Fast, Precise, and Reliable Multiplex Detection of Potato Viruses by Loop-Mediated Isothermal Amplification

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Prioritization of Loop-Mediated Isothermal Amplification (LAMP) as an Alternative Nucleic Acid Amplification Test (NAAT) for Potato Virus Detection
2.2. Iterative LAMP Optimization for the Improved Detection of Potato Viruses
2.2.1. Comparison of Commercial LAMP Kits and Final Selection for Assay Development
2.2.2. Comparison and Optimization of LAMP Primer Sets
2.2.3. General Optimization of LAMP Reactions
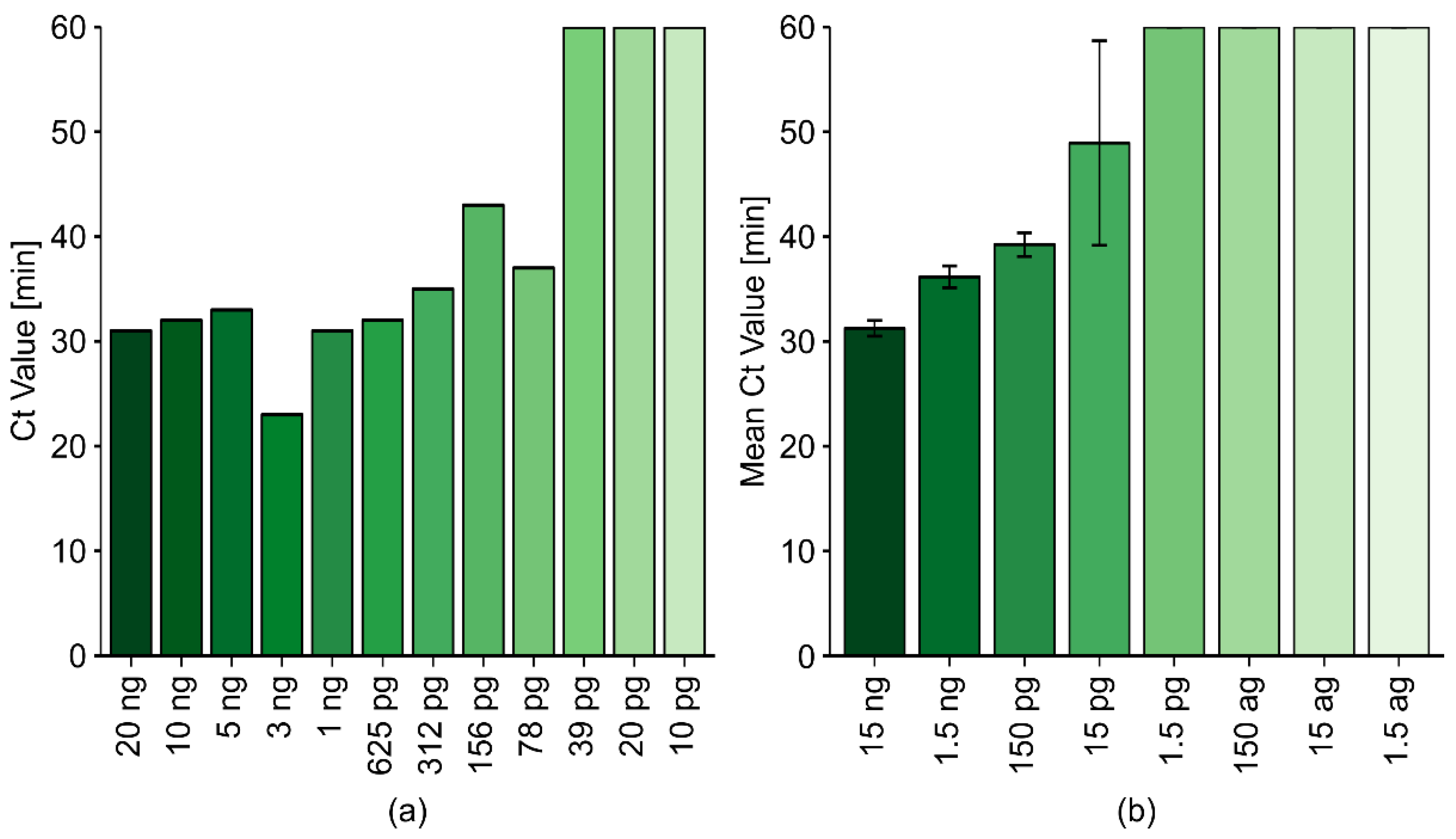
2.2.4. The Sensitivity of the Optimized LAMP Reaction Protocol
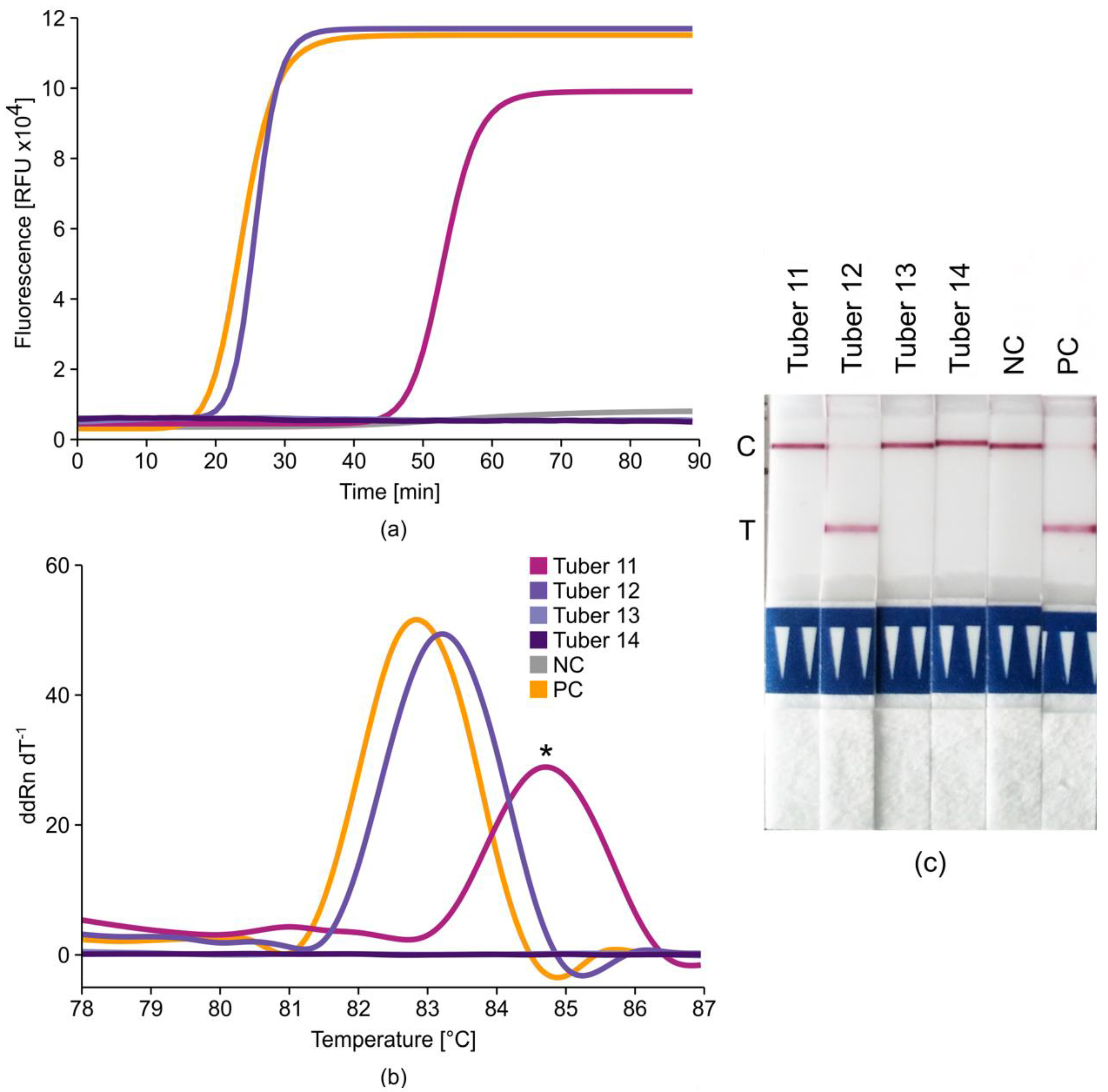
2.3. Lateral-Flow Dipsticks for Post-LAMP Result Verification
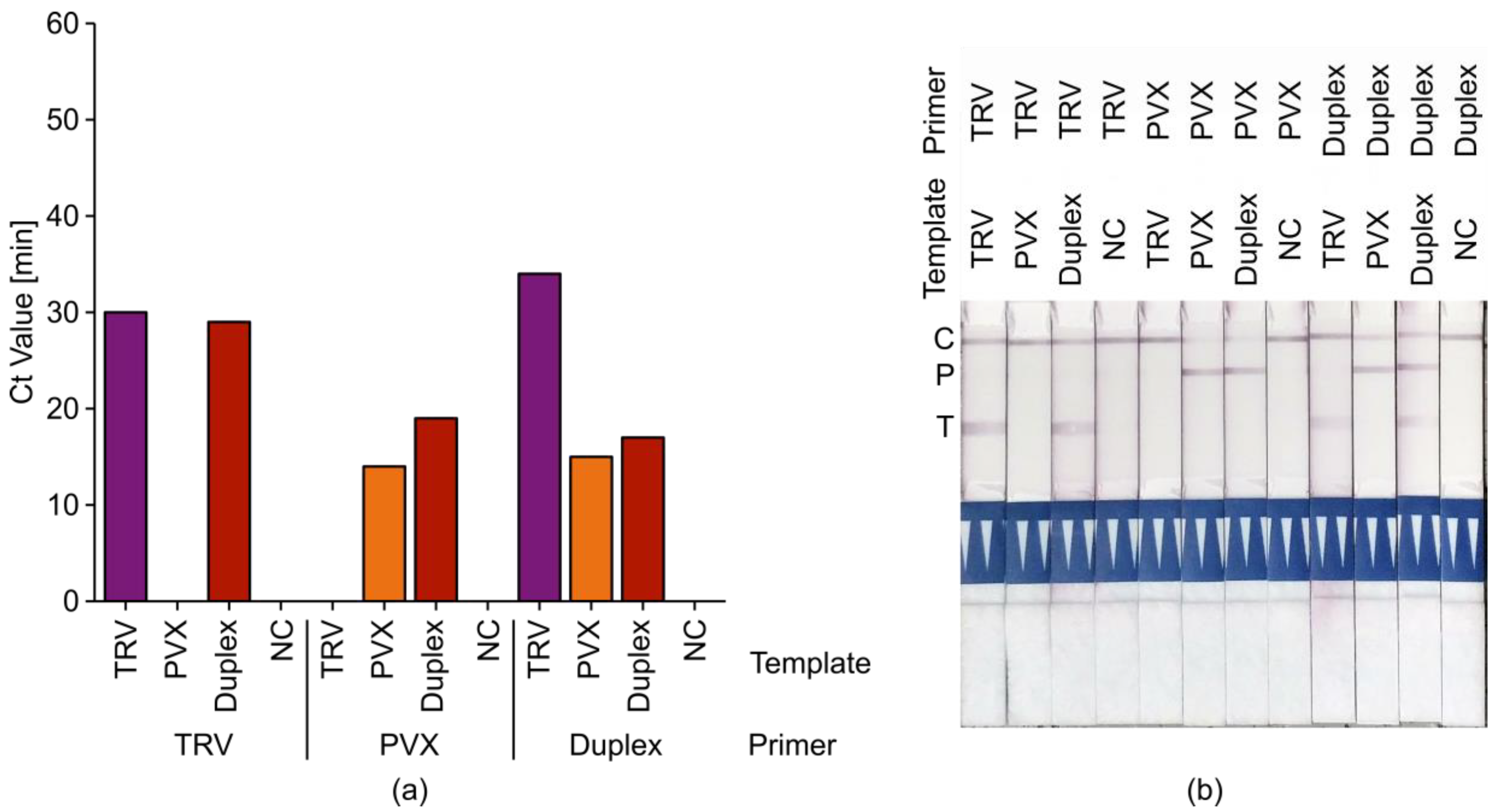
2.4. Tobacco Rattle Virus (TRV)+ Potato Virus X (PVX) Duplex LAMP Assays
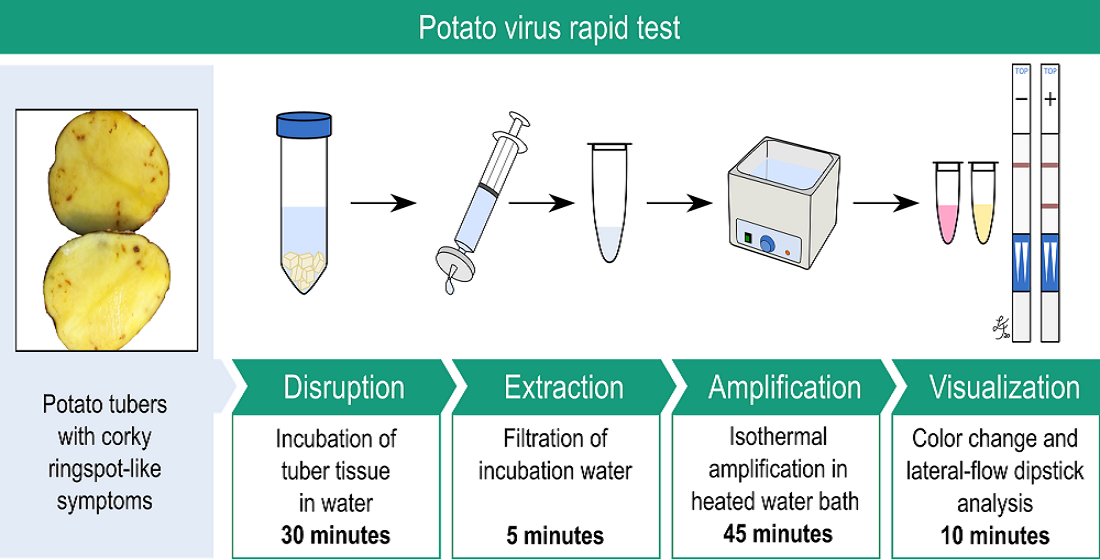
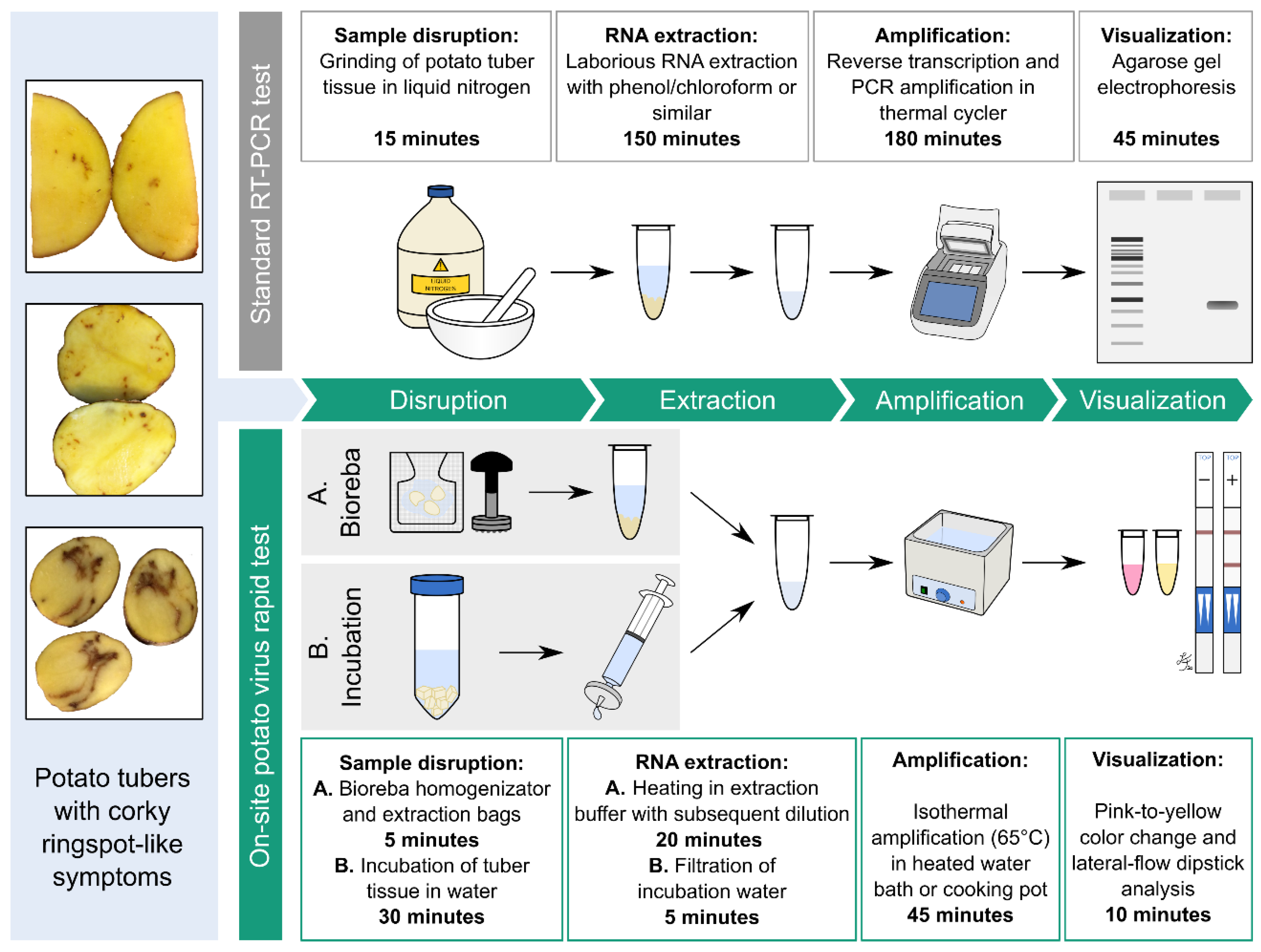
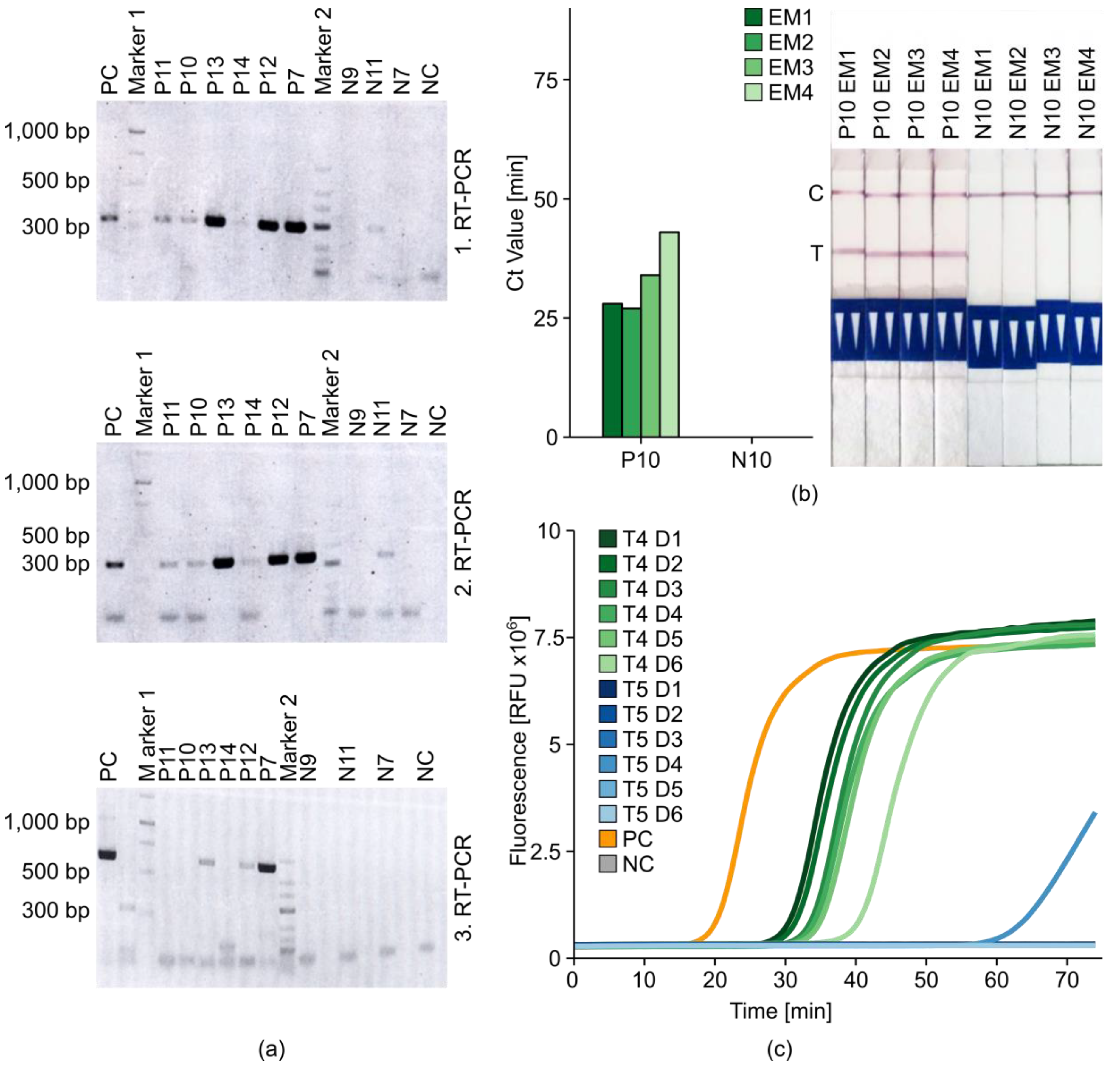
2.5. Introduction of Minimal Sampling Methods for the Potential On-Site Screening of Potato Viruses
Rapid Extraction of Potato Nucleic Acids Using Bioreba Components
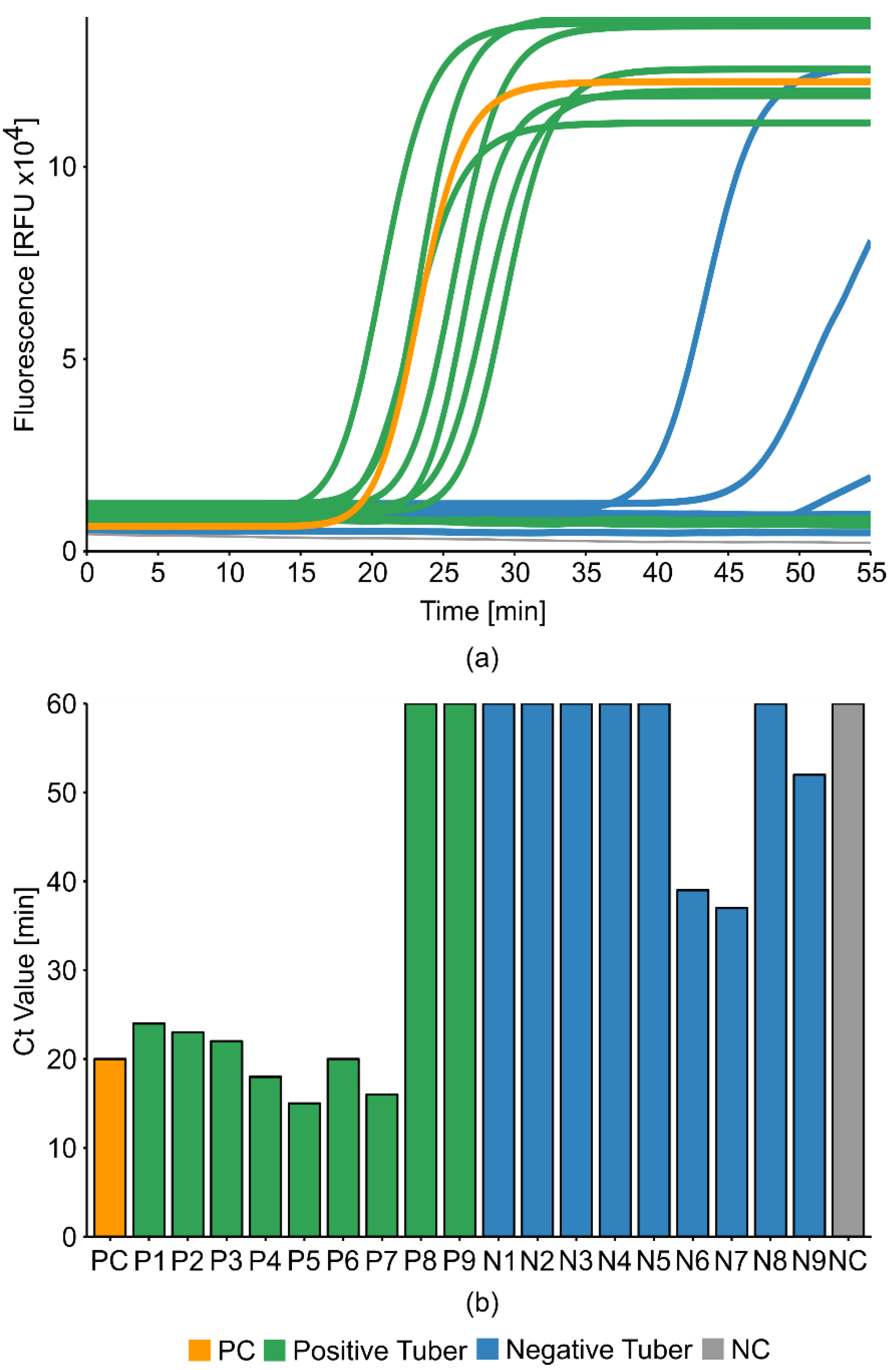
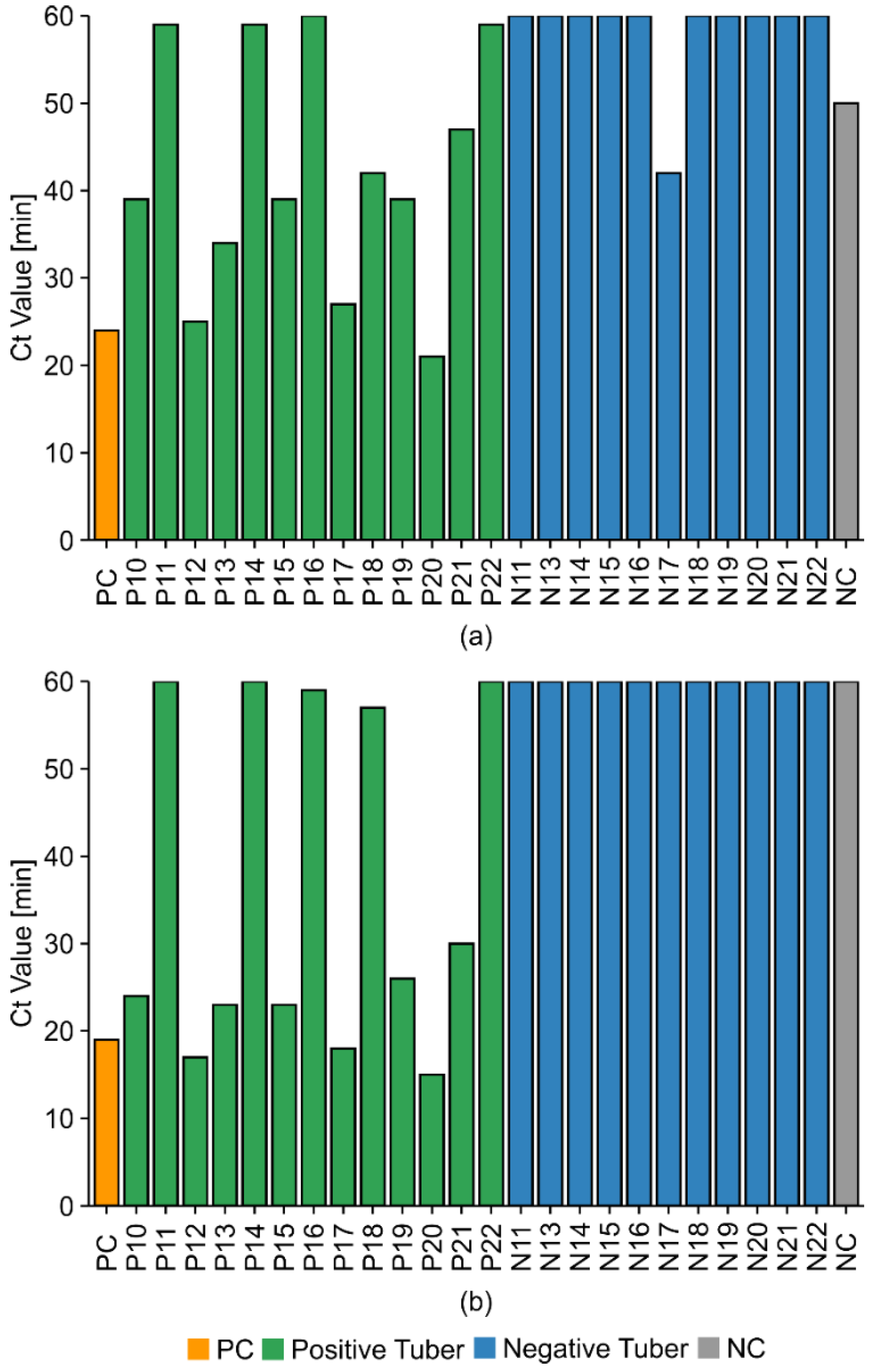
2.6. Potato Tuber Incubation Samples (InCus)
3. Discussion
4. Materials and Methods
4.1. Potato Tuber Tissue and Controls
4.2. Extraction Methods
4.3. RT-PCR Amplification Protocol
4.4. Alternative Nucleic Acid Amplification Tests (NAATs)
4.5. Optimization of RT-LAMP Reactions
4.5.1. Primer Design
4.5.2. Additives
4.6. Lateral-Flow Dipstick (LFD) Analysis
4.7. Sensitivity and Applicability of Optimized RT-LAMP
4.8. Duplex RT-LAMP-LFD Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| InCus | Incubation samples |
| LAMP | Loop-mediated isothermal amplification |
| LFD | Lateral-flow dipstick |
| PVX | Potato virus X |
| RT-PCR | Reverse transcription polymerase chain reaction |
| TRV | Tobacco rattle virus |
References
- Olmstead, R.G.; Bohs, L.; Migid, H.A.; Santiago-Valentin, E.; Garcia, V.F.; Collier, S.M. A molecular phylogeny of the Solanaceae. Taxon 2008, 57, 1159–1181. [Google Scholar] [CrossRef]
- Graeme, T.; Sansonetti, G. New Light on a Hidden Treasure: International Year of the Potato 2008, an End-of-Year Review; Food and Agriculture Organization of the United Nations: Rome, Italy, 2009. [Google Scholar]
- FAOSTAT. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 29 September 2020).
- Devaux, A.; Kromann, P.; Ortiz, O. Potatoes for Sustainable Global Food Security. Potato Res. 2014, 57, 185–199. [Google Scholar] [CrossRef]
- Hafez, S.L.; Sundararaj, P. Management of Corky Ringspot Disease of Potatoes in the Pacific Northwest; University of Idaho: Moscow, ID, USA, 2009; pp. 1–6. [Google Scholar]
- Crosslin, J.M.; Hamm, P.B.; Kirk, W.W.; Hammond, R.W. Complete genomic sequence of a Tobacco rattle virus isolate from Michigan-grown potatoes. Arch. Virol. 2010, 155, 621–625. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, S.A. Tobraviruses-plant pathogens and tools for biotechnology. Mol. Plant Pathol. 2012, 11, 577–583. [Google Scholar] [CrossRef]
- Ghazala, W.; Varrelmann, M. Tobacco Rattle virus 29K Movement Protein is the Elicitor of Extreme and Hypersensitive-like Resistance in Two Cultivars of Solanum tuberosum. Mol. Plant Microbe Interact. 2007, 20, 1396–1405. [Google Scholar] [CrossRef] [Green Version]
- Thiel, W.; Heeren, F.; Fricke, H. Nur die guten Qualitäten kommen zum Zuge. Kartoffelbau 2015, 1–2, 48–52. [Google Scholar]
