ANK2 Hypermethylation in Canine Mammary Tumors and Human Breast Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
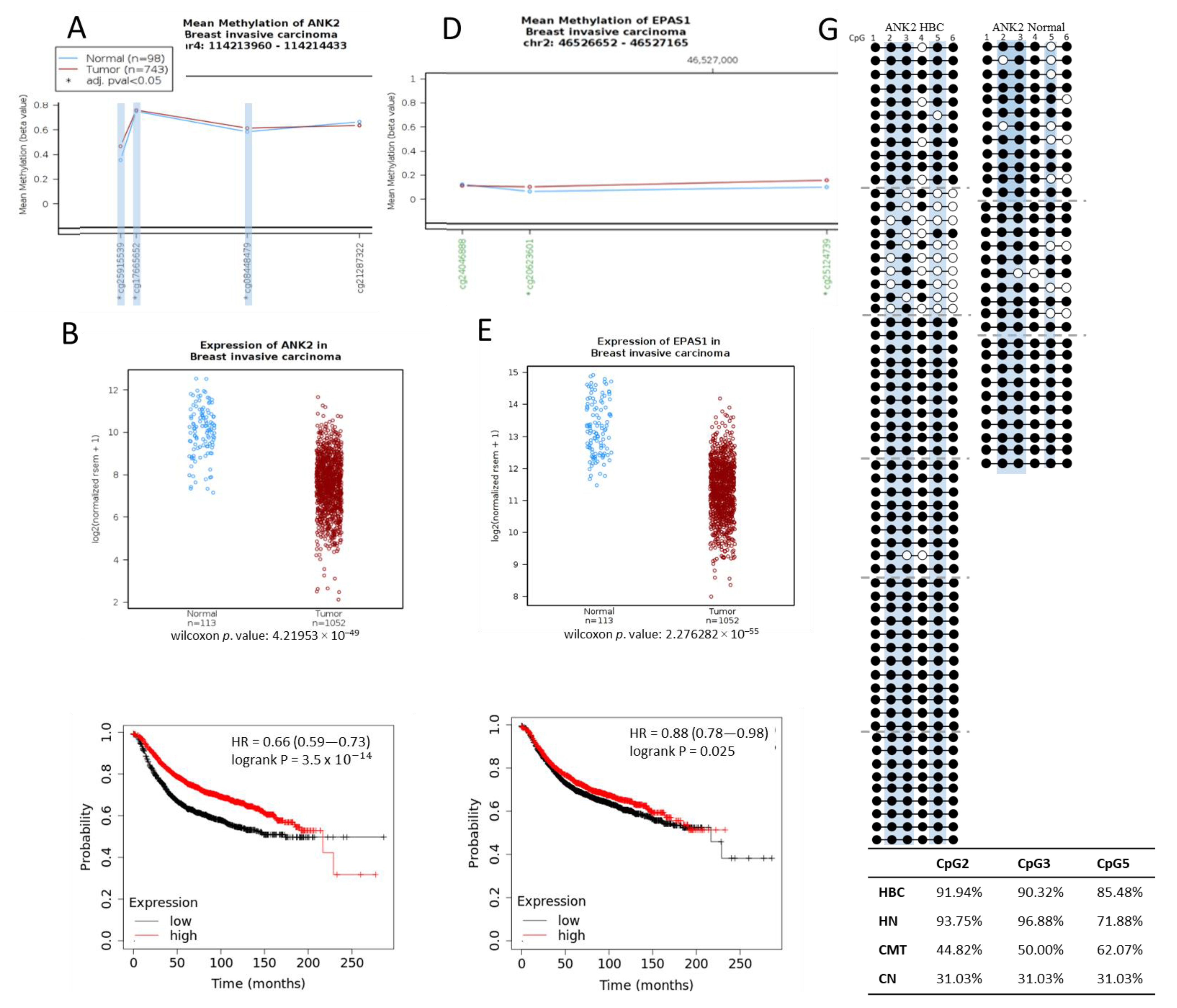
2. Results
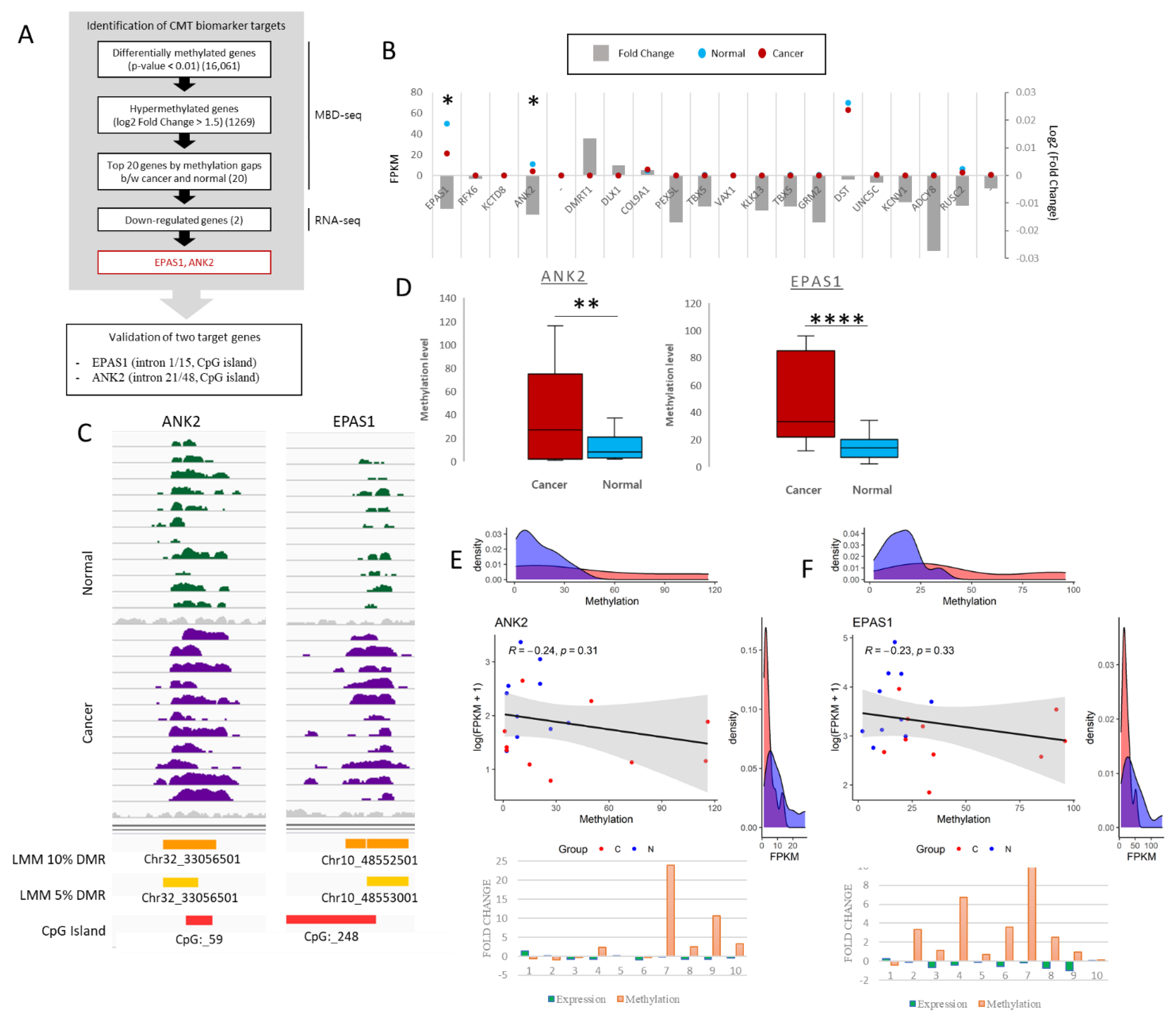
2.1. Identification of Differentially Methylated Regions
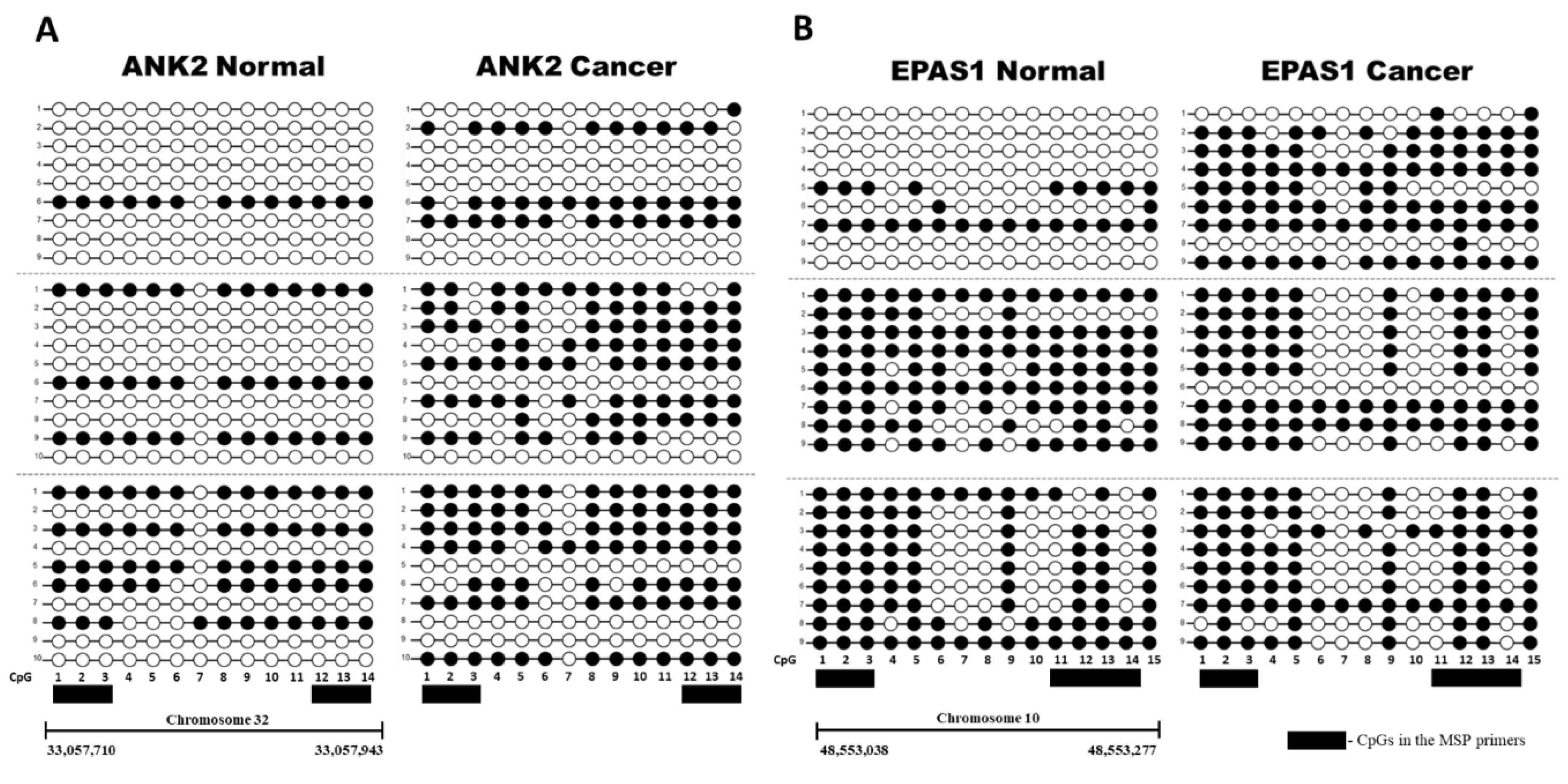
2.2. Evaluation of Differentially Methylated Regions in CMT and Adjacent Normal Tissue
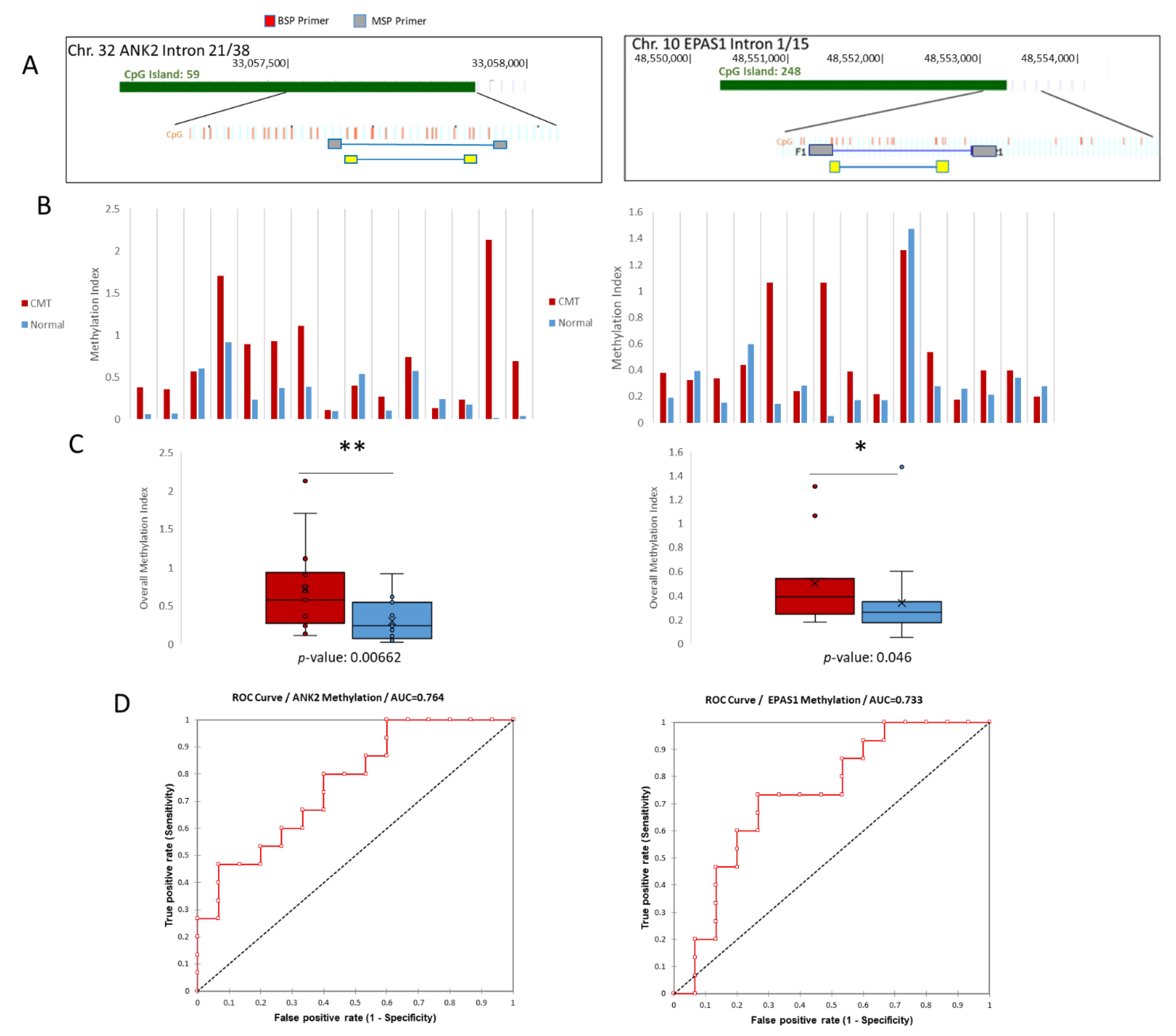
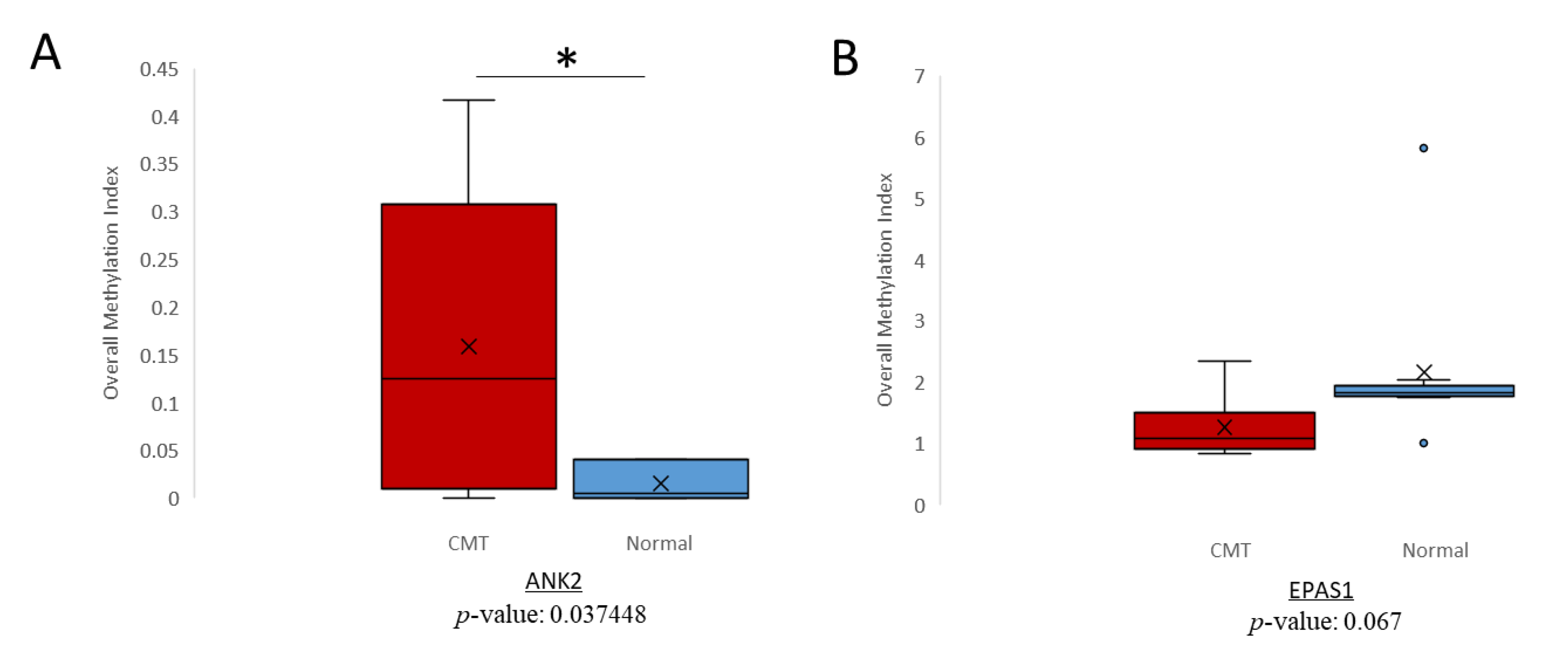
2.3. Detection of Differential Methylation in Canine Plasma cfDNA
2.4. Orthologous Human Regions Analyzed from TCGA Data
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Tissue and Plasma Samples
4.3. Correlation Analysis between Methylation and Gene Expression
4.4. DNA Isolation
4.5. Bisulfite Sequencing
4.6. Quantitative Methylation-Specific PCR (qMSP)
4.7. Human TCGA Data
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moe, L. Population-based incidence of mammary tumours in some dog breeds. J. Reprod. Fertil. Suppl. 2001, 57, 439–443. [Google Scholar]
- Sorenmo, K.U. Canine mammary gland tumors. Vet. Clin. N. Am. Small Anim. Pract. 2003, 33, 573–596. [Google Scholar] [CrossRef]
- Salas, Y.; Márquez, A.; Diaz, D.; Romero, L. Epidemiological Study of Mammary Tumors in Female Dogs Diagnosed during the Period 2002–2012: A Growing Animal Health Problem. PLoS ONE 2015, 10, e0127381. [Google Scholar] [CrossRef] [Green Version]
- Sleeckx, N.; de Rooster, H.; Kroeze, E.J.B.V.; van Ginneken, C.; van Brantegem, L. Canine Mammary Tumours, an Overview. Reprod. Domest. Anim. 2011, 46, 1112–1131. [Google Scholar] [CrossRef]
