Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HDACs
2.1. HDACs Substrates
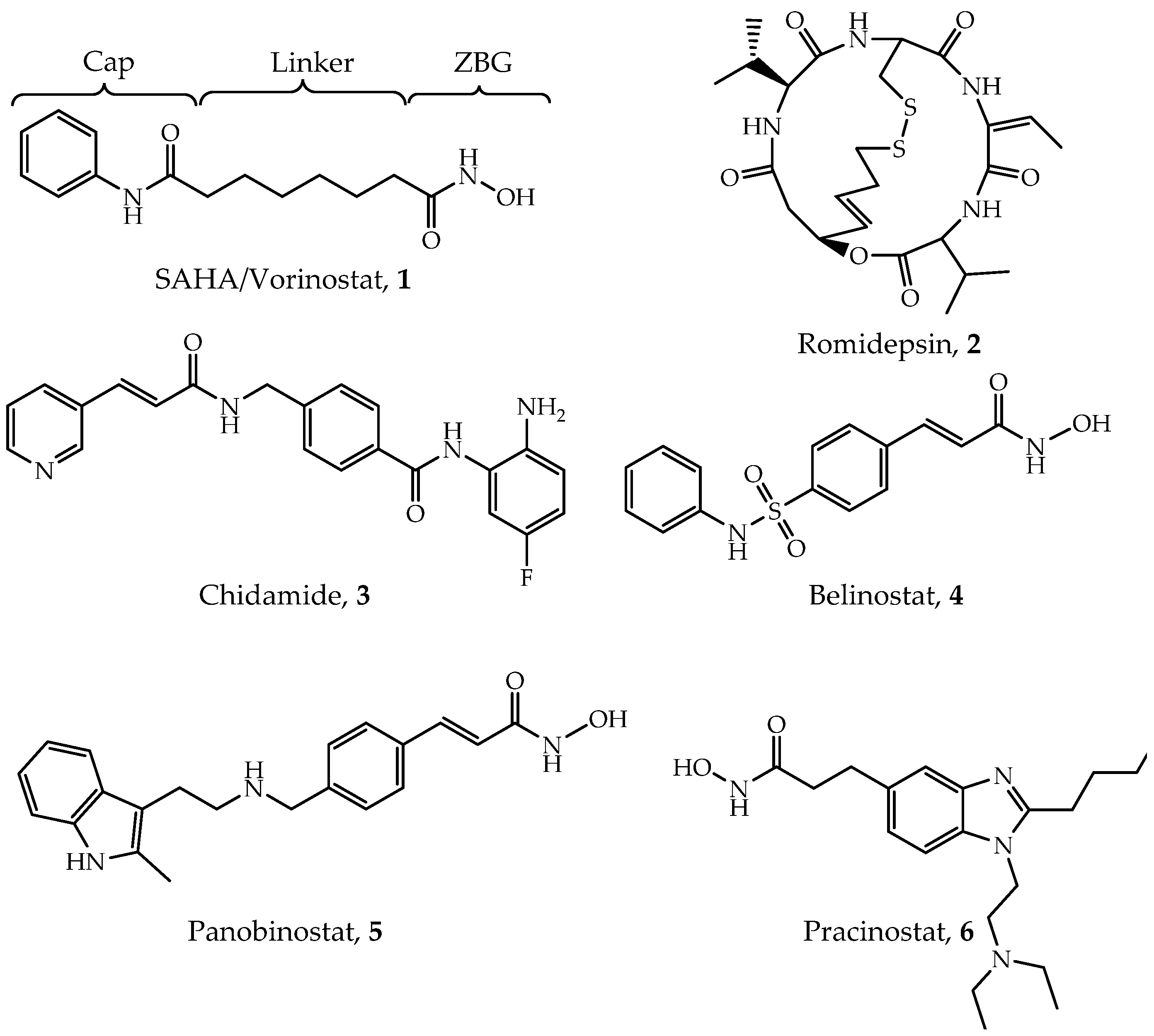
2.2. FDA Approved HDAC Inhibitors
3. Structures of Class I HDACs
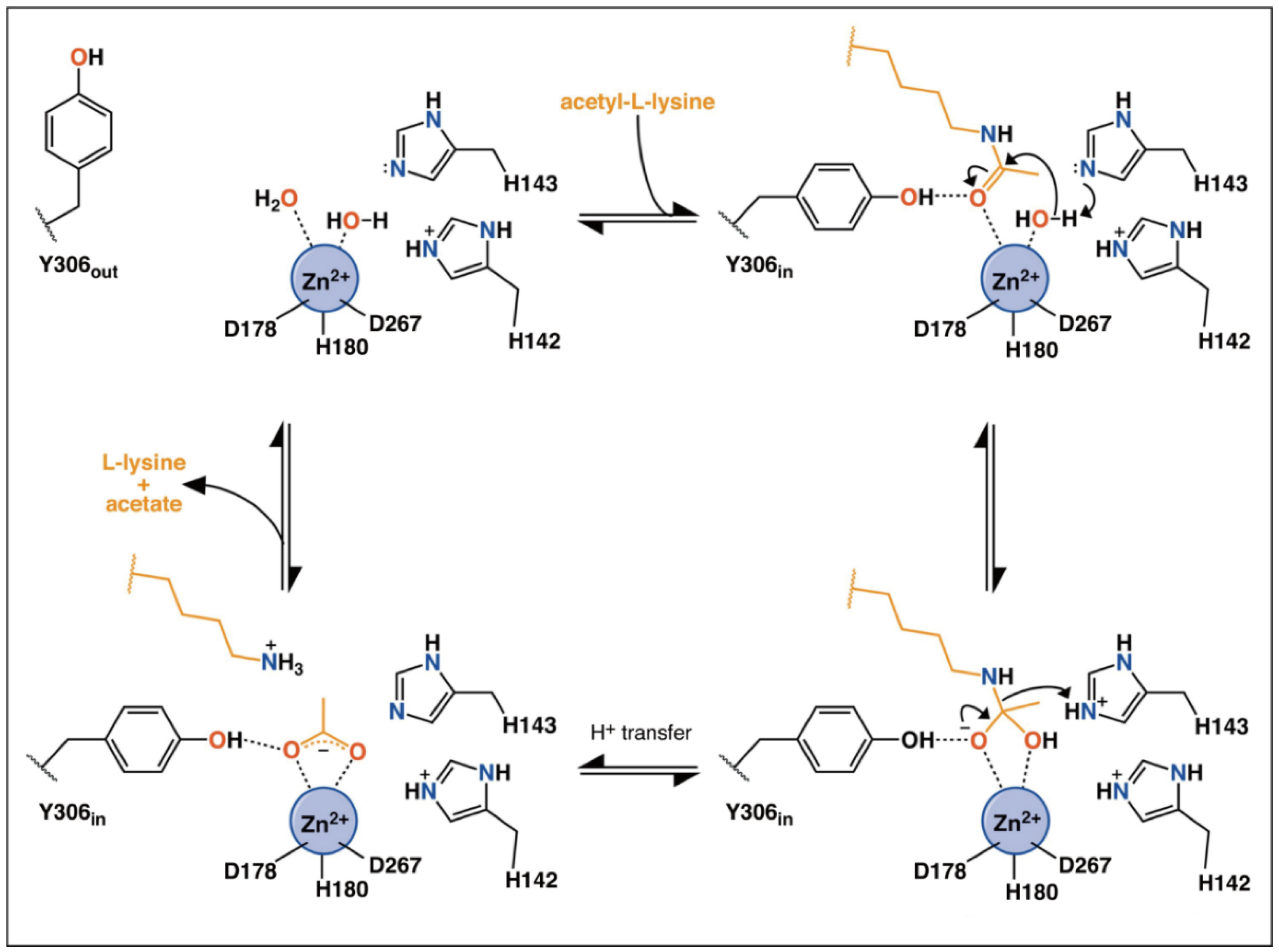
3.1. Structure and Catalytic Mechanism of Monomeric HDACs
3.2. Structure of HDAC Complexes
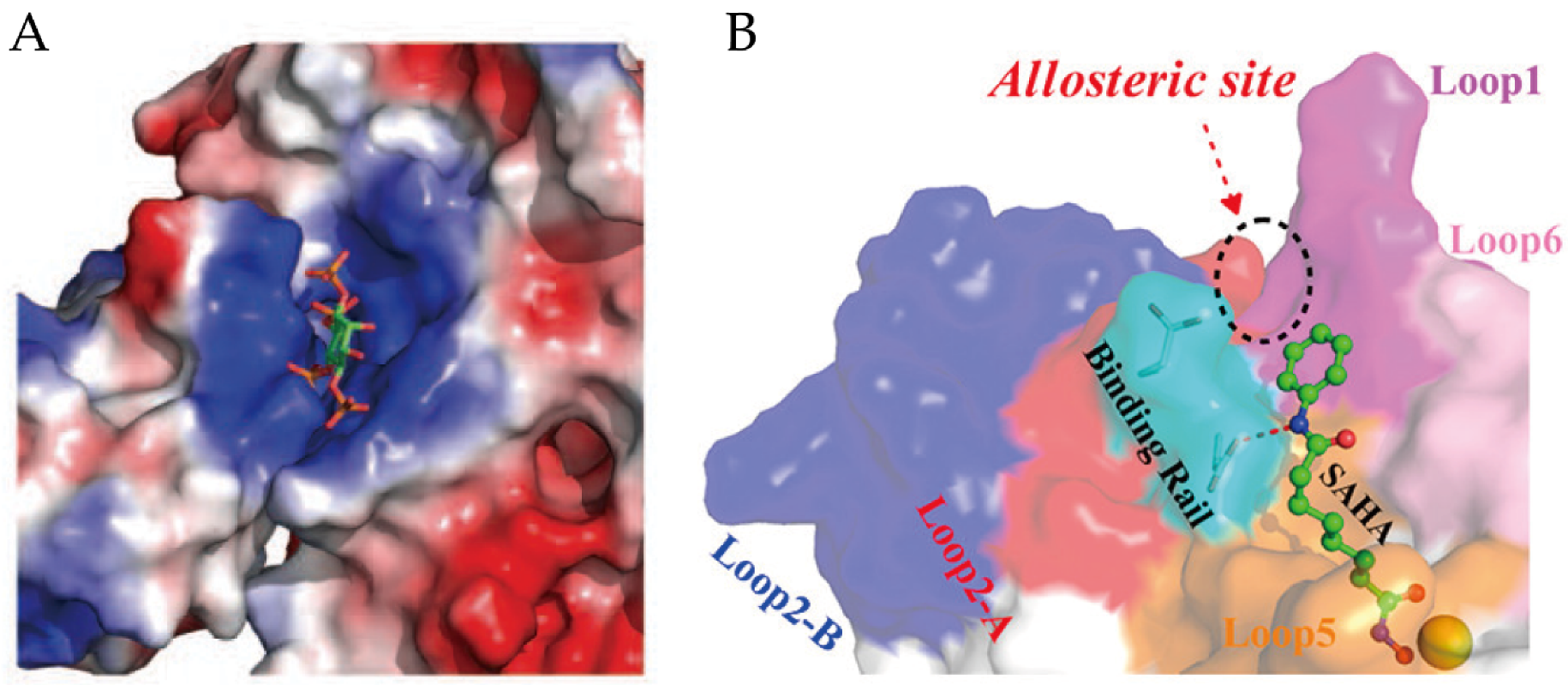
3.3. Allosteric Sites and Regulations
4. Pan-Inhibitors
4.1. Hydroxamic Acids
4.2. Benzamides
4.3. Cyclic Peptides
4.4. Aliphatic Carboxylic Acids
5. Isoform-Selective Inhibitors
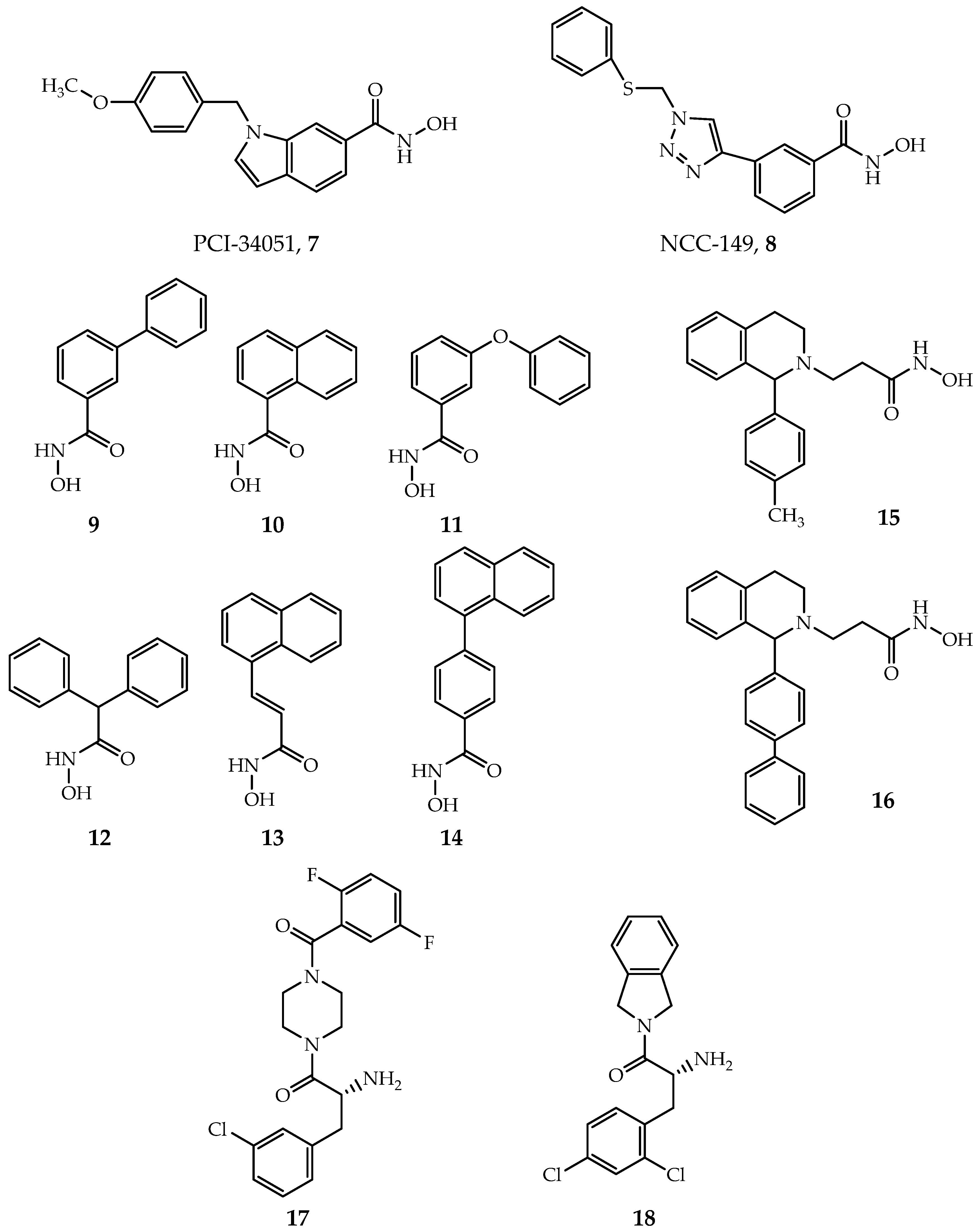
5.1. HDAC8-Selective Inhibitors
5.2. HDAC1/2-Selective Inhibitors
5.3. HDAC3-Selective Inhibitors
6. Complex-Specific Inhibitors
7. Conclusions and Perspectives
Funding
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| AP-MS | Affinity purification mass spectrometry |
| CDK2AP1 | Cyclin-dependent kinase 2-associated protein 1 |
| CFDA | Chinese Food and Drug Administration |
| CHD | Chromodomain helicase DNA‑binding protein |
| CNS | Central nervous system |
| CoREST | Corepressor of RE1-silencing transcription |
| Cryo-EM | Cryo-electron microscopy |
| DLBCL | Diffuse large B-cell lymphoma |
| DNTTIP1 | Deoxynucleotidyl-transferase terminal-interacting protein 1 |
| EM | Electron microscopy |
| FDA | Food and Drug Administration |
| GPS | G-protein pathway suppressor |
| HDAC | Histone deacetylase |
| HDX-MS | Hydrogen-deuterium exchange MS |
| HL | Hodgkin lymphoma |
| Hsp90 | heat shock protein 90 |
| LSD | Lysine-specific demethylase |
| MBD | Methylated CpG-binding domain protein |
| MiDAC | Mitotic deacetylase |
| MIDEAS | Mitotic deacetylase-associated SANT domain |
| MTA | Metastasis tumor-associated protein |
| NCoR | Nuclear receptor corepressor |
| NMPA | National Medical Products Administration |
| NMR | Nuclear magnetic resonance |
| NuRD | Nucleosome remodeling and deacetylase |
| PTMs | Post-translational modifications |
| QM/MM | Quantum mechanical/molecular mechanical |
| RBBP | Retinoblastoma-binding protein |
| SAHA | Suberoylanilide hydroxamic acid |
| SAP30 | Sin3-associated protein p30 |
| SAXS | Small-angle X-ray scattering |
| Sin3 | Switch intensive 3 |
| SIRT | Sirtuin |
| SMRT | Silencing mediator of retinoic acid and thyroid hormone receptors |
| STAT3 | Signal transducers and activators of transcription 3 |
| SUD3 | Suppressor of defective silencing 3 |
| TBL1 | Transducin beta-like 1 |
| XL-MS | Chemical crosslinking mass spectrometry |
| ZBG | Zinc binding group |
References
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Froehlich, L.F. Epigenetic Targeting of Autophagy via HDAC Inhibition in Tumor Cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef]
- Emmett, M.J.; Lazar, M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.L.; Dong, H.; Xu, Q.F.; Zhang, Y.J. Histone deacetylase (HDAC) inhibitors in cancer: A patent review (2017–present). Expert Opin. Ther. Pat. 2020, 30, 263–274. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Welker Leng, K.R.; Castañeda, C.A.; Decroos, C.; Islam, B.; Haider, S.M.; Christianson, D.W.; Fierke, C.A. Phosphorylation of Histone Deacetylase 8: Structural and Mechanistic Analysis of the Phosphomimetic S39E Mutant. Biochemistry 2019, 58, 4480–4493. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Fedele, P.; Orlando, L.; Cinieri, S. Targeting triple negative breast cancer with histone deacetylase inhibitors. Expert Opin. Investig. Drugs 2017, 26, 1199–1206. [Google Scholar] [CrossRef]
- Cao, L.L.; Song, X.; Pei, L.; Liu, L.; Wang, H.; Jia, M. Histone deacetylase HDAC1 expression correlates with the progression and prognosis of lung cancer: A meta-analysis. Medcine (Baltim.) 2017, 96, e7663. [Google Scholar] [CrossRef]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone Deacetylase Inhibitor Romidepsin Induces HIV Expression in CD4 T Cells from Patients on Suppressive Antiretroviral Therapy at Concentrations Achieved by Clinical Dosing. PLoS Path. 2014, 10, e1004071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, P. Therapeutic effects of histone deacetylase inhibitors on kidney disease. Arch. Pharm. Res. 2018, 41, 162–183. [Google Scholar] [CrossRef]
- Das Gupta, K.; Shakespear, M.R.; Iyer, A.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases in monocyte/macrophage development, activation and metabolism: Refining HDAC targets for inflammatory and infectious diseases. Clin. Transl. Immunol. 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef]
- Watson, P.J.; Millard, C.J.; Riley, A.M.; Robertson, N.S.; Wright, L.C.; Godage, H.Y.; Cowley, S.M.; Jamieson, A.G.; Potter, B.V.L.; Schwabe, J.W.R. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun. 2016, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.A.; White, D.A.; Lavender, J.S.; O’Neill, L.P.; Turner, B.M. Human class I histone deacetylase complexes show enhanced catalytic activity in the presence of ATP and co-immunoprecipitate with the ATP-dependent chaperone protein Hsp70. J. Biol. Chem. 2002, 277, 9590–9597. [Google Scholar] [CrossRef] [Green Version]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef]
- Gong, P.; Wang, Y.; Jing, Y. Apoptosis Induction byHistone Deacetylase Inhibitors in Cancer Cells: Role of Ku70. Int. J. Mol. Sci. 2019, 20, 1601. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Kleinheinz, J.; Froehlich, L.F. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.; Han, J.J.; Stenson, M.; Wellik, