Ferroptosis Mechanisms Involved in Neurodegenerative Diseases

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ferroptosis
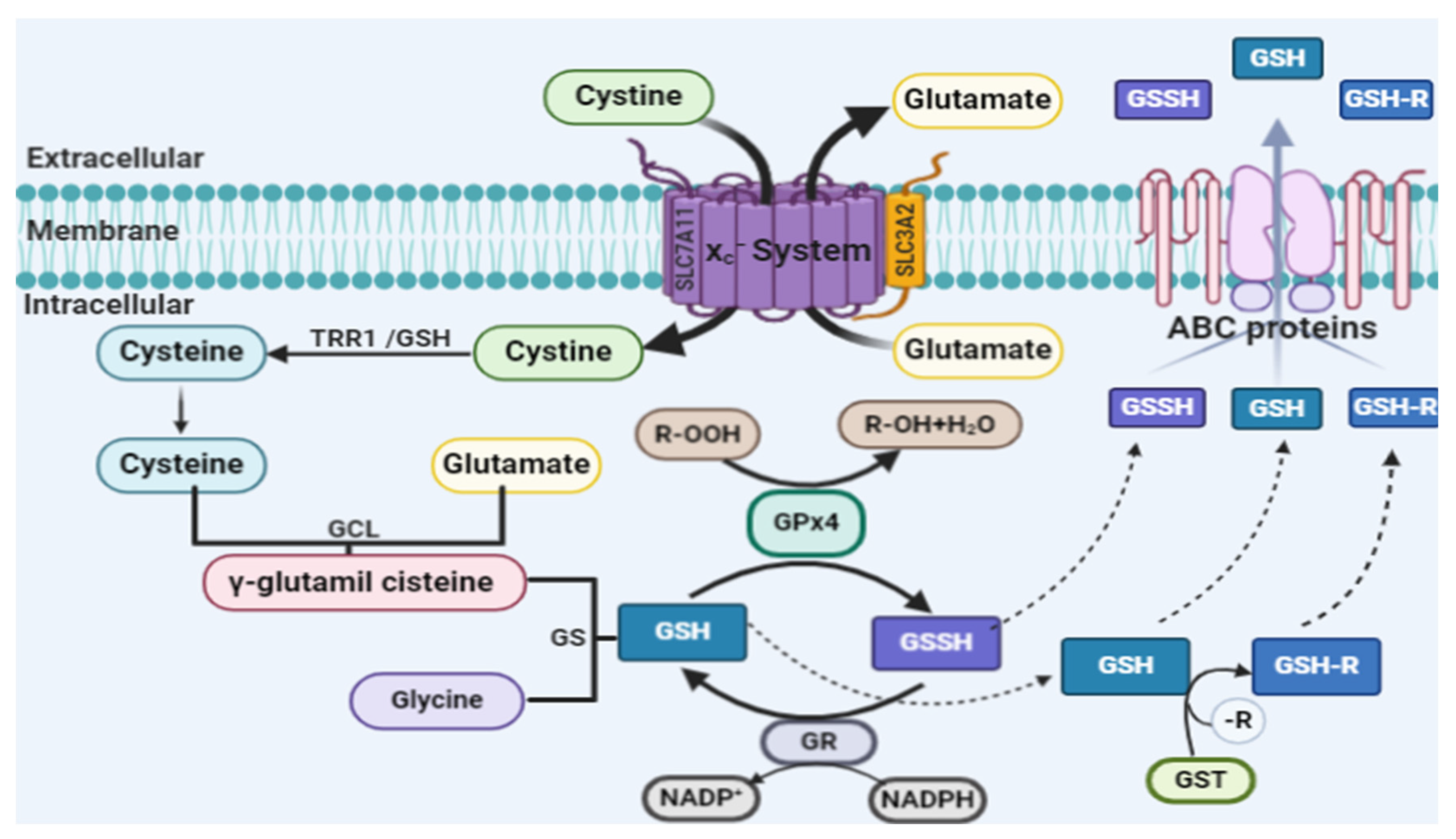
2.1. Lipid Peroxidation and Ferroptosis
2.2. Glutathione Peroxidase 4 and Ferroptosis
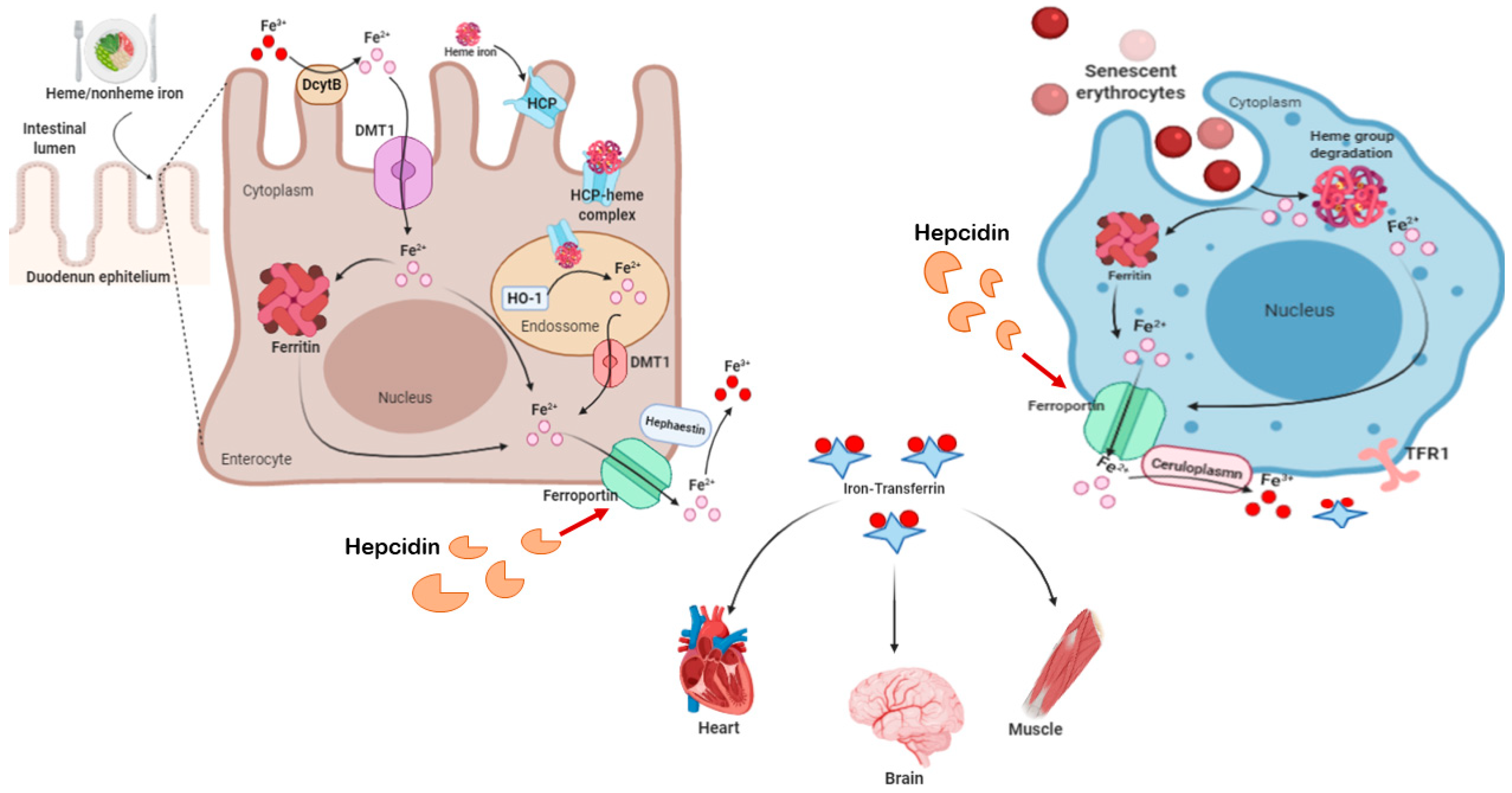
2.3. Iron and Ferroptosis
3. Ferroptosis in Neurodegenerative Diseases
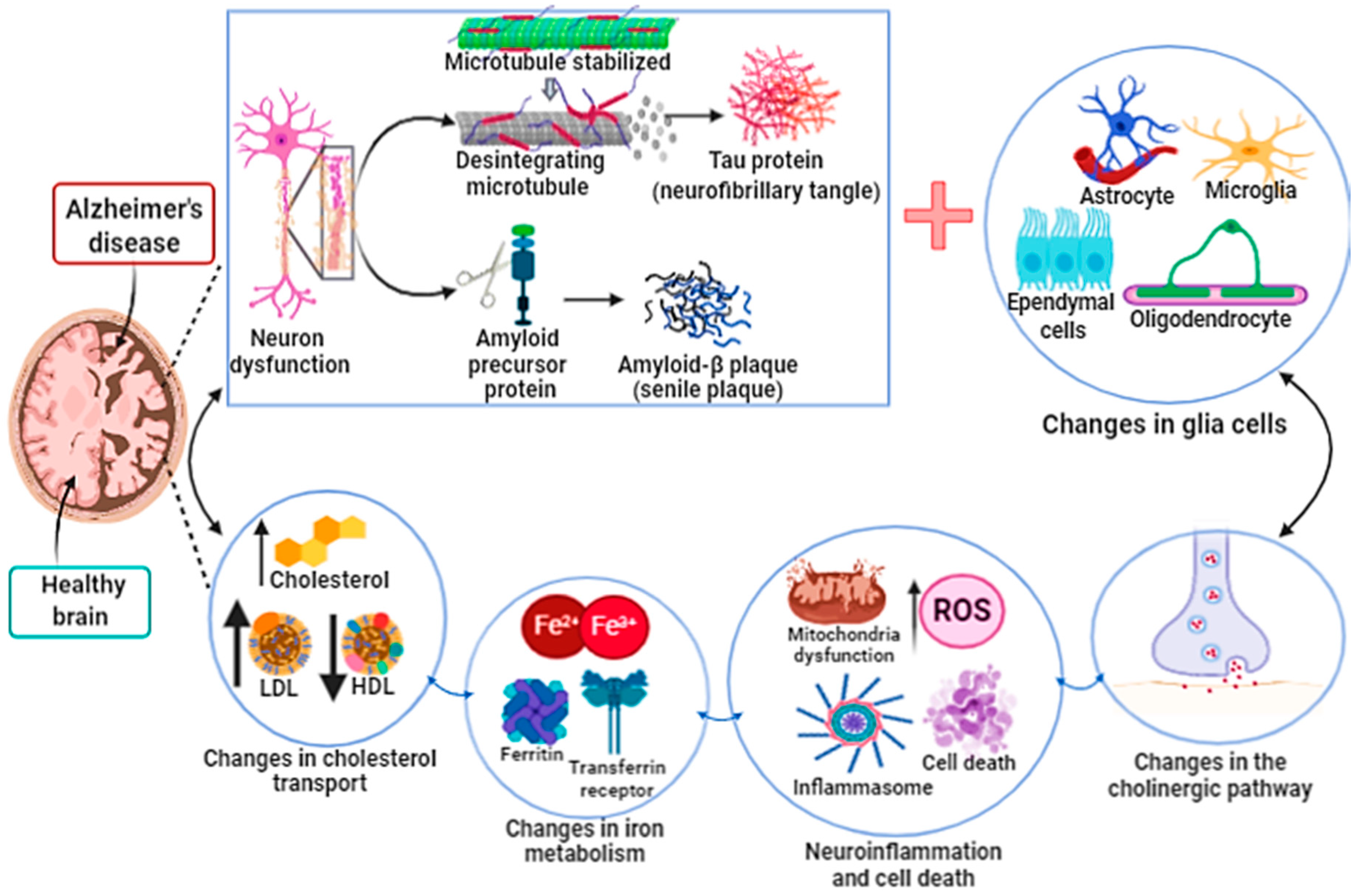
3.1. Ferroptosis in Alzheimer’s Disease
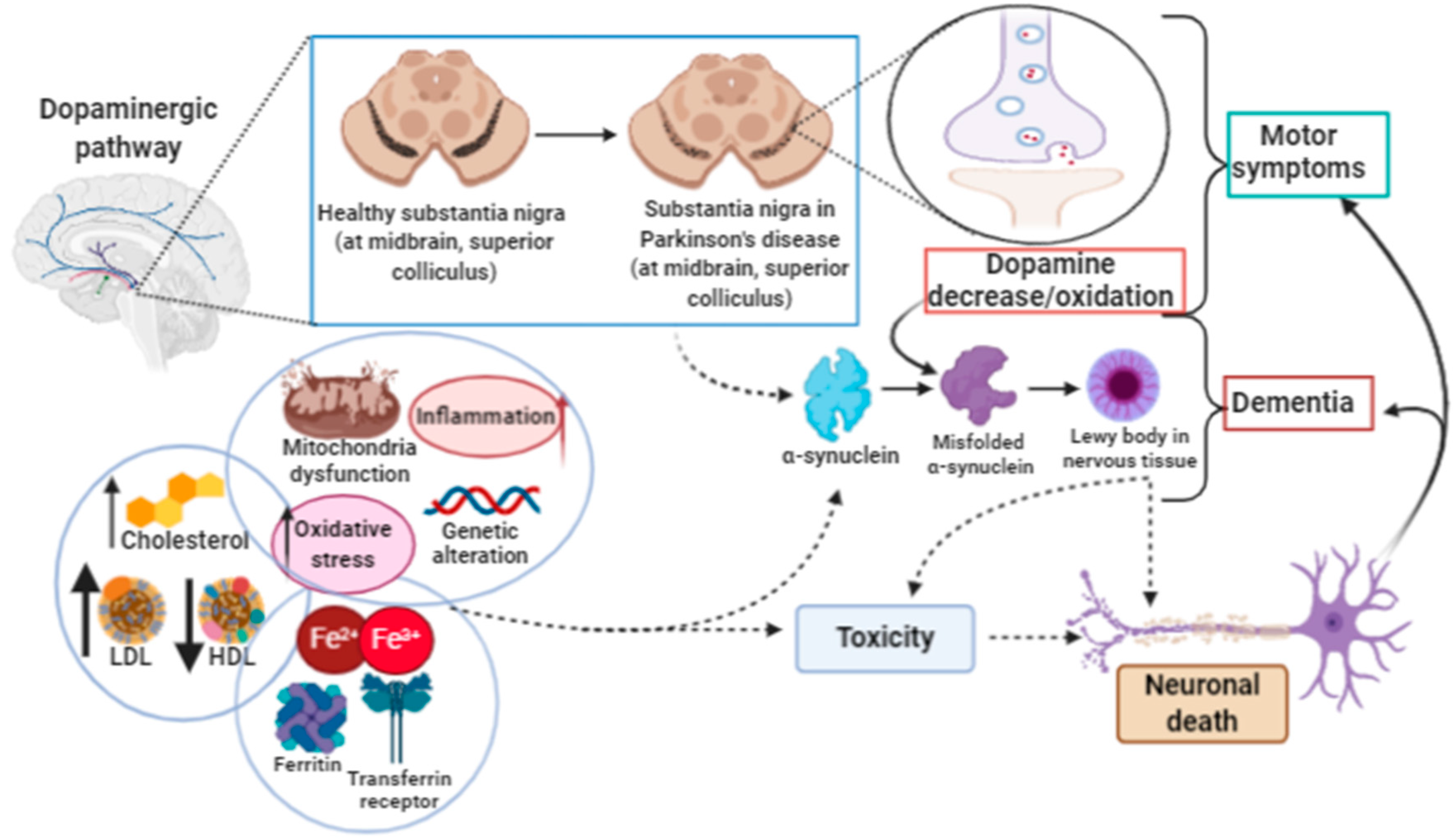
3.2. Ferroptosis in Parkinson’s Disease
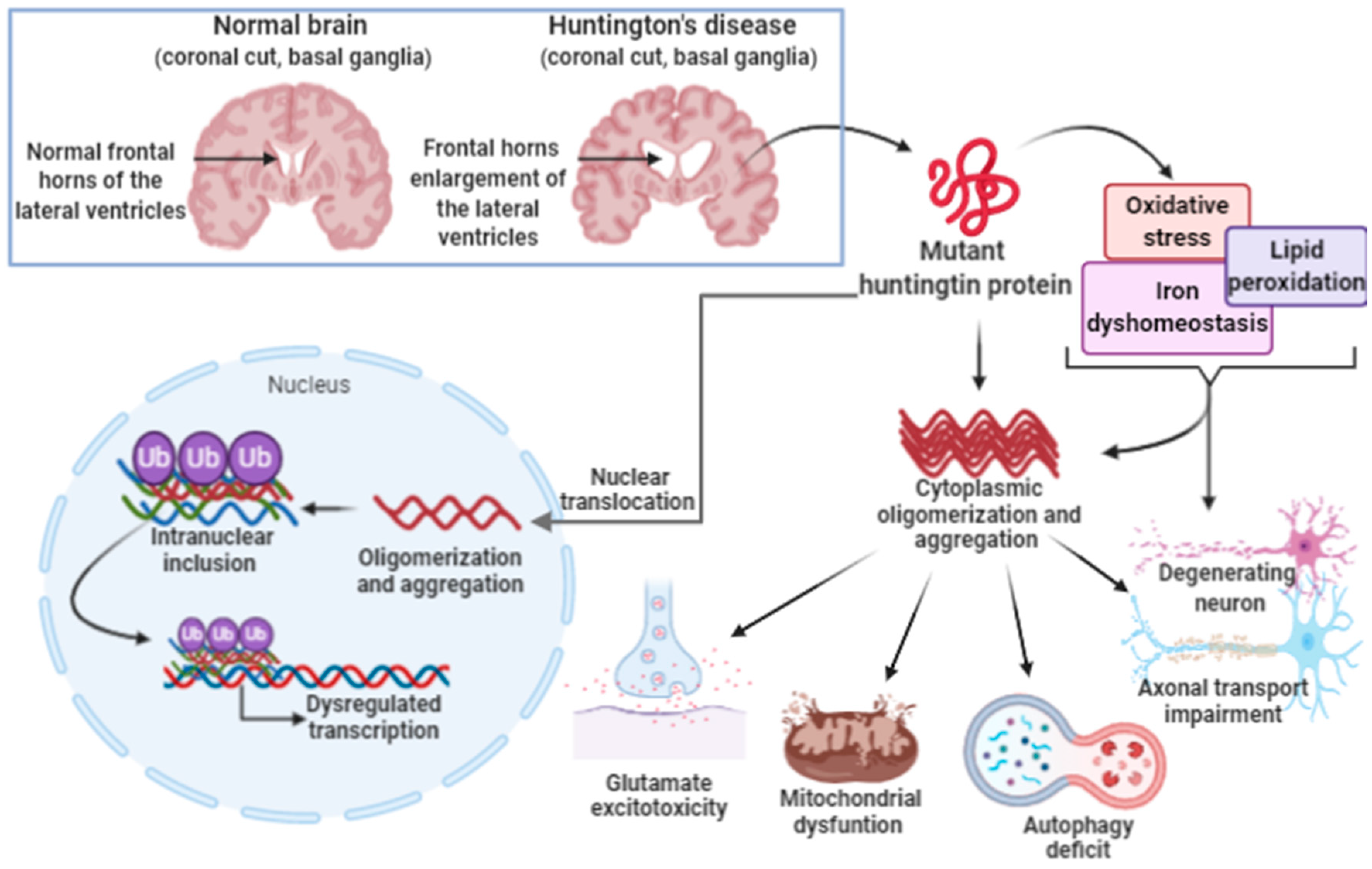
3.3. Ferroptosis in Huntington’s Disease
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Sachet, M.; Oehler, R. Circulating biomarkers of cell death. Clin. Chim. Acta 2020, 500, 87–97. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pedro, J.M.B.-S.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell. Mol. Life Sci. 2016, 73, 2405–2410. [Google Scholar] [CrossRef]
- Levy, D.; de Melo, T.C.; Oliveira, B.A.; Paz, J.L.; de Freitas, F.A.; Reichert, C.O.; Rodrigues, A.; Bydlowski, S.P. 7-Ketocholesterol and cholestane-triol increase expression of SMO and LXRα signaling pathways in a human breast cancer cell line. Biochem. Biophys. Rep. 2018, 19. [Google Scholar] [CrossRef]
- Paz, J.L.; Levy, D.; Oliveira, B.A.; de Melo, T.C.; de Freitas, F.A.; Reichert, C.O.; Rodrigues, A.; Pereira, J.; Bydlowski, S.P. 7-Ketocholesterol Promotes Oxiapoptophagy in Bone Marrow Mesenchymal Stem Cell from Patients with Acute Myeloid Leukemia. Cells 2019, 8, 482. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012. [Google Scholar] [CrossRef] [Green Version]
- Bayır, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The emerging role of ferroptosis in inflammation. Biomed. Pharmacother. 2020, 127, 11010. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Zhao, Y.; Li, H.; Lei, L. Ferroptosis as an emerging target in inflammatory diseases. Prog. Biophys. Mol. Biol. 2020, 155, 20–28. [Google Scholar] [CrossRef]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System x c- cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Jiang, X. The Chemistry and Biology of Ferroptosis. Cell Chem. Biol. 2020, 27, 365–375. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, Z.; Pan, K.; Li, J.; Chen, Q. The function and mechanism of ferroptosis in cancer. Apoptosis 2020, 25, 1–13. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.N.; Farris, F.F.; Ray, S.D. Lipid Peroxidation. In Encyclopedia of Toxicology, 3rd ed.; Elsevier: Amsterdam, The Netherland, 2014; ISBN 9780123864543. [Google Scholar]
- Tyurina, Y.Y.; Croix, C.M.S.; Watkins, S.C.; Watson, A.M.; Epperly, M.W.; Anthonymuthu, T.S.; Kisin, E.R.; Vlasova, I.I.; Krysko, O.; Krysko, D.V.; et al. Redox (phospho)lipidomics of signaling in inflammation and programmed cell death. J. Leukoc. Biol. 2019, 106, 57–81. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Konstorum, A.; Tesfay, L.; Paul, B.T.; Torti, F.M.; Laubenbacher, R.C.; Torti, S.V. Systems biology of ferroptosis: A modeling approach. J. Theor. Biol. 2020, 493, 110222. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Anthonymuthu, T.S.; Kenny, E.M.; Shrivastava, I.; Tyurina, Y.Y.; Hier, Z.E.; Ting, H.C.; Dar, H.H.; Tyurin, V.A.; Nesterova, A.; Amoscato, A.A.; et al. Empowerment of 15-Lipoxygenase Catalytic Competence in Selective Oxidation of Membrane ETE-PE to Ferroptotic Death Signals, HpETE-PE. J. Am. Chem. Soc. 2018, 140, 17835–17839. [Google Scholar] [CrossRef]
- Dar, H.H.; Tyurina, Y.Y.; Mikulska-Ruminska, K.; Shrivastava, I.; Ting, H.C.; Tyurin, V.A.; Krieger, J.; Croix, C.M.S.; Watkins, S.; Bayir, E.; et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Investig. 2018, 128, 4639–4653. [Google Scholar] [CrossRef]
- Kuhn, H.; Banthiya, S.; Van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 308–330. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H.; Walther, M.; Kuban, R.J. Mammalian arachidonate 15-lipoxygenases: Structure, function, and biological implications. Prostaglandins Other Lipid Mediat. 2002, 68, 263–290. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; Croix, C.M.S.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Sheng, X.H.; Cui, C.C.; Shan, C.; Li, Y.Z.; Sheng, D.H.; Sun, B.; Chen, D.Z. O-Phenylenediamine: A privileged pharmacophore of ferrostatins for radical-trapping reactivity in blocking ferroptosis. Org. Biomol. Chem. 2018, 16, 3952–3960. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Angeli, J.P.F. Glutathione peroxidase 4 (Gpx4) and ferroptosis: What’s so special about it? Mol. Cell. Oncol. 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [Green Version]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef]
- Chu, F.F. The human glutathione peroxidase genes GPX2, GPX3, and GPX4 map to chromosomes 14, 5, and 19, respectively. Cytogenet. Genome Res. 1994, 66, 96–98. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchert, A.; Kalms, J.; Roth, S.R.; Rademacher, M.; Schmidt, A.; Holzhutter, H.G.; Kuhn, H.; Scheerer, P. Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Conrad, M. Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Imai, H.; Nakagawa, Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003, 34, 145–169. [Google Scholar] [CrossRef]
- Conrad, M.; Schneider, M.; Seiler, A.; Bornkamm, G.W. Physiological role of phospholipid hydroperoxide glutathione peroxidase in mammals. Biol. Chem. 2007, 388, 1019–1025. [Google Scholar] [CrossRef]
- Toppo, S.; Vanin, S.; Bosello, V.; Tosatto, S.C.E. Evolutionary and structural insights into the multifaceted glutathione peroxidase (Gpx) superfamily. Antioxid. Redox Signal. 2008, 10, 1501–1514. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bhadhadhara, K.; Govil, S.; Mathur, N.; Pathak, A.N. In-silico studies for comparative analysis of glutathione peroxidaes isozymes in Homo sapiens. Int. J. Pharma Bio Sci. 2014, 5, B352–B363. [Google Scholar]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. In Current Topics in Microbiology and Immunology; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Meister, A. Glutathione metabolism. Methods Enzymol. 1995, 251, 3–7. [Google Scholar]
- Kalinina, E.V.; Chernov, N.N.; Novichkova, M.D. Role of glutathione, glutathione transferase, and glutaredoxin in regulation of redox-dependent processes. Biochemistry 2014, 79, 1562–1583. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 drives ferroptosis through GPX4 inactivation and ros production in colorectal cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [Green Version]
- Eaton, J.; Furst, L.; Ruberto, R.; Moosmayer, D.; Hillig, R.; Hilpmann, A.; Zimmermann, K.; Ryan, M.; Niehues, M.; Badock, V.; et al. Targeting a Therapy-Resistant Cancer Cell State Using Masked Electrophiles as GPX4 Inhibitors. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Eaton, J.K.; Furst, L.; Cai, L.L.; Viswanathan, V.S.; Schreiber, S.L. Structure-activity relationships of GPX4 inhibitor warheads. Bioorganic Med. Chem. Lett. 2020, 30, 127538. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef]
- Vučković, A.M.; Travain, V.B.; Bordin, L.; Cozza, G.; Miotto, G.; Rossetto, M.; Toppo, S.; Venerando, R.; Zaccarin, M.; Maiorino, M.; et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3ε. FEBS Lett. 2020, 594, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.-Y.; Pang, Y.-L.; Li, W.-X.; Zhao, C.-X.; Zhang, Y.; Wang, X.; Ning, G.-Z.; Kong, X.-H.; Liu, C.; Yao, X.; et al. Liproxstatin-1 is an effective inhibitor of oligodendrocyte ferroptosis induced by inhibition of glutathione peroxidase 4. Neural Regen. Res. 2020, 16, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, H.; Xu, X.; Liu, H.; Wu, C.; Zhao, L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol. Lett. 2020, 19, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Takahashi, H.; Rajabi, H.; Alam, M.; Suzuki, Y.; Yin, L.; Tagde, A.; Maeda, T.; Hiraki, M.; Sukhatme, V.P.; et al. Functional interactions of the cystine/glutamate antiporter, CD44V and MUC1-C oncoprotein in triple-negative breast cancer cells. Oncotarget 2016, 7, 11756–11769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, X.; Liu, L.; Yu, B.; Xue, Y.; Liu, Y. Erastin sensitizes Glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol. Rep. 2015, 33, 1465–1474. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.F.; Chen, M.S.; Chou, Y.C.; Ueng, Y.F.; Yin, P.H.; Yeh, T.S.; Lee, H.C. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2α-ATF4-xCT pathway. Oncotarget 2016, 7, 74132–74151. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, M.; Li, Y.; Shen, M.; Kong, D.; Shao, J.; Ding, H.; Tan, S.; Chen, A.; Zhang, F.; et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 2020, 16, 1482–1505. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, C.; Jian, L.; Guo, S.; Chen, R.; Li, K.; Qu, F.; Tao, K.; Fu, Y.; Luo, F.; et al. Sulfasalazine-induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol. Rep. 2019, 42, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The role of Erastin in ferroptosis and its prospects in cancer therapy. Onco. Targets. Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc- in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballatori, N.; Hammond, C.L.; Cunningham, J.B.; Krance, S.M.; Marchan, R. Molecular mechanisms of reduced glutathione transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol. Appl. Pharmacol. 2005, 204, 238–255. [Google Scholar] [CrossRef]
- Lorendeau, D.; Dury, L.; Nasr, R.; Boumendjel, A.; Teodori, E.; Gutschow, M.; Falson, P.; Di Pietro, A.; Baubichon-Cortay, H. MRP1-dependent Collateral Sensitivity of Multidrug-resistant Cancer Cells: Identifying Selective Modulators Inducing Cellular Glutathione Depletion. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Shelton, P.; Jaiswal, A.K. The transcription factor NF-E2-related factor 2 (nrf2): A protooncogene? FASEB J. 2013, 27, 414–423. [Google Scholar] [CrossRef] [Green Version]
- Reisman, S.A.; Csanaky, I.I.; Yeager, R.I.; Klaassen, C.D. Nrf2 activation enhances biliary excretion of sulfobromophthalein by inducing glutathione-S-Transferase activity. Toxicol. Sci. 2009, 109, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, A.C.; Mulcahy, R.T. Regulation of γ-Glutamylcysteine synthetase subunit gene expression: Insights into transcriptional control of antioxidant defenses. Free Radic. Res. 2000, 32, 281–301. [Google Scholar] [CrossRef] [PubMed]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc- are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, H. Targeting the NF-E2-Related Factor 2 Pathway: A Novel Strategy for Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 1773–1785. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, D.; Giustarini, D.; Pietrella, D.; Rossi, R.; Galli, F. Glutathione S-transferase P influences the Nrf2-dependent response of cellular thiols to seleno-compounds. Cell Biol. Toxicol. 2020, 36, 379–386. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of human coenzyme q10 metabolism: An overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef] [PubMed]
- Suresh, P.K.; Sah, A.K.; Daharwal, S.J. Role of free radicals in ocular diseases: An overview. Res. J. Pharm. Technol. 2014, 7, 1330–1344. [Google Scholar]
- Venkatesh, D.; O’Brien, N.A.; Zandkarimi, F.; Tong, D.R.; Stokes, M.E.; Dunn, D.E.; Kengmana, E.S.; Aron, A.T.; Klein, A.M.; Csuka, J.M.; et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 2020, 34, 526–543. [Google Scholar] [CrossRef]
- Reichert, C.O.; Da Cunha, J.; Levy, D.; Maselli, L.M.F.; Bydlowski, S.P.; Spada, C. Hepcidin: Homeostasis and Diseases Related to Iron Metabolism. Acta Haematol. 2017, 137. [Google Scholar] [CrossRef]
- Reichert, C.O.; Da Cunha, J.; Levy, D.; Maselli, L.M.F.; Bydlowski, S.P.; Spada, C. Hepcidin: SNP-Like Polymorphisms Present in Iron Metabolism and Clinical Complications of Iron Accumulation and Deficiency. In Genetic Polymorphisms; Intechopen: London, UK, 2017. [Google Scholar]
- Reichert, C.O.; Marafon, F.; Levy, D.; Maselli, L.M.F.; Bagatini, M.D.; Blatt, S.L.; Bydlowski, S.P.; Spada, C. Influence of Hepcidin in the Development of Anemia. In Current Topics in Anemia; Intechopen: London, UK, 2018. [Google Scholar]
- Lane, D.J.R.; Bae, D.H.; Merlot, A.M.; Sahni, S.; Richardson, D.R. Duodenal cytochrome b (DCYTB) in Iron metabolism: An update on function and regulation. Nutrients 2015, 7, 2274–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T. Iron Homeostasis: Fitting the Puzzle Pieces Together. Cell Metab. 2008, 7, 288–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D. Steap proteins: Implications for iron and copper metabolism. Nutr. Rev. 2007, 65, 335–340. [Google Scholar]
