The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. 14-3-3 Proteins
3. 14-3-3 Protein-Dependent Regulation of Selected Kinases
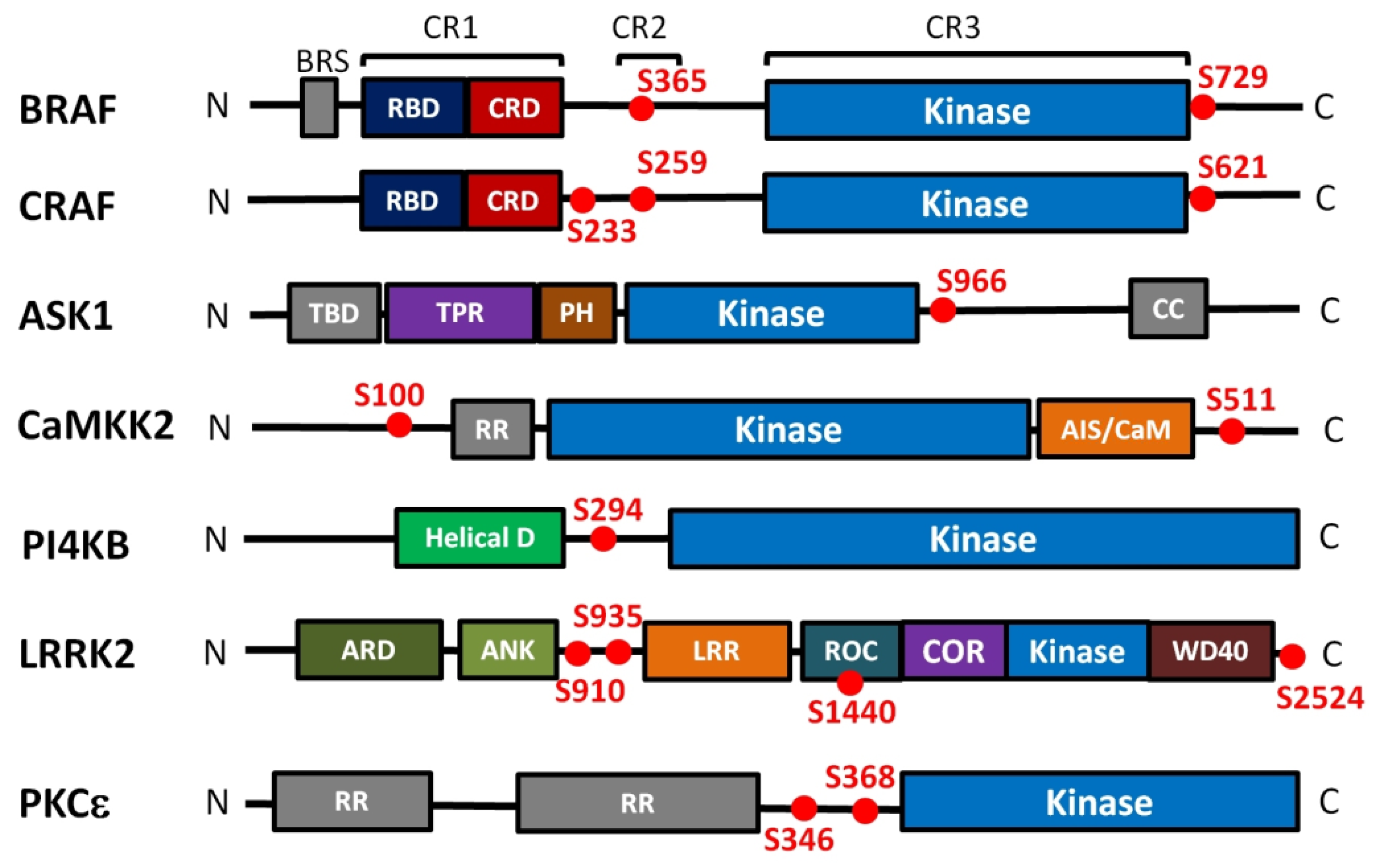
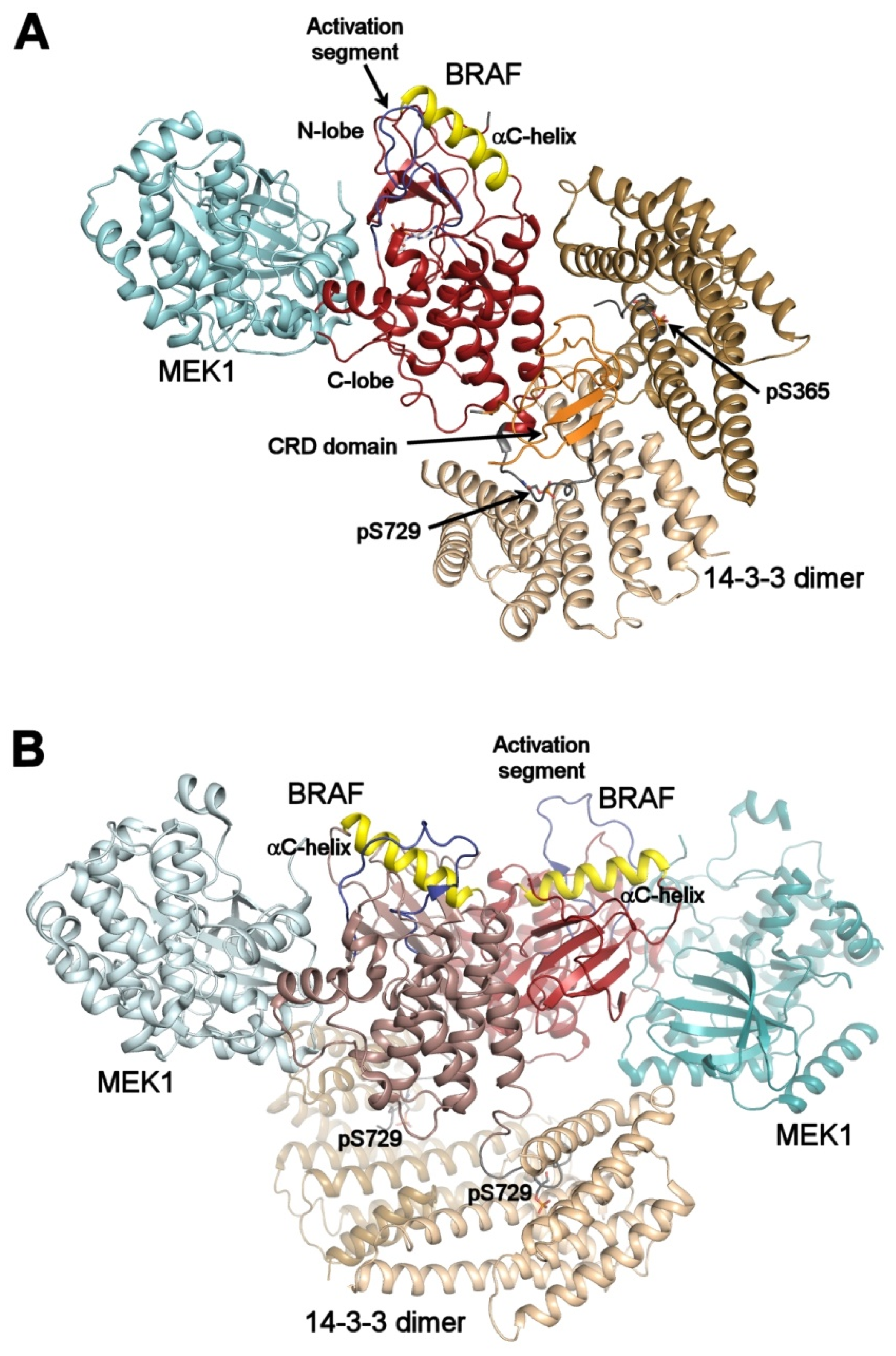
3.1. RAF Kinases
3.2. Apoptosis Signal-Regulating Kinase 1 (ASK1)
3.3. Calcium/Calmodulin-Dependent Protein Kinase Kinases (CaMKK)
3.4. Phosphatidylinositol 4-Kinases
3.5. Leucine-Rich Repeat Protein Kinase-2 (LRRK2)
3.6. Protein Kinase C (PKC)
4. Conclusions and Challenges
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACBD3 | Acyl-CoA-binding protein 3 |
| AMPK | AMP-activated protein kinase |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| CaMKK | Calcium/calmodulin-dependent protein kinase kinase |
| GSK3 | Glycogen synthase kinase 3 |
| LRRK2 | Leucine-rich repeat protein kinase-2 |
| MAP | Mitogen-activated protein |
| MAPK | Mitogen-activated protein kinase |
| MAP2K | Mitogen-activated protein kinase kinase |
| MAP3K | Mitogen-activated protein kinase kinase kinase |
| PD | Parkinson’s disease |
| PI | Phosphatidylinositol |
| PIP | Phosphoinositides |
| PI4K | Phosphoinositide 4-kinases |
| PI4KB | Phosphatidylinositol-4-kinase-IIIβ |
| PKC | Protein kinase C |
| RAF | Rapidly Accelerated Fibrosarcoma |
| RhoA | Ras Homolog Family Member A |
References
- Tinti, M.; Madeira, F.; Murugesan, G.; Hoxhaj, G.; Toth, R.; Mackintosh, C. ANIA: ANnotation and Integrated Analysis of the 14-3-3 interactome. Database 2014, 2014, bat085. [Google Scholar] [CrossRef] [PubMed]
- Petrvalska, O.; Kosek, D.; Kukacka, Z.; Tosner, Z.; Man, P.; Vecer, J.; Herman, P.; Obsilova, V.; Obsil, T. Structural Insight into the 14-3-3 Protein-dependent Inhibition of Protein Kinase ASK1 (Apoptosis Signal-regulating kinase 1). J. Biol. Chem. 2016, 291, 20753–20765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psenakova, K.; Petrvalska, O.; Kylarova, S.; Lentini Santo, D.; Kalabova, D.; Herman, P.; Obsilova, V.; Obsil, T. 14-3-3 protein directly interacts with the kinase domain of calcium/calmodulin-dependent protein kinase kinase (CaMKK2). Biochim. Biophys. Acta 2018, 1862, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- Chalupska, D.; Eisenreichova, A.; Rozycki, B.; Rezabkova, L.; Humpolickova, J.; Klima, M.; Boura, E. Structural analysis of phosphatidylinositol 4-kinase IIIbeta (PI4KB)—14-3-3 protein complex reveals internal flexibility and explains 14-3-3 mediated protection from degradation in vitro. J. Struct. Biol. 2017, 200, 36–44. [Google Scholar] [CrossRef]
- Kondo, Y.; Ognjenovic, J.; Banerjee, S.; Karandur, D.; Merk, A.; Kulhanek, K.; Wong, K.; Roose, J.P.; Subramaniam, S.; Kuriyan, J. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 2019, 366, 109–115. [Google Scholar] [CrossRef]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Pino, G.G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Wendorff, T.J.; Quinn, J.G.; Steffek, M.; Phung, W.; Liu, P.; Tang, J.; Irudayanathan, F.J.; Izadi, S.; Shaw, A.S.; et al. Negative regulation of RAF kinase activity by ATP is overcome by 14-3-3-induced dimerization. Nat. Struct. Mol. Biol. 2020, 27, 134–141. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Venkatanarayan, A.; Quinn, J.G.; Phung, W.; Malek, S.; Hymowitz, S.G.; Sudhamsu, J. Dimerization Induced by C-Terminal 14-3-3 Binding Is Sufficient for BRAF Kinase Activation. Biochemistry 2020, 59, 3982–3992. [Google Scholar] [CrossRef]
- Aitken, A.; Amess, B.; Howell, S.; Jones, D.; Martin, H.; Patel, Y.; Robinson, K.; Toker, A. The role of specific isoforms of 14-3-3 protein in regulating protein kinase activity in the brain. Biochem. Soc. Trans. 1992, 20, 607–611. [Google Scholar] [CrossRef] [Green Version]
- Bridges, D.; Moorhead, G.B. 14-3-3 proteins: A number of functions for a numbered protein. Sci. STKE 2005, 2005, re10. [Google Scholar] [CrossRef]
- Aitken, A. Post-translational modification of 14-3-3 isoforms and regulation of cellular function. Semin. Cell Dev. Biol. 2011, 22, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Sluchanko, N.N. Association of Multiple Phosphorylated Proteins with the 14-3-3 Regulatory Hubs: Problems and Perspectives. J. Mol. Biol. 2018, 430, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Aitken, A.; Howell, S.; Jones, D.; Madrazo, J.; Patel, Y. 14-3-3 alpha and delta are the phosphorylated forms of raf-activating 14-3-3 beta and zeta. In vivo stoichiometric phosphorylation in brain at a Ser-Pro-Glu-Lys MOTIF. J. Biol. Chem. 1995, 270, 5706–5709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hemert, M.J.; van Heusden, G.P.; Steensma, H.Y. Yeast 14-3-3 proteins. Yeast 2001, 18, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Sehnke, P.C.; Rosenquist, M.; Alsterfjord, M.; DeLille, J.; Sommarin, M.; Larsson, C.; Ferl, R.J. Evolution and isoform specificity of plant 14-3-3 proteins. Plant. Mol. Biol. 2002, 50, 1011–1018. [Google Scholar] [CrossRef]
- Xiao, B.; Smerdon, S.J.; Jones, D.H.; Dodson, G.G.; Soneji, Y.; Aitken, A.; Gamblin, S.J. Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature 1995, 376, 188–191. [Google Scholar] [CrossRef]
- Liu, D.; Bienkowska, J.; Petosa, C.; Collier, R.J.; Fu, H.; Liddington, R. Crystal structure of the zeta isoform of the 14-3-3 protein. Nature 1995, 376, 191–194. [Google Scholar] [CrossRef]
- Ma, Y.; Pitson, S.; Hercus, T.; Murphy, J.; Lopez, A.; Woodcock, J. Sphingosine activates protein kinase A type II by a novel cAMP-independent mechanism. J. Biol. Chem. 2005, 280, 26011–26017. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, J.M.; Murphy, J.; Stomski, F.C.; Berndt, M.C.; Lopez, A.F. The dimeric versus monomeric status of 14-3-3zeta is controlled by phosphorylation of Ser58 at the dimer interface. J. Biol. Chem. 2003, 278, 36323–36327. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, J.M.; Coolen, C.; Goodwin, K.L.; Baek, D.J.; Bittman, R.; Samuel, M.S.; Pitson, S.M.; Lopez, A.F. Destabilisation of dimeric 14-3-3 proteins as a novel approach to anti-cancer therapeutics. Oncotarget 2015, 6, 14522–14536. [Google Scholar] [CrossRef] [Green Version]