- Dale, M.F.B.; Robinson, D.J.; Todd, D. Effects of systemic infections with Tobacco rattle virus on agronomic and quality traits of a range of potato cultivars. Plant Pathol. 2004, 53, 788–793. [Google Scholar] [CrossRef]
- Benker, M.; Heupel, M. Kleine braune Flecken bereiten Probleme. Kartoffelbau 2014, 6, 24–28. [Google Scholar]
- Harrison, B.D.; Robinson, D.J. The Tobraviruses. Adv. Virus Res. 1978, 23, 25–77. [Google Scholar]
- Mumford, R.A.; Walsh, K.; Barker, I.; Boonham, N. Detection of Potato mop top virus and Tobacco rattle virus Using a Multiplex Real-Time Fluorescent Reverse-Transcription Polymerase Chain Reaction Assay. Phytopathology 2000, 90, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Macdonald, J. Advances in isothermal amplification: Novel strategies inspired by biological processes. Biosens. Bioelectron. 2015, 64, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Park, K.; Kim, D.E. DNA amplification in vitro: The helicase-dependent amplification system. Cell. Mol. Life Sci. 2009, 66, 3325–3336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Hu, L.; Ying, L.; Zhao, Z.; Chu, P.K.; Yu, X.F. A CRISPR–Cas9-triggered strand displacement amplification method for ultrasensitive DNA detection. Nat. Commun. 2018, 9, 5012. [Google Scholar] [CrossRef] [Green Version]
- Notomi, T.; Okayam, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef] [Green Version]
- FAOSTAT: Sustainable Potato Production. Available online: http://www.fao.org/3/a-i1127e.pdf (accessed on 29 September 2020).
- Primer Explorer: LAMP Primer Designing Software. Available online: https://primerexplorer.jp/e/ (accessed on 4 April 2019).
- Rockweiler, T. Loop-Mediated Isothermal Amplification (LAMP) and Primer Optimization. Available online: https://www.lucigen.com/docs/slide-decks/Loop-Mediated-Isothermal-Amplification-Design-Primers-Webinar-March-2018.pdf (accessed on 18 March 2019).