- Lv, H.; Shang, P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of ferroptosis and relations with regulated cell death: A review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The hallmarks of ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Piccinelli, P.; Samuelsson, T. Evolution of the iron-responsive element. RNA 2007, 13, 952–966. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Hintze, K.J.; Theil, E.C. Cellular regulation and molecular interactions of the ferritins. Cell. Mol. Life Sci. 2006, 63, 591–600. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2019, 66, 89–100. [Google Scholar] [CrossRef]
- Hamaï, A.; Mehrpour, M. Autophagy and iron homeostasis. Medecine/Sciences 2017, 33, 260–267. [Google Scholar] [CrossRef]
- Li, N.; Wang, W.; Zhou, H.; Wu, Q.; Duan, M.; Liu, C.; Wu, H.; Deng, W.; Shen, D.; Tang, Q. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 2020, 160, 303–318. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [Green Version]
- Bai, T.; Lei, P.; Zhou, H.; Liang, R.; Zhu, R.; Wang, W.; Zhou, L.; Sun, Y. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J. Cell. Mol. Med. 2019, 23, 7349–7359. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [Green Version]
- DeGregorio-Rocasolano, N.; Martí-Sistac, O.; Gasull, T. Deciphering the iron side of stroke: Neurodegeneration at the crossroads between iron dyshomeostasis, excitotoxicity, and ferroptosis. Front. Neurosci. 2019, 13, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichert, C.O.; de Macedo, C.G.; Levy, D.; Sini, B.C.; Monteiro, A.M.; Gidlund, M.; Maselli, L.M.F.; Gualandro, S.F.M.; Bydlowski, S.P. Paraoxonases (PON) 1, 2, and 3 polymorphisms and PON-1 activities in patients with sickle cell disease. Antioxidants 2019, 8, 252. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Reichert, C.O.; Bydlowski, S.P. Paraoxonases activities and polymorphisms in elderly and old-age diseases: An overview. Antioxidants 2019, 8, 118. [Google Scholar] [CrossRef] [Green Version]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in parkinson’s disease. J. Parkinson Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Peng, Y.; Xie, Y.; Zhou, B.; Sun, X.; Kang, R.; Tang, D. Antiferroptotic activity of non-oxidative dopamine. Biochem. Biophys. Res. Commun. 2016, 480, 602–607. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Roveri, A.; Peng, X.; Freitas, F.P.; Aichler, M.; Jastroch, M.; et al. Selenium utilization by GPX4 was an evolutionary requirement to prevent hydroperoxide-induced ferroptosis. Free Radic. Biol. Med. 2017, 112, 24. [Google Scholar] [CrossRef]
- Wu, C.; Zhao, W.; Yu, J.; Li, S.; Lin, L.; Chen, X. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep. 2018, 8, 574. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Bai, L.L.; Zou, Z.; Meng, P.; Xia, Y.; Cheng, S.; Mu, S.; Zhou, J.; Wang, X.; Qin, X.; et al. Ferroptosis is newly characterized form of neuronal cell death in response to arsenite exposure. Neurotoxicology 2018, 67, 27–36. [Google Scholar] [CrossRef]
- Yang, Y.W.; Liou, S.H.; Hsueh, Y.M.; Lyu, W.S.; Liu, C.S.; Liu, H.J.; Chung, M.C.; Hung, P.H.; Chung, C.J. Risk of Alzheimer’s disease with metal concentrations in whole blood and urine: A case-control study using propensity score matching. Toxicol. Appl. Pharmacol. 2018, 356, 8–14. [Google Scholar] [CrossRef]
- García-Chávez, E.; Segura, B.; Merchant, H.; Jiménez, I.; Del Razo, L.M. Functional and morphological effects of repeated sodium arsenite exposure on rat peripheral sensory nerves. J. Neurol. Sci. 2007, 258, 104–110. [Google Scholar] [CrossRef]
- Muhammad, A.; Odunola, O.A.; Gbadegesin, M.A.; Sallau, A.B.; Ndidi, U.S.; Ibrahim, M.A. Inhibitory effects of sodium arsenite and acacia honey on acetylcholinesterase in rats. Int. J. Alzheimers Dis. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampayo-Reyes, A.; Zakharyan, R.A. Inhibition of human glutathione S-transferase omega by tocopherol succinate. Biomed. Pharmacother. 2006, 60, 238–244. [Google Scholar] [CrossRef]
- Patti, F.; Fiore, M.; Chisari, C.G.; D’Amico, E.; Lo Fermo, S.; Toscano, S.; Copat, C.; Ferrante, M.; Zappia, M. CSF neurotoxic metals/metalloids levels in amyotrophic lateral sclerosis patients: Comparison between bulbar and spinal onset. Environ. Res. 2020, 188, 109820. [Google Scholar] [CrossRef]
- Bello, A.; Woskie, S.R.; Gore, R.; Sandler, D.P.; Schmidt, S.; Kamel, F. Retrospective assessment of occupational exposures for the GENEVA study of ALS among military veterans. Ann. Work Expo. Heal. 2017, 61, 299–310. [Google Scholar] [CrossRef]
- De Benedetti, S.; Lucchini, G.; Del Bò, C.; Deon, V.; Marocchi, A.; Penco, S.; Lunetta, C.; Gianazza, E.; Bonomi, F.; Iametti, S. Blood trace metals in a sporadic amyotrophic lateral sclerosis geographical cluster. BioMetals 2017, 30, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Huang, R.; Sun, F.; Zhang, L.; Wang, Q. NADPH oxidase regulates paraquat and maneb-induced dopaminergic neurodegeneration through ferroptosis. Toxicology 2019, 417, 64–73. [Google Scholar] [CrossRef]
- Hu, C.L.; Nydes, M.; Shanley, K.L.; Pantoja, I.E.M.; Howard, T.A.; Bizzozero, O.A. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurochem. 2019, 148, 426–439. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 1–12. [Google Scholar]
- Koukoulitsa, C.; Villalonga-Barber, C.; Csonka, R.; Alexi, X.; Leonis, G.; Dellis, D.; Hamelink, E.; Belda, O.; Steele, B.R.; Micha-Screttas, M.; et al. Biological and computational evaluation of resveratrol inhibitors against Alzheimers disease. J. Enzym. Inhib. Med. Chem. 2016, 31, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Zeitschrift, A. About a peculiar disease of the cerebral cortex. By Alois Alzheimer, 1907 (Translated by L. Jarvik and H. Greenson). Alzheimer Dis. Assoc. Disord. 1987, 1, 3–8. [Google Scholar]
- Butterfield, D.A.; Lauderback, M.C. Serial Review: Causes and Consequences of Oxidative Stress in Alzheimer’s Disease. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Carney, J.M. Oxidative alterations in Alzheimer’s disease. Brain Pathol. 1999, 9, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; Laferla, F.M. Mechanisms of disease Alzheimer’s Disease. N. Engl. J. Med. 2010, 326, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Hallgren, B.; Sourander, P. The Non-Haemin Iron in the Cerebral Cortex in Alzheimer’s Disease. J. Neurochem. 1960, 5, 307–310. [Google Scholar] [CrossRef]
- Dedman, D.J.; Treffry, A.; Candy, J.M.; Taylor, G.A.A.; Morris, C.M.; Bloxham, C.A.; Perry, R.H.; Edwardson, J.A.; Harrison, P.M. Iron and aluminium in relation to brain ferritin in normal individuals and Alzheimer’s-disease and chronic renal-dialysis patients. Biochem. J. 1992, 287, 509–514. [Google Scholar] [CrossRef]
- Svobodová, H.; Kosnáč, D.; Balázsiová, Z.; Tanila, H.; Miettinen, P.O.; Sierra, A.; Vitovič, P.; Wagner, A.; Polák, S.; Kopáni, M. Elevated age-related cortical iron, ferritin and amyloid plaques in APPswe/PS1ΔE9 transgenic mouse model of Alzheimer’s disease. Physiol. Res. 2019. [Google Scholar] [CrossRef]
- Yan, N.; Zhang, J.J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. Front. Neurosci. 2020, 13, 1443. [Google Scholar] [CrossRef]
- Nikseresht, S.; Bush, A.I.; Ayton, S. Treating Alzheimer’s disease by targeting iron. Br. J. Pharmacol. 2019, 176, 3622–3635. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, 1–23. [Google Scholar] [CrossRef]
- Bonda, D.J.; Castellani, R.J.; Zhu, X.; Nunomura, A.; Lee, H.-G.; Perry, G.; Smith, M.A. A Novel Perspective on Tau in Alzheimers Disease. Curr. Alzheimer Res. 2011, 8, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Council, M.R.; Road, H.; Kingdom, U. N Eurodegenerative auopathies. Genetics 2001, 24, 1121–1161. [Google Scholar]
- Pohanka, M. Alzheimer’s Disease and Oxidative Stress: A Review. Curr. Med. Chem. 2013, 21, 356–364. [Google Scholar] [CrossRef]
- Rao, S.S.; Lago, L.; De Vega, R.G.; Bray, L.; Hare, D.J.; Clases, D.; Doble, P.A.; Adlard, P.A. Characterising the spatial and temporal brain metal profile in a mouse model of tauopathy. Metallomics 2020, 12, 301–313. [Google Scholar] [CrossRef]
- Rao, S.S.; Portbury, S.D.; Lago, L.; Bush, A.I.; Adlard, P.A. The Iron Chelator Deferiprone Improves the Phenotype in a Mouse Model of Tauopathy. J. Alzheimers Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Walter, J. Twenty years of presenilins—Important proteins in health and disease. Mol. Med. 2015, 21, S41–S48. [Google Scholar] [CrossRef] [PubMed]
- Rees, T.; Hammond, P.I.; Soreq, H.; Younkin, S.; Brimijoin, S. Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiol. Aging 2003, 24, 777–787. [Google Scholar] [CrossRef]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1976–1990. [Google Scholar] [CrossRef] [Green Version]
- Bortolami, M.; Pandolfi, F.; De Vita, D.; Carafa, C.; Messore, A.; Di Santo, R.; Feroci, M.; Costi, R.; Chiarotto, I.; Bagetta, D.; et al. New deferiprone derivatives as multi-functional cholinesterase inhibitors: Design, synthesis and in vitro evaluation. Eur. J. Med. Chem. 2020, 198, 112350. [Google Scholar] [CrossRef]
- Neuner, S.M.; Tcw, J.; Goate, A.M. Neurobiology of Disease Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 2020, 143, 104976. [Google Scholar] [CrossRef] [PubMed]
- Tsatsanis, A.; Wong, B.X.; Gunn, A.P.; Ayton, S.; Bush, A.I.; Devos, D.; Duce, J.A. Amyloidogenic processing of Alzheimer’s disease β-amyloid precursor protein induces cellular iron retention. Mol. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Chen, W.Y.; Huang, X.T.; Xu, Y.C.; Zhang, H.Y. Iron dysregulates APP processing accompanying with sAPPα cellular retention and β-secretase inhibition in rat cortical neurons. Acta Pharmacol. Sin. 2017, 39, 177–183. [Google Scholar] [CrossRef]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 2019, 25, 2932–2941. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Y.; Zhang, J.H.; Han, K.; Zhang, X.; Bai, X.; You, L.H.; Yu, P.; Shi, Z.; Chang, Y.Z.; et al. Astrocyte hepcidin ameliorates neuronal loss through attenuating brain iron deposition and oxidative stress in APP/PS1 mice. Free Radic. Biol. Med. 2020, 158, 84–95. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.D.; Donovan, A.; Ward, D.M.V.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004, 360, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Maher, P. Potentiation of glutathione loss and nerve cell death by the transition metals iron and copper: Implications for age-related neurodegenerative diseases. Free Radic. Biol. Med. 2018, 115, 92–104. [Google Scholar] [CrossRef]
- Hirata, Y.; Yamada, C.; Ito, Y.; Yamamoto, S.; Nagase, H.; Oh-hashi, K.; Kiuchi, K.; Suzuki, H.; Sawada, M.; Furuta, K. Novel oxindole derivatives prevent oxidative stress-induced cell death in mouse hippocampal HT22 cells. Neuropharmacology 2018, 135, 242–252. [Google Scholar] [CrossRef]
- Yagami, T.; Yamamoto, Y.; Koma, H. Pathophysiological Roles of Intracellular Proteases in Neuronal Development and Neurological Diseases. Mol. Neurobiol. 2019, 56, 3090–3112. [Google Scholar] [CrossRef]
- Kostandy, B.B. The role of glutamate in neuronal ischemic injury: The role of spark in fire. Neurol. Sci. 2012, 33, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Currais, A.; Liang, Z.; Pinto, A.; Maher, P. Old age-associated phenotypic screening for Alzheimer’s disease drug candidates identifies sterubin as a potent neuroprotective compound from Yerba santa. Redox Biol. 2019, 21, 101089. [Google Scholar] [CrossRef]
- Gunesch, S.; Hoffmann, M.; Kiermeier, C.; Fischer, W.; Pinto, A.F.M.; Maurice, T.; Maher, P.; Decker, M. 7-O-Esters of taxifolin with pronounced and overadditive effects in neuroprotection, anti-neuroinflammation, and amelioration of short-term memory impairment in vivo. Redox Biol. 2020, 29, 101378. [Google Scholar] [CrossRef]
- Cong, L.; Dong, X.; Wang, Y.; Deng, Y.; Li, B.; Dai, R. On the role of synthesized hydroxylated chalcones as dual functional amyloid-β aggregation and ferroptosis inhibitors for potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 166, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-B.; Chai, R.; Zhang, S.; Xu, S.-F.; Zhang, Y.-H.; Li, H.-L.; Fan, Y.-G.; Guo, C. Iron Exposure and the Cellular Mechanisms Linked to Neuron Degeneration in Adult Mice. Cells 2019, 8, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, J.; Brooks, J.; Lermyte, F.; O’Connor, P.B.; Sadler, P.J.; Dobson, J.; Collingwood, J.F.; Telling, N.D. Iron stored in ferritin is chemically reduced in the presence of aggregating Aβ(1-42). Sci. Rep. 2020. [Google Scholar] [CrossRef]
- Ates, G.; Goldberg, J.; Currais, A.; Maher, P. CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimer’s disease. Redox Biol. 2020. [Google Scholar] [CrossRef]
- Ashraf, A.; So, P.W. Spotlight on Ferroptosis: Iron-Dependent Cell Death in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 196. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Schneider, R.B.; Iourinets, J.; Richard, I.H. Parkinson’s disease psychosis: Presentation, diagnosis and management. Neurodegener. Dis. Manag. 2017, 7, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Bayulkem, K.; Lopez, G. Nonmotor fluctuations in Parkinson’s disease: Clinical spectrum and classification. J. Neurol. Sci. 2010, 289, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Witjas, T.; Kaphan, E.; Azulay, J.P.; Blin, O.; Ceccaldi, M.; Pouget, J.; Poncet, M.; Ali Chérif, A. Nonmotor fluctuations in Parkinson’s disease: Frequent and disabling. Neurology 2002, 59, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.V. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.V.; Bezard, E.; Brotchie, J.; Calon, F.; Collingridge, G.L.; Ferger, B.; Hengerer, B.; Hirsch, E.; Jenner, P.; Le Novère, N.; et al. Novel pharmacological targets for the treatment of Parkinson’s disease. Nat. Rev. Drug Discov. 2006, 5, 845–854. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Lee, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased Nigral Iron Content and Alterations in Other Metal Ions Occurring in Brain in Parkinson’s Disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Jenner, P.; Schapira, A.H.V.; Marsden, C.D. Alterations in levels of iron, ferritin, and other trace metals in neurodegenerative diseases affecting the basal ganglia. Ann. Neurol. 1992, 32, S94–S100. [Google Scholar] [CrossRef] [PubMed]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 1997, 48, 650–658. [Google Scholar] [CrossRef]
- Graham, J.M.; Paley, M.N.J.; Grünewald, R.A.; Hoggard, N.; Griffiths, P.D. Brain iron deposition in Parkinson’s disease imaged using the PRIME magnetic resonance sequence. Brain 2000, 123, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Ruottinen, H.; Soimakallio, S.; Elovaara, I.; Dastidar, P. Clinical MRI for iron detection in Parkinson’s disease. Clin. Imaging 2013, 37, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Grolez, G.; Moreau, C.; Sablonnière, B.; Garçon, G.; Devedjian, J.C.; Meguig, S.; Gelé, P.; Delmaire, C.; Bordet, R.; Defebvre, L.; et al. Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol. 2015, 15, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef]
- Vassiliev, V.; Harris, Z.L.; Zatta, P. Ceruloplasmin in neurodegenerative diseases. Brain Res. Rev. 2005, 49, 633–640. [Google Scholar] [CrossRef]
- Zhao, Z.; Bao, X.Q.; Zhang, D. Mechanisms of ferroptosis and its involvement in Parkinson’s disease. Yaoxue Xuebao 2019, 54, 399–406. [Google Scholar]
- Rhodes, S.L.; Ritz, B. Genetics of iron regulation and the possible role of iron in Parkinson’s disease. Neurobiol. Dis. 2008, 32, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuča, K.; Musílek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha synuclein aggregation drives ferroptosis: An interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020, 27, 2781–2796. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef]
- Kaidery, N.A.; Ahuja, M.; Thomas, B. Crosstalk between Nrf2 signaling and mitochondrial function in Parkinson’s disease. Mol. Cell. Neurosci. 2019, 101, 103413. [Google Scholar] [CrossRef]
- Fan, Z.; Wirth, A.-K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; He, L.; Wang, T.; Hua, W.; Qin, H.; Wang, J.; Wang, L.; Gu, W.; Li, T.; Li, N.; et al. Activation of p62-Keap1-Nrf2 Pathway Protects 6-Hydroxydopamine-Induced Ferroptosis in Dopaminergic Cells. Mol. Neurobiol. 2020, 57, 4628–4641. [Google Scholar] [CrossRef]
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in neurodegeneration. Front. Neurosci. 2018, 12, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, D.; Andersen, J.K. Ironing out Parkinson’s disease: Is therapeutic treatment with iron chelators a real possibility? Aging Cell 2002, 1, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Huang, C.; Luo, Q.; Xia, Y.; Liu, W.; Zeng, W.; Cheng, A.; Shi, R.; Zhengli, C. Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson’s disease through AKT/mTOR pathway. Aging (Albany N. Y.) 2020, 12, 9515–9533. [Google Scholar] [CrossRef] [PubMed]
- Levy, O.A.; Malagelada, C.; Greene, L.A. Cell death pathways in Parkinson’s disease: Proximal triggers, distal effectors, and final steps. Apoptosis 2009, 14, 478–500. [Google Scholar] [CrossRef]
- Gellera, C.; Meoni, C.; Castellotti, B.; Zappacosta, B.; Girotti, F.; Taroni, F.; DiDonato, S. Errors in Huntington disease diagnostic test caused by trinucleotide deletion in the IT15 gene. Am. J. Hum. Genet. 1996, 59, 475–477. [Google Scholar] [PubMed]
- Roos, R.A.C. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wexler, N.S. Huntington’s disease: Advocacy driving science. Annu. Rev. Med. 2012, 63, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Wexler, A.; Wild, E.J.; Tabrizi, S.J. George Huntington: A legacy of inquiry, empathy and hope. Brain 2016, 139, 2326–2333. [Google Scholar] [CrossRef] [Green Version]
- Nopoulos, P.C. Huntington disease: A single-gene degenerative disorder of the striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar]
- Caterino, M.; Squillaro, T.; Montesarchio, D.; Giordano, A.; Giancola, C.; Melone, M.A.B. Huntingtin protein: A new option for fixing the Huntington’s disease countdown clock. Neuropharmacology 2018, 135, 126–138. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 1–21. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.; Schwarz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.P.; Carraway, R.; Reeves, S.A.; et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 2017, 7, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Marks, E.; Lai, B.; Zhang, Z.; Duce, J.A.; Lam, L.Q.; Volitakis, I.; Bush, A.I.; Hersch, S.; Fox, J.H. Iron Accumulates in Huntington’s Disease Neurons: Protection by Deferoxamine. PLoS ONE 2013, 8, e77023. [Google Scholar] [CrossRef]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef] [Green Version]
- Rosas, H.D.; Chen, Y.I.; Doros, G.; Salat, D.H.; Chen, N.K.; Kwong, K.K.; Bush, A.; Fox, J.; Hersch, S.M. Alterations in brain transition metals in Huntington disease: An evolving and intricate story. Arch. Neurol. 2012, 69, 887–893. [Google Scholar] [CrossRef]
- Magtanong, L.; Dixon, S.J. Ferroptosis and Brain Injury. Dev. Neurosci. 2019, 40, 382–395. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Choo, Y.S.; Johnson, G.V.W.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Gao, X.; Xu, H.; Cui, Y.; Zhang, Y.; Gou, X. The Emerging Roles of Ferroptosis in Huntington’s Disease. NeuroMol. Med. 2019, 21, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.P.; Rubinsztein, D.C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 2002, 11, 1137–1151. [Google Scholar] [CrossRef]
- Klepac, N.; Relja, M.; Klepac, R.; Hećimović, S.; Babić, T.; Trkulja, V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol. 2007, 254, 1676–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Csobonyeiova, M.; Polak, S.; Danisovic, L. Recent overview of the use of iPSCs huntington’s disease modeling and therapy. Int. J. Mol. Sci. 2020, 21, 2239. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.d.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. https://doi.org/10.3390/ijms21228765
Reichert CO, de Freitas FA, Sampaio-Silva J, Rokita-Rosa L, Barros PdL, Levy D, Bydlowski SP. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(22):8765. https://doi.org/10.3390/ijms21228765
Chicago/Turabian StyleReichert, Cadiele Oliana, Fábio Alessandro de Freitas, Juliana Sampaio-Silva, Leonardo Rokita-Rosa, Priscila de Lima Barros, Debora Levy, and Sérgio Paulo Bydlowski. 2020. "Ferroptosis Mechanisms Involved in Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 22: 8765. https://doi.org/10.3390/ijms21228765
APA StyleReichert, C. O., de Freitas, F. A., Sampaio-Silva, J., Rokita-Rosa, L., Barros, P. d. L., Levy, D., & Bydlowski, S. P. (2020). Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(22), 8765. https://doi.org/10.3390/ijms21228765