W.; Chen, C.; Kou, X.; Zhao, Y.; Chen, J.; et al. Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol. 2018, 20, 620–631. [Google Scholar] [CrossRef]
- Ninova, M.; Fejes Toth, K.; Aravin, A.A. The control of gene expression and cell identity by H3K9 trimethylation. Development 2019, 146, dev181180. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.T.; Heyne, S.; Dror, E.; Casas, E.; Leonhardt, L.; Boenke, T.; Yang, C.H.; Sagar; Arrigoni, L.; Dalgaard, K.; et al. The Polycomb-Dependent Epigenome Controls beta Cell Dysfunction, Dedifferentiation, and Diabetes. Cell Metab. 2018, 27, 1294–1308.e7. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.G.; Iglesias, A.H.; Lizcano, F.; Villanueva, R.; Camelo, S.; Jingu, H.; Teh, B.T.; Koibuchi, N.; Chin, W.W.; Kokkotou, E.; et al. Functional characterization of JMJD2A, a histone deacetylase- and retinoblastoma-binding protein. J. Biol. Chem. 2005, 280, 28507–28518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales, W.; Lizcano, F. The Histone Demethylase JMJD2A Modulates the Induction of Hypertrophy Markers in iPSC-Derived Cardiomyocytes. Front. Genet. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, D.; Bracken, A.P.; Agger, K.; Christensen, J.; Hansen, K.; Cloos, P.A.; Helin, K. Regulation of stem cell differentiation by histone methyltransferases and demethylases. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 253–263. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, G.W.; Jeon, Y.H.; Yoo, J.; Lee, S.W.; Kwon, S.H. Advances in histone demethylase KDM4 as cancer therapeutic targets. FASEB J. 2020, 34, 3461–3484. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; Lizcano, F. Kdm4c is Recruited to Mitotic Chromosomes and Is Relevant for Chromosomal Stability, Cell Migration and Invasion of Triple Negative Breast Cancer Cells. Breast Cancer (Auckl) 2018, 12, 1178223418773075. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Huang, K.; Zhu, Y.; Wang, T.; Shan, Y.; Long, B.; Li, Y.; Chen, Q.; Wang, P.; Zhao, S.; et al. Vitamin C-dependent lysine demethylase 6 (KDM6)-mediated demethylation promotes a chromatin state that supports the endothelial-to-hematopoietic transition. J. Biol. Chem. 2019, 294, 13657–13670. [Google Scholar] [CrossRef]
- Coskun, E.; Ercin, M.; Gezginci-Oktayoglu, S. The Role of Epigenetic Regulation and Pluripotency-Related MicroRNAs in Differentiation of Pancreatic Stem Cells to Beta Cells. J. Cell Biochem. 2018, 119, 455–467. [Google Scholar] [CrossRef]
- LaPierre, M.P.; Stoffel, M. MicroRNAs as stress regulators in pancreatic beta cells and Diabetes. Mol. Metab. 2017, 6, 1010–1023. [Google Scholar] [CrossRef]
- Dhawan, S.; Tschen, S.I.; Zeng, C.; Guo, T.; Hebrok, M.; Matveyenko, A.; Bhushan, A. DNA methylation directs functional maturation of pancreatic beta cells. J. Clin. Investig. 2015, 125, 2851–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avrahami, D.; Li, C.; Zhang, J.; Schug, J.; Avrahami, R.; Rao, S.; Stadler, M.B.; Burger, L.; Schubeler, D.; Glaser, B.; et al. Aging-Dependent Demethylation of Regulatory Elements Correlates with Chromatin State and Improved beta Cell Function. Cell Metab. 2015, 22, 619–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhawan, S.; Georgia, S.; Tschen, S.I.; Fan, G.; Bhushan, A. Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Dev. Cell 2011, 20, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saber, N.; Bruin, J.E.; O’Dwyer, S.; Schuster, H.; Rezania, A.; Kieffer, T.J. Sex Differences in Maturation of Human Embryonic Stem Cell-Derived beta Cells in Mice. Endocrinology 2018, 159, 1827–1841. [Google Scholar] [CrossRef]
- Abazari, M.F.; Nasiri, N.; Nejati, F.; Zare Karizi, S.; Amini Faskhodi, M.; Saburi, E.; Aghapur, N.; Mahdavi, M.R.; Ardeshirylajimi, A.; Enderami, S.E.; et al. Comparison of human-induced pluripotent stem cells and mesenchymal stem cell differentiation potential to insulin producing cells in 2D and 3D culture systems in vitro. J. Cell Physiol. 2020, 235, 4239–4246. [Google Scholar] [CrossRef]
- Odorico, J.; Markmann, J.; Melton, D.; Greenstein, J.; Hwa, A.; Nostro, C.; Rezania, A.; Oberholzer, J.; Pipeleers, D.; Yang, L.; et al. Report of the Key Opinion Leaders Meeting on Stem Cell-derived Beta Cells. Transplantation 2018, 102, 1223–1229. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroyave, F.; Montaño, D.; Lizcano, F. Diabetes Mellitus Is a Chronic Disease that Can Benefit from Therapy with Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 8685. https://doi.org/10.3390/ijms21228685
Arroyave F, Montaño D, Lizcano F. Diabetes Mellitus Is a Chronic Disease that Can Benefit from Therapy with Induced Pluripotent Stem Cells. International Journal of Molecular Sciences. 2020; 21(22):8685. https://doi.org/10.3390/ijms21228685
Chicago/Turabian StyleArroyave, Felipe, Diana Montaño, and Fernando Lizcano. 2020. "Diabetes Mellitus Is a Chronic Disease that Can Benefit from Therapy with Induced Pluripotent Stem Cells" International Journal of Molecular Sciences 21, no. 22: 8685. https://doi.org/10.3390/ijms21228685
APA StyleArroyave, F., Montaño, D., & Lizcano, F. (2020). Diabetes Mellitus Is a Chronic Disease that Can Benefit from Therapy with Induced Pluripotent Stem Cells. International Journal of Molecular Sciences, 21(22), 8685. https://doi.org/10.3390/ijms21228685