- Paoloni, M.; Khanna, C. Translation of new cancer treatments from pet dogs to humans. Nat. Rev. Cancer 2008, 8, 147–156. [Google Scholar] [CrossRef]
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.E.; Meehan, J.; Martínez-Pérez, C.; Kay, C.; Turnbull, A.K.; Morrison, L.R.; Pang, L.Y.; Argyle, D. Naturally Occurring Canine Mammary Tumors as a Translational Model for Human Breast Cancer. Front. Oncol. 2020, 10, 617. [Google Scholar] [CrossRef]
- Strandberg, J.D.; Goodman, D.G. Animal model of human disease: Canine mammary neoplasia. Am. J. Pathol. 1974, 75, 225–228. [Google Scholar]
- Visan, S.; Balacescu, O.; Berindan-Neagoe, I.; Catoi, C. In vitro comparative models for canine and human breast cancers. Clujul. Med. 2016, 89, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Abdelmegeed, S.M.; Mohammed, S. Canine mammary tumors as a model for human disease. Oncol. Lett. 2018, 15, 8195–8205. [Google Scholar] [CrossRef] [Green Version]
- Wartenberg, J.A.B.D. Environmental causes for sinonasal cancers in pet dogs, and their usefulness as sentinels of indoor cancer risk. J. Toxicol. Environ. Health Part A 1998, 54, 579–591. [Google Scholar] [CrossRef]
- Lin, C.-H.; Lo, P.-Y.; Wu, H.-D.; Chang, C.; Wang, L.-C. Association between indoor air pollution and respiratory disease in companion dogs and cats. J. Vet. Intern. Med. 2018, 32, 1259–1267. [Google Scholar] [CrossRef]