- Molzan, M.; Kasper, S.; Roglin, L.; Skwarczynska, M.; Sassa, T.; Inoue, T.; Breitenbuecher, F.; Ohkanda, J.; Kato, N.; Schuler, M.; et al. Stabilization of physical RAF/14-3-3 interaction by cotylenin A as treatment strategy for RAS mutant cancers. ACS Chem. Biol. 2013, 8, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumaz, N.; Marais, R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J. Biol. Chem. 2003, 278, 29819–29823. [Google Scholar] [CrossRef] [Green Version]
- Stanton, V.P., Jr.; Nichols, D.W.; Laudano, A.P.; Cooper, G.M. Definition of the human raf amino-terminal regulatory region by deletion mutagenesis. Mol. Cell Biol. 1989, 9, 639–647. [Google Scholar] [CrossRef] [Green Version]
- Tzivion, G.; Luo, Z.; Avruch, J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 1998, 394, 88–92. [Google Scholar] [CrossRef]
- Luo, Z.; Tzivion, G.; Belshaw, P.J.; Vavvas, D.; Marshall, M.; Avruch, J. Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature 1996, 383, 181–185. [Google Scholar] [CrossRef]
- Fantl, W.J.; Muslin, A.J.; Kikuchi, A.; Martin, J.A.; MacNicol, A.M.; Gross, R.W.; Williams, L.T. Activation of Raf-1 by 14-3-3 proteins. Nature 1994, 371, 612–614. [Google Scholar] [CrossRef]
- Freed, E.; Symons, M.; Macdonald, S.G.; McCormick, F.; Ruggieri, R. Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science 1994, 265, 1713–1716. [Google Scholar] [CrossRef]
- Irie, K.; Gotoh, Y.; Yashar, B.M.; Errede, B.; Nishida, E.; Matsumoto, K. Stimulatory effects of yeast and mammalian 14-3-3 proteins on the Raf protein kinase. Science 1994, 265, 1716–1719. [Google Scholar] [CrossRef]
- Fu, H.; Xia, K.; Pallas, D.C.; Cui, C.; Conroy, K.; Narsimhan, R.P.; Mamon, H.; Collier, R.J.; Roberts, T.M. Interaction of the protein kinase Raf-1 with 14-3-3 proteins. Science 1994, 266, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Petosa, C.; Masters, S.C.; Bankston, L.A.; Pohl, J.; Wang, B.; Fu, H.; Liddington, R.C. 14-3-3zeta binds a phosphorylated Raf peptide and an unphosphorylated peptide via its conserved amphipathic groove. J. Biol. Chem. 1998, 273, 16305–16310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molzan, M.; Ottmann, C. Synergistic binding of the phosphorylated S233- and S259-binding sites of C-RAF to one 14-3-3zeta dimer. J. Mol. Biol. 2012, 423, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Molzan, M.; Schumacher, B.; Ottmann, C.; Baljuls, A.; Polzien, L.; Weyand, M.; Thiel, P.; Rose, R.; Rose, M.; Kuhenne, P.; et al. Impaired binding of 14-3-3 to C-RAF in Noonan syndrome suggests new approaches in diseases with increased Ras signaling. Mol. Cell Biol. 2010, 30, 4698–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. The importance of Raf dimerization in cell signaling. Small Gtpases 2013, 4, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Durrant, D.E.; Morrison, D.K. Targeting the Raf kinases in human cancer: The Raf dimer dilemma. Br. J. Cancer 2018, 118, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, S.; Fujisawa, T.; Ichijo, H. Apoptosis signal-regulating kinase 1 (ASK1) as a therapeutic target for neurological diseases. Expert Opin. Ther. Targets 2020, 1061–1064. [Google Scholar] [CrossRef]
- Psenakova, K.; Hexnerova, R.; Srb, P.; Obsilova, V.; Veverka, V.; Obsil, T. The redox-active site of thioredoxin is directly involved in apoptosis signal-regulating kinase 1 binding that is modulated by oxidative stress. FEBS J. 2020, 287, 1626–1644. [Google Scholar] [CrossRef]