- Watkins, N.E., Jr.; SantaLucia, J., Jr. Nearest-neighbor thermodynamics of deoxyinosine pairs in DNA duplexes. Nucl. Acids Res. 2005, 33, 6258–6267. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, R.; Kangayan, M.; Subramaniam, S.; Mohapatra, J.K.; Biswal, J.K.; Sharma, G.K.; Sanyal, A.; Pattnaik, B. Development and evaluation of a one step reverse transcription-loop mediated isothermal amplification assay (RT-LAMP) for rapid detection of foot and mouth disease virus in India. Virusdisease 2014, 25, 358–364. [Google Scholar] [CrossRef]
- Rajendrakumar, C.S.; Suryanarayana, T.; Reddy, A.R. DNA helix destabilization by proline and betaine: Possible role in the salinity tolerance process. FEBS Lett. 1997, 410, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.G.; Brewster, J.D.; Paul, M.; Tomasula, P.M. Two methods for increased specificity and sensitivity in loop-mediated isothermal amplification. Molecules 2015, 20, 6048–6059. [Google Scholar] [CrossRef] [Green Version]
- Hardinge, P.; Kiddle, G.; Tisi, L.; Murray, J.A.H. Optimised LAMP allows single copy detection of 35Sp and NOSt in transgenic maize using Bioluminescent Assay in Real Time (BART). Sci. Rep. 2018, 8, 17590. [Google Scholar] [CrossRef]
- Rees, W.A.; Yager, T.D.; Korte, J.; Von Hippel, P.H. Betaine can eliminate the base pair composition dependence of DNA melting. Biochemistry 1993, 32, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wan, Z.; Yang, S.; Li, Y.; Li, M.; Wang, B.; Hu, Y.; Xia, X.; Jin, X.; Yu, N.; et al. A Mismatch-Tolerant Reverse Transcription Loop-Mediated Isothermal Amplification Method and Its Application on Simultaneous Detection of All Four Serotype of Dengue Viruses. Front. Microbiol. 2019, 10, 1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, L.; Tang, W.; Ranalli, T.A.; Kim, H.J.; Wytiaz, J.; Kong, H. Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J. Biol. Chem. 2005, 280, 28952–28958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NEB—Tte UvrD Helicase. Available online: https://international.neb.com/products/m1202-tte-uvrd-helicase#Product%20Information (accessed on 24 October 2020).
- Kim, E.M.; Jeon, H.S.; Kim, J.J.; Shin, Y.K.; Lee, Y.J.; Yeo, S.G.; Park, C.K. Uracil-DNA glycosylase-treated reverse transcription loop-mediated isothermal amplification for rapid detection of avian influenza virus preventing carry-over contamination. J. Vet. Sci. 2016, 30, 421–425. [Google Scholar] [CrossRef]
- NEB—UDG-FAQ. Available online: https://international.neb.com/faqs/2016/04/25/how-do-i-use-antarctic-thermolabile-udg-for-carryover-prevention-in-lamp-reactions (accessed on 24 October 2020).
- Hardinge, P.; Murray, J.A.H. Reduced False Positives and Improved Reporting of Loop-Mediated Isothermal Amplification using Quenched Fluorescent Primers. Sci. Rep. 2019, 9, 7400. [Google Scholar] [CrossRef]
- Koczula, K.M.; Gallotta, A. Lateral flow assays. Essays Biochem. 2016, 60, 111–120. [Google Scholar]
- Wang, J.; Wang, X.; Li, Y.; Yan, S.; Zhou, Q.; Gao, B.; Peng, J.; DU, J.; Fu, Q.; Jia, S.; et al. A novel, universal and sensitive lateral-flow based method for the detection of multiple bacterial contamination in platelet concentrations. Anal. Sci. 2012, 28, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Chen, T.; Gao, J.; Dong, C.; Wong, A.H.; Jia, Y.; Mak, P.I.; Deng, C.X.; Martins, R.P. A digital microfluidic system for loop-mediated isothermal amplification and sequence specific pathogen detection. Sci. Rep. 2017, 6, 14586. [Google Scholar] [CrossRef] [Green Version]
- Vogt, E.I.; Kupfer, V.M.; Bechtner, J.D.; Frisch, L.M.; Vogel, R.F.; Niessen, L. Detection of Penicllium oxalicum in Grapes with a Loop Mediated Isothermal Amplification (Lamp) Assay. J. Microbiol. Biotech. Food Sci. 2017, 7, 265–270. [Google Scholar] [CrossRef]
- Verma, G.; Raigond, B.; Pathania, S.; Kochhar, T.; Naga, K. Development and comparison of reverse transcription-loop-mediated isothermal amplification assay (RT-LAMP), RT-PCR and real time PCR for detection of Potato spindle tuber viroid in potato. Eur. J. Plant Pathol. 2020, 158, 951–964. [Google Scholar] [CrossRef]
- Kokane, A.; Kokane, S.; Warghane, A.; Gubyad, M.G.; Sharma, A.K.; Reddy, K.M.; Ghosh, D.K. A Rapid and Sensitive Reverse Transcription-Loop-Mediated Isothermal Amplification (RT-LAMP) Assay for the Detection of Indian Citrus Ringspot Virus. Plant Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Trzmiel, K.; Hasiów-Jaroszewska, B. Development of loop-mediated isothermal amplification assay for rapid detection of genetically different wheat dwarf virus isolates. Mol. Biol. Rep. 2020, 47, 8325–8329. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myhrvold, C.; Freije, C.A.; Gootenberg, J.S.; Abudayyeh, O.O.; Metsky, H.C.; Durbin, A.F.; Kellner, M.J.; Tan, A.L.; Paul, L.M.; Parham, L.A.; et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. [Google Scholar] [CrossRef] [Green Version]
- Rolando, J.C.; Jue, E.; Barlow, J.T.; Ismagilov, R.F. Real-time kinetics and high-resolution melt curves in single-molecule digital LAMP to differentiate and study specific and non-specific amplification. Nucleic Acids Res. 2020, 48, e42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senarath, K.; Usgodaarachchi, R.; Navaratne, V.; Nagahawatte, A.; Wijayarathna, C.; Alvitigala, J.; Goonasekara, C. Non Specific Amplification with the LAMP Technique in the Diagnosis of Tuberculosis in Sri Lankan Settings. J. Tuberc. Res. 2014, 2, 168–172. [Google Scholar] [CrossRef] [Green Version]
- Treder, K.; Chołuj, J.; Zacharzewska, B.; Babujee, L.; Mielczarek, M.; Burzynski, A.; Rakotondrafara, A.M. Optimization of a magnetic capture RT-LAMP assay for fast and real-time detection of potato virus Y and differentiation of N and O serotypes. Arch. Virol. 2018, 163, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Okiro, L.A.; Tancos, M.A.; Nyanjom, S.G.; Smart, C.D.; Parker, M.L. Comparative Evaluation of LAMP, qPCR, Conventional PCR, and ELISA to Detect Ralstonia solanacearum in Kenyan Potato Fields. Plant Dis. 2019, 103, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Raigond, B.; Verma, A.; Roach, S.; Kochhar, T.; Arjunan, J.S.; Kumar, R.; Sharma, S.; Chakrabarti, S.K. One-step reverse transcription loop-mediated isothermal amplification: A simple, sensitive and rapid assay for detection of potato virus X in potato leaves and tubers. Indian Phytopathol. 2019, 72, 321–328. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Y.; Sun, H.H.; Yin, B.C.; Ye, B.C. An RNA-Guided Cas9 Nickase-Based Method for Universal Isothermal DNA Amplification. Angew. Chem. Int. Ed. 2019, 58, 65382–65386. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dilution Factor | Template | Ct Value 1 (min) | |||||
|---|---|---|---|---|---|---|---|
| PVX-PM4 (Standard Desalted) | PVX-PM4 (HPLC-Purified) | PVX-PM4 + Loop Primer (HPLC-Purified) | |||||
| 450 mM Betaine | 7.5% DMSO | 450 mM Betaine | 7.5% DMSO | 450 mM Betaine | 7.5% DMSO | ||
| 1/200 | PV-0014 | 33.79 | 49.99 | 28.50 | 43.29 | 14.76 | 33.79 |
| PV-0020 | 26.65 | 51.79 | 22.70 | 42.84 | 12.70 | 26.65 | |
| PV-0847 | 21.15 | 43.69 | 18.59 | 36.44 | 10.65 | 21.15 | |
| PV-1101 | 28.85 | 57.05 | 27.00 | 42.52 | 24.08 | 28.85 | |
| mean | 27.61 | 50.63 | 24.20 | 41.30 | 15.55 | 27.61 | |
| NC-0017 | No Ct | ||||||
| N11 | |||||||
| N12 | |||||||
| 1/1600 | PV-0014 | 36.81 | 55.01 | 31.11 | 43.42 | 16.86 | 36.81 |
| PV-0020 | 26.60 | 51.07 | 23.36 | 43.57 | 14.88 | 26.60 | |
| PV-0847 | 27.47 | 46.88 | 24.21 | 39.61 | 11.66 | 27.47 | |
| PV-1101 | 28.68 | 25.23 | 27.43 | 35.14 | 20.48 | 28.68 | |
| mean | 29.89 | 44.55 | 26.53 | 40.44 | 15.97 | 29.89 | |
| NC-0017 | No Ct | ||||||
| Template | Ct 1 Value (min) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1/100 | 1/200 | 1/400 | 1/800 | 1/1600 | 1/3200 | Mean Ct | SD 2 Ct | |
| T1 | No Ct | |||||||
| T2 | 27.43 | 33.05 | 35.80 | 54.73 | No Ct | 35.30 | 32.15 | n/a 3 |
| T3 | 37.46 | 40.14 | 40.63 | 41.58 | 50.66 | 41.98 | 42.08 | 4.50 |
| T4 | 32.64 | 33.49 | 35.65 | 36.14 | 36.98 | 42.73 | 36.27 | 3.57 |
| T5 | No Ct | |||||||
| T6 | 34.85 | 42.43 | 32.89 | No Ct | 35.84 | No Ct | 37.19 | n/a 3 |
| T7 | No Ct | |||||||
| T8 | No Ct | |||||||
| T9 | 40.21 | 40.56 | 41.05 | 44.56 | 53.87 | No Ct | 44.05 | n/a 3 |
| T10 | 34.82 | 36.23 | 38.84 | 41.48 | 39.16 | 54.96 | 40.91 | 7.27 |
| N11 | No Ct | no data | ||||||
| N12 | No Ct | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edgü, G.; Freund, L.J.; Hartje, S.; Tacke, E.; Hofferbert, H.-R.; Twyman, R.M.; Noll, G.A.; Muth, J.; Prüfer, D. Fast, Precise, and Reliable Multiplex Detection of Potato Viruses by Loop-Mediated Isothermal Amplification. Int. J. Mol. Sci. 2020, 21, 8741. https://doi.org/10.3390/ijms21228741
Edgü G, Freund LJ, Hartje S, Tacke E, Hofferbert H-R, Twyman RM, Noll GA, Muth J, Prüfer D. Fast, Precise, and Reliable Multiplex Detection of Potato Viruses by Loop-Mediated Isothermal Amplification. International Journal of Molecular Sciences. 2020; 21(22):8741. https://doi.org/10.3390/ijms21228741
Chicago/Turabian StyleEdgü, Güven, Lena Julie Freund, Stefanie Hartje, Eckhard Tacke, Hans-Reinhard Hofferbert, Richard M. Twyman, Gundula A. Noll, Jost Muth, and Dirk Prüfer. 2020. "Fast, Precise, and Reliable Multiplex Detection of Potato Viruses by Loop-Mediated Isothermal Amplification" International Journal of Molecular Sciences 21, no. 22: 8741. https://doi.org/10.3390/ijms21228741
APA StyleEdgü, G., Freund, L. J., Hartje, S., Tacke, E., Hofferbert, H. -R., Twyman, R. M., Noll, G. A., Muth, J., & Prüfer, D. (2020). Fast, Precise, and Reliable Multiplex Detection of Potato Viruses by Loop-Mediated Isothermal Amplification. International Journal of Molecular Sciences, 21(22), 8741. https://doi.org/10.3390/ijms21228741