- Bollati, V.; Baccarelli, A. Environmental epigenetics. Heredity 2010, 105, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Baccarelli, A.; Bollati, V. Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 2009, 21, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, G.P. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Brandão, Y.D.O.; Toledo, M.B.; Chequin, A.; Cristo, T.G.; Sousa, R.S.; Ramos, E.A.S.; Klassen, G. DNA Methylation Status of the Estrogen Receptor α Gene in Canine Mammary Tumors. Vet. Pathol. 2018, 55, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Li, H.; Song, X.; Wu, Y.; Liu, Y. 5-Azacytidine treatment induces demethylation of DAPK1 and MGMT genes and inhibits growth in canine mammary gland tumor cells. OncoTargets Ther. 2018, 11, 2805–2813. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Lin, D. Roles of DNA mutation in the coding region and DNA methylation in the 5′ flanking region of BRCA1 in canine mammary tumors. J. Vet. Med. Sci. 2016, 78, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Perakis, S.; Speicher, M.R. Emerging concepts in liquid biopsies. BMC Med. 2017, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Board, R.E.; Knight, L.; Greystoke, A.; Blackhall, F.H.; Hughes, A.; Dive, C.; Ranson, M. DNA Methylation in Circulating Tumour DNA as a Biomarker for Cancer. Biomark. Insights 2007, 2, 307–319. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L. Cell-Free DNA Methylation Profiling Analysis—Technologies and Bioinformatics. Cancers 2019, 11, 1741. [Google Scholar] [CrossRef] [Green Version]
- Constâncio, V.; Nunes, S.P.; Henrique, R.; Jerónimo, C. DNA Methylation-Based Testing in Liquid Biopsies as Detection and Prognostic Biomarkers for the Four Major Cancer Types. Cells 2020, 9, 624. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-H.; Shin, T.-J.; Kim, W.-H.; Cho, J.-Y. Methylation of LINE-1 in cell-free DNA serves as a liquid biopsy biomarker for human breast cancers and dog mammary tumors. Sci. Rep. 2019, 9, 175. [Google Scholar] [CrossRef]
- Nam, A.-R.; Lee, K.-H.; Hwang, H.-J.; Schabort, J.J.; An, J.-H.; Won, S.-H.; Cho, J.-Y. Alternative methylation of intron motifs is associated with cancer-related gene expression in both canine mammary tumor and human breast cancer. Clin. Epigenet. 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [Green Version]
- Akirav, E.M.; Lebastchi, J.; Galvan, E.M.; Henegariu, O.; Akirav, M.; Ablamunits, V.; Lizardi, P.M.; Herold, K.C. Detection of cell death in diabetes using differentially methylated circulating DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 19018–19023. [Google Scholar] [CrossRef] [Green Version]
- Eissa, M.A.L.; Lerner, L.; Abdelfatah, E.; Shankar, N.; Canner, J.K.; Hasan, N.M.; Yaghoobi, V.; Huang, B.; Kerner, Z.; Takaesu, F.; et al. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin. Epigenet. 2019, 11, 1–10. [Google Scholar] [CrossRef]
- Giannopoulou, L.; Mastoraki, S.; Buderath, P.; Strati, A.; Pavlakis, K.; Kasimir-Bauer, S.; Lianidou, E.S. ESR1 methylation in primary tumors and paired circulating tumor DNA of patients with high-grade serous ovarian cancer. Gynecol. Oncol. 2018, 150, 355–360. [Google Scholar] [CrossRef]
- Giannopoulou, L.; Chebouti, I.; Pavlakis, K.; Kasimir-Bauer, S.; Lianidou, E. RASSF1A promoter methylation in high-grade serous ovarian cancer: A direct comparison study in primary tumors, adjacent morphologically tumor cell-free tissues and paired circulating tumor DNA. Oncotarget 2017, 8, 21429–21443. [Google Scholar] [CrossRef] [Green Version]
- Díez-Villanueva, A.; Mallona, I.; Peinado, M.A. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenet. Chromatin 2015, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Schabort, J.J.; Nam, A.R.; Lee, K.H.; Kim, S.W.; Lee, J.E.; Cho, J.Y. ANK2 Hypermethylation in Canine Mammary Tumors and Human Breast Cancer. Unpublished work. 2020. [Google Scholar]
- Lambert, S.; Bennett, V. Postmitotic expression of ankyrinR and beta R-spectrin in discrete neuronal populations of the rat brain. J. Neurosci. 1993, 13, 3725–3735. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Zhu, H.; Shao, L.; Zhu, D.; Chen, Y.W. Rho-kinase (ROK) promotes CD44v(3,8-10)-ankyrin interaction and tumor cell migration in metastatic breast cancer cells. Cell Motil. Cytoskelet. 1999, 43, 269–287. [Google Scholar] [CrossRef]
- Zhu, D.; Bourguignon, L.Y. Interaction between CD44 and the repeat domain of ankyrin promotes hyaluronic acid-mediated ovarian tumor cell migration. J. Cell. Physiol. 2000, 183, 182–195. [Google Scholar] [CrossRef]
- Chen, Y.; Löhr, J.-M.; Jesnowski, R. Inhibition of Ankyrin-B Expression Reduces Growth and Invasion of Human Pancreatic Ductal Adenocarcinoma. Pancreatology 2010, 10, 586–596. [Google Scholar] [CrossRef]
- Liao, C.; Huang, X.; Gong, Y.; Lin, Q. Discovery of core genes in colorectal cancer by weighted gene co-expression network analysis. Oncol. Lett. 2019, 18, 3137–3149. [Google Scholar] [CrossRef]