- Tobiume, K.; Saitoh, M.; Ichijo, H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J. Cell Physiol. 2002, 191, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Bunkoczi, G.; Salah, E.; Filippakopoulos, P.; Fedorov, O.; Muller, S.; Sobott, F.; Parker, S.A.; Zhang, H.; Min, W.; Turk, B.E.; et al. Structural and functional characterization of the human protein kinase ASK1. Structure 2007, 15, 1215–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevelyan, S.J.; Brewster, J.L.; Burgess, A.E.; Crowther, J.M.; Cadell, A.L.; Parker, B.L.; Croucher, D.R.; Dobson, R.C.J.; Murphy, J.M.; Mace, P.D. Structure-based mechanism of preferential complex formation by apoptosis signal-regulating kinases. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Fu, H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 8511–8515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federspiel, J.D.; Codreanu, S.G.; Palubinsky, A.M.; Winland, A.J.; Betanzos, C.M.; McLaughlin, B.; Liebler, D.C. Assembly Dynamics and Stoichiometry of the Apoptosis Signal-regulating Kinase (ASK) Signalosome in Response to Electrophile Stress. Mol. Cell Proteom. 2016, 15, 1947–1961. [Google Scholar] [CrossRef] [Green Version]
- Cockrell, L.M.; Puckett, M.C.; Goldman, E.H.; Khuri, F.R.; Fu, H. Dual engagement of 14-3-3 proteins controls signal relay from ASK2 to the ASK1 signalosome. Oncogene 2010, 29, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Goldman, E.H.; Chen, L.; Fu, H. Activation of apoptosis signal-regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14-3-3 dissociation. J. Biol. Chem. 2004, 279, 10442–10449. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Fujino, G.; Noguchi, T.; Matsuzawa, A.; Yamauchi, S.; Saitoh, M.; Takeda, K.; Ichijo, H. Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell Biol. 2007, 27, 8152–8163. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Nishitoh, H.; Ichijo, H.; Kyriakis, J.M. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol. Cell Biol. 2000, 20, 2198–2208. [Google Scholar] [CrossRef] [Green Version]
- Weijman, J.F.; Kumar, A.; Jamieson, S.A.; King, C.M.; Caradoc-Davies, T.T.; Ledgerwood, E.C.; Murphy, J.M.; Mace, P.D. Structural basis of autoregulatory scaffolding by apoptosis signal-regulating kinase 1. Proc. Natl. Acad. Sci. USA 2017, 114, E2096–E2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obsil, T.; Ghirlando, R.; Klein, D.C.; Ganguly, S.; Dyda, F. Crystal structure of the 14-3-3zeta:serotonin N-acetyltransferase complex. A role for scaffolding in enzyme regulation. Cell 2001, 105, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Alblova, M.; Smidova, A.; Docekal, V.; Vesely, J.; Herman, P.; Obsilova, V.; Obsil, T. Molecular basis of the 14-3-3 protein-dependent activation of yeast neutral trehalase Nth1. Proc. Natl. Acad. Sci. USA 2017, 114, E9811–E9820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obsil, T.; Obsilova, V. Structural aspects of protein kinase ASK1 regulation. Adv. Biol. Regul. 2017, 66, 31–36. [Google Scholar] [CrossRef]
- Kaplan, A.; Ottmann, C.; Fournier, A.E. 14-3-3 adaptor protein-protein interactions as therapeutic targets for CNS diseases. Pharm. Res. 2017, 125, 114–121. [Google Scholar] [CrossRef]
- Lentini Santo, D.; Petrvalska, O.; Obsilova, V.; Ottmann, C.; Obsil, T. Stabilization of Protein-Protein Interactions between CaMKK2 and 14-3-3 by Fusicoccins. ACS Chem. Biol. 2020, 15, 3060–3071. [Google Scholar] [CrossRef]
- Stevers, L.M.; Sijbesma, E.; Botta, M.; MacKintosh, C.; Obsil, T.; Landrieu, I.; Cau, Y.; Wilson, A.J.; Karawajczyk, A.; Eickhoff, J.; et al. Modulators of 14-3-3 Protein-Protein Interactions. J. Med. Chem. 2018, 61, 3755–3778. [Google Scholar] [CrossRef] [Green Version]
- Wurtele, M.; Jelich-Ottmann, C.; Wittinghofer, A.; Oecking, C. Structural view of a fungal toxin acting on a 14-3-3 regulatory complex. EMBO J. 2003, 22, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca2+/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef] [Green Version]
- Racioppi, L.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2: Roles in signaling and pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. [Google Scholar] [CrossRef] [Green Version]
- Soderling, T.R. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem. Sci. 1999, 24, 232–236. [Google Scholar] [CrossRef]
- Tokumitsu, H.; Wayman, G.A.; Muramatsu, M.; Soderling, T.R. Calcium/calmodulin-dependent protein kinase kinase: Identification of regulatory domains. Biochemistry 1997, 36, 12823–12827. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Nairn, A.C.; Kuriyan, J. Structural basis for the autoinhibition of calcium/calmodulin-dependent protein kinase I. Cell 1996, 84, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Wayman, G.A.; Kaech, S.; Grant, W.F.; Davare, M.; Impey, S.; Tokumitsu, H.; Nozaki, N.; Banker, G.; Soderling, T.R. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J. Neurosci. 2004, 24, 3786–3794. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Marcelo, K.L.; Rajapakshe, K.; Coarfa, C.; Dean, A.; Wilganowski, N.; Robinson, H.; Sevick, E.; Bissig, K.D.; Goldie, L.C.; et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology 2015, 62, 505–520. [Google Scholar] [CrossRef]
- Edelman, A.M.; Mitchelhill, K.I.; Selbert, M.A.; Anderson, K.A.; Hook, S.S.; Stapleton, D.; Goldstein, E.G.; Means, A.R.; Kemp, B.E. Multiple Ca2+-calmodulin-dependent protein kinase kinases from rat brain. Purification, regulation by Ca2+-calmodulin, and partial amino acid sequence. J. Biol. Chem. 1996, 271, 10806–10810. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.A.; Ribar, T.J.; Lin, F.; Noeldner, P.K.; Green, M.F.; Muehlbauer, M.J.; Witters, L.A.; Kemp, B.E.; Means, A.R. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008, 7, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Chen, Z.; Zhang, F.; Cui, X.; Sun, W.; Geary, G.G.; Wang, Y.; Johnson, D.A.; Zhu, Y.; Chien, S.; et al. Ca2+/calmodulin-dependent protein kinase kinase beta phosphorylation of Sirtuin 1 in endothelium is atheroprotective. Proc. Natl. Acad. Sci. USA 2013, 110, E2420–E2427. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Widmer, J.; Stapleton, D.; Teh, T.; Cox, T.; Kemp, B.E.; Witters, L.A. Catalytic subunits of the porcine and rat 5’-AMP-activated protein kinase are members of the SNF1 protein kinase family. Biochim. Biophys. Acta 1995, 1266, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Davare, M.A.; Saneyoshi, T.; Guire, E.S.; Nygaard, S.C.; Soderling, T.R. Inhibition of calcium/calmodulin-dependent protein kinase kinase by protein 14-3-3. J. Biol. Chem. 2004, 279, 52191–52199. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, T.; Taoka, M.; Hozumi, Y.; Goto, K.; Tokumitsu, H. 14-3-3 Proteins directly regulate Ca2+/calmodulin-dependent protein kinase kinase alpha through phosphorylation-dependent multisite binding. FEBS Lett. 2008, 582, 661–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, M.; Nairn, A.C. Inhibition of the Ca2+/calmodulin-dependent protein kinase I cascade by cAMP-dependent protein kinase. J. Biol. Chem. 1999, 274, 10086–10093. [Google Scholar] [CrossRef] [Green Version]
- Wayman, G.A.; Tokumitsu, H.; Soderling, T.R. Inhibitory cross-talk by cAMP kinase on the calmodulin-dependent protein kinase cascade. J. Biol. Chem. 1997, 272, 16073–16076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langendorf, C.G.; O’Brien, M.T.; Ngoei, K.R.W.; McAloon, L.M.; Dhagat, U.; Hoque, A.; Ling, N.X.Y.; Dite, T.A.; Galic, S.; Loh, K.; et al. CaMKK2 is inactivated by cAMP-PKA signaling and 14-3-3 adaptor proteins. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Spengler, K.; Zibrova, D.; Woods, A.; Langendorf, C.G.; Scott, J.W.; Carling, D.; Heller, R. Protein kinase A negatively regulates VEGF-induced AMPK activation by phosphorylating CaMKK2 at serine 495. Biochem. J. 2020, 477, 3453–3469. [Google Scholar] [CrossRef] [PubMed]
- De Boer, A.H.; de Vries-van Leeuwen, I.J. Fusicoccanes: Diterpenes with surprising biological functions. Trends Plant. Sci. 2012, 17, 360–368. [Google Scholar] [CrossRef]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef]
- Burke, J.E. Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol. Cell 2018, 71, 653–673. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef]
- Balla, A.; Balla, T. Phosphatidylinositol 4-kinases: Old enzymes with emerging functions. Trends Cell Biol. 2006, 16, 351–361. [Google Scholar] [CrossRef]
- Boura, E.; Nencka, R. Phosphatidylinositol 4-kinases: Function, structure, and inhibition. Exp. Cell Res. 2015, 337, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, J.; Ishikawa, K.; Arita, M.; Taniguchi, K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2012, 31, 754–766. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, P.; Zwart, W.T.; van Dijken, R.A.; Deneka, M.; Schulz, T.K.; Geijsen, N.; Coffer, P.J.; Gadella, B.M.; Verkleij, A.J.; van der Sluijs, P.; et al. Phosphatidylinositol 4-kinasebeta is critical for functional association of rab11 with the Golgi complex. Mol. Biol. Cell 2004, 15, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Hausser, A.; Link, G.; Hoene, M.; Russo, C.; Selchow, O.; Pfizenmaier, K. Phospho-specific binding of 14-3-3 proteins to phosphatidylinositol 4-kinase III beta protects from dephosphorylation and stabilizes lipid kinase activity. J. Cell Sci. 2006, 119, 3613–3621. [Google Scholar] [CrossRef] [Green Version]