- Stein, L.; Rothschild, J.; Luce, J.; Cowell, J.K.; Thomas, G.; Bogdanova, T.I.; Tronko, M.D.; Hawthorn, L. Copy Number and Gene Expression Alterations in Radiation-Induced Papillary Thyroid Carcinoma from Chernobyl Pediatric Patients. Thyroid 2010, 20, 475–487. [Google Scholar] [CrossRef]
- Wu, Z.-H.; Zhou, T.; Sun, H.-Y. DNA methylation-based diagnostic and prognostic biomarkers of nasopharyngeal carcinoma patients. Medicine 2020, 99, e20682. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Lau, K.W.; Tian, Y.-M.; Raval, R.R.; Ratcliffe, P.J.; Pugh, C.W. Target gene selectivity of hypoxia-inducible factor-α in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br. J. Cancer 2007, 96, 1284–1292. [Google Scholar] [CrossRef]
- Wallace, E.M.; Rizzi, J.P.; Han, G.; Wehn, P.M.; Cao, Z.; Du, X.; Cheng, T.; Czerwinski, R.M.; Dixon, D.D.; Goggin, B.S.; et al. A Small-Molecule Antagonist of HIF2α Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016, 76, 5491–5500. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-X.; Xu, Y.; Yang, X.-R.; Wang, W.-M.; Bai, H.; Shi, R.-Y.; Nayar, S.K.; Devbhandari, R.P.; He, Y.-Z.; Zhu, Q.; et al. Hypoxia inducible factor 2 alpha inhibits hepatocellular carcinoma growth through the transcription factor dimerization partner 3/ E2F transcription factor 1-dependent apoptotic pathway. Hepatology 2013, 57, 1088–1097. [Google Scholar] [CrossRef]
- Imamura, T.; Kikuchi, H.; Herraiz, M.-T.; Park, D.-Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1α and HIF-2α have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Rawłuszko, A.A.; Horbacka, K.; Krokowicz, P.; Misztal, M.; Jagodziński, P.P. Prognostic Potential of DNA Methylation and Transcript Levels of HIF1A and EPAS1 in Colorectal Cancer. Mol. Cancer Res. 2014, 12, 1112–1127. [Google Scholar] [CrossRef] [Green Version]
- Westerlund, I.; Shi, Y.; Toskas, K.; Fell, S.M.; Li, S.; Surova, O.; Södersten, E.; Kogner, P.; Nyman, U.; Schlisio, S.; et al. Combined epigenetic and differentiation-based treatment inhibits neuroblastoma tumor growth and links HIF2α to tumor suppression. Proc. Natl. Acad. Sci. USA 2017, 114, E6137–E6146. [Google Scholar] [CrossRef] [Green Version]
- Klahan, S.; Wong, H.S.-C.; Tu, S.-H.; Chou, W.-H.; Zhang, Y.; Ho, T.-F.; Liu, C.-Y.; Yih, S.-Y.; Lu, H.-F.; Chen, S.C.-C.; et al. Identification of genes and pathways related to lymphovascular invasion in breast cancer patients: A bioinformatics analysis of gene expression profiles. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Fuady, J.H.; Gutsche, K.; Santambrogio, S.; Varga, Z.; Hoogewijs, D.; Wenger, R.H. Estrogen-dependent downregulation of hypoxia-inducible factor (HIF)-2α in invasive breast cancer cells. Oncotarget 2016, 7, 31153–31165. [Google Scholar] [CrossRef] [Green Version]
- Jarman, E.J.; Ward, C.; Turnbull, A.K.; Martínez-Pérez, C.; Meehan, J.; Xintaropoulou, C.; Sims, A.H.; Langdon, S.P. HER2 regulates HIF-2α and drives an increased hypoxic response in breast cancer. Breast Cancer Res. 2019, 21, 10. [Google Scholar] [CrossRef]
- Xu, X.-H.; Bao, Y.; Wang, X.; Yan, F.; Guo, S.; Ma, Y.; Xu, D.; Jin, L.; Xu, J.; Wang, J. Hypoxic-stabilized EPAS1 proteins transactivate DNMT1 and cause promoter hypermethylation and transcription inhibition of EPAS1 in non-small cell lung cancer. FASEB J. 2018, 32, 6694–6705. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Zhou, C.; Dai, J.; Ying, X.; Yu, H.; Zhong, J.; Zhang, Y.; Wu, B.; Mao, Y.; Wu, D.; et al. Endothelial PAS domain protein 1 gene hypomethylation is associated with colorectal cancer in Han Chinese. Exp. Ther. Med. 2018, 16, 4983–4990. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar]
- Győrffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schabort, J.J.; Nam, A.-R.; Lee, K.-H.; Kim, S.W.; Lee, J.E.; Cho, J.-Y. ANK2 Hypermethylation in Canine Mammary Tumors and Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8697. https://doi.org/10.3390/ijms21228697
Schabort JJ, Nam A-R, Lee K-H, Kim SW, Lee JE, Cho J-Y. ANK2 Hypermethylation in Canine Mammary Tumors and Human Breast Cancer. International Journal of Molecular Sciences. 2020; 21(22):8697. https://doi.org/10.3390/ijms21228697
Chicago/Turabian StyleSchabort, Johannes J., A-Reum Nam, Kang-Hoon Lee, Seok Won Kim, Jeong Eon Lee, and Je-Yoel Cho. 2020. "ANK2 Hypermethylation in Canine Mammary Tumors and Human Breast Cancer" International Journal of Molecular Sciences 21, no. 22: 8697. https://doi.org/10.3390/ijms21228697
APA StyleSchabort, J. J., Nam, A. -R., Lee, K. -H., Kim, S. W., Lee, J. E., & Cho, J. -Y. (2020). ANK2 Hypermethylation in Canine Mammary Tumors and Human Breast Cancer. International Journal of Molecular Sciences, 21(22), 8697. https://doi.org/10.3390/ijms21228697