- Demmel, L.; Beck, M.; Klose, C.; Schlaitz, A.L.; Gloor, Y.; Hsu, P.P.; Havlis, J.; Shevchenko, A.; Krause, E.; Kalaidzidis, Y.; et al. Nucleocytoplasmic shuttling of the Golgi phosphatidylinositol 4-kinase Pik1 is regulated by 14-3-3 proteins and coordinates Golgi function with cell growth. Mol. Biol. Cell 2008, 19, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.E.; Inglis, A.J.; Perisic, O.; Masson, G.R.; McLaughlin, S.H.; Rutaganira, F.; Shokat, K.M.; Williams, R.L. Structures of PI4KIIIbeta complexes show simultaneous recruitment of Rab11 and its effectors. Science 2014, 344, 1035–1038. [Google Scholar] [CrossRef] [Green Version]
- Eisenreichova, A.; Klima, M.; Boura, E. Crystal structures of a yeast 14-3-3 protein from Lachancea thermotolerans in the unliganded form and bound to a human lipid kinase PI4KB-derived peptide reveal high evolutionary conservation. Acta Cryst. F Struct. Biol. Commun. 2016, 72, 799–803. [Google Scholar] [CrossRef]
- Johnson, C.; Crowther, S.; Stafford, M.J.; Campbell, D.G.; Toth, R.; MacKintosh, C. Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem. J. 2010, 427, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Hausser, A.; Storz, P.; Martens, S.; Link, G.; Toker, A.; Pfizenmaier, K. Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIbeta at the Golgi complex. Nat. Cell Biol. 2005, 7, 880–886. [Google Scholar] [CrossRef]
- Szivak, I.; Lamb, N.; Heilmeyer, L.M. Subcellular localization and structural function of endogenous phosphorylated phosphatidylinositol 4-kinase (PI4K92). J. Biol. Chem. 2006, 281, 16740–16749. [Google Scholar] [CrossRef] [Green Version]
- Valente, C.; Turacchio, G.; Mariggio, S.; Pagliuso, A.; Gaibisso, R.; Di Tullio, G.; Santoro, M.; Formiggini, F.; Spano, S.; Piccini, D.; et al. A 14-3-3gamma dimer-based scaffold bridges CtBP1-S/BARS to PI(4)KIIIbeta to regulate post-Golgi carrier formation. Nat. Cell Biol. 2012, 14, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Chalupska, D.; Rozycki, B.; Humpolickova, J.; Faltova, L.; Klima, M.; Boura, E. Phosphatidylinositol 4-kinase IIIbeta (PI4KB) forms highly flexible heterocomplexes that include ACBD3, 14-3-3, and Rab11 proteins. Sci. Rep. 2019, 9, 567. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Hanoch, T.; Porat, Z.; Hausser, A.; Seger, R. Mitotic Golgi translocation of ERK1c is mediated by a PI4KIIIbeta-14-3-3gamma shuttling complex. J. Cell Sci. 2015, 128, 4083–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.K. The 14-3-3 proteins: Integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009, 19, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, I.; van Egmond, W.N.; van Haastert, P.J. The Roco protein family: A functional perspective. FASEB J. 2008, 22, 3103–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.; Alessi, D.R. Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Dzamko, N.; Morrice, N.A.; Campbell, D.G.; Deak, M.; Ordureau, A.; Macartney, T.; Tong, Y.; Shen, J.; Prescott, A.R.; et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 2010, 430, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Stevers, L.M.; de Vries, R.M.; Doveston, R.G.; Milroy, L.G.; Brunsveld, L.; Ottmann, C. Structural interface between LRRK2 and 14-3-3 protein. Biochem. J. 2017, 474, 1273–1287. [Google Scholar] [CrossRef] [Green Version]
- Manschwetus, J.T.; Wallbott, M.; Fachinger, A.; Obergruber, C.; Pautz, S.; Bertinetti, D.; Schmidt, S.H.; Herberg, F.W. Binding of the Human 14-3-3 Isoforms to Distinct Sites in the Leucine-Rich Repeat Kinase 2. Front. Neurosci. 2020, 14, 302. [Google Scholar] [CrossRef]
- Muda, K.; Bertinetti, D.; Gesellchen, F.; Hermann, J.S.; von Zweydorf, F.; Geerlof, A.; Jacob, A.; Ueffing, M.; Gloeckner, C.J.; Herberg, F.W. Parkinson-related LRRK2 mutation R1441C/G/H impairs PKA phosphorylation of LRRK2 and disrupts its interaction with 14-3-3. Proc. Natl. Acad. Sci. USA 2014, 111, E34–E43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, Q.J.; Pan, N.; Lee, S.; Zhao, Y.; Chait, B.T.; Yue, Z. Phosphorylation-dependent 14-3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS ONE 2011, 6, e17153. [Google Scholar]
- Deniston, C.K.; Salogiannis, J.; Mathea, S.; Snead, D.M.; Lahiri, I.; Matyszewski, M.; Donosa, O.; Watanabe, R.; Bohning, J.; Shiau, A.K.; et al. Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Buschauer, R.; Bohning, J.; Audagnotto, M.; Lasker, K.; Lu, T.W.; Boassa, D.; Taylor, S.; Villa, E. The In Situ Structure of Parkinson’s Disease-Linked LRRK2. Cell 2020, 182, 1508–1518.e16. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984, 308, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010, 11, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Poised to signal. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E395–E402. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [Green Version]
- Glotzer, M. The molecular requirements for cytokinesis. Science 2005, 307, 1735–1739. [Google Scholar] [CrossRef] [Green Version]
- Saurin, A.T.; Durgan, J.; Cameron, A.J.; Faisal, A.; Marber, M.S.; Parker, P.J. The regulated assembly of a PKCepsilon complex controls the completion of cytokinesis. Nat. Cell Biol. 2008, 10, 891–901. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Kostelecky, B.; Saurin, A.T.; Purkiss, A.; Parker, P.J.; McDonald, N.Q. Recognition of an intra-chain tandem 14-3-3 binding site within PKCepsilon. EMBO Rep. 2009, 10, 983–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obsilova, V.; Obsil, T. The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases. Int. J. Mol. Sci. 2020, 21, 8824. https://doi.org/10.3390/ijms21228824
Obsilova V, Obsil T. The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases. International Journal of Molecular Sciences. 2020; 21(22):8824. https://doi.org/10.3390/ijms21228824
Chicago/Turabian StyleObsilova, Veronika, and Tomas Obsil. 2020. "The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases" International Journal of Molecular Sciences 21, no. 22: 8824. https://doi.org/10.3390/ijms21228824
APA StyleObsilova, V., & Obsil, T. (2020). The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases. International Journal of Molecular Sciences, 21(22), 8824. https://doi.org/10.3390/ijms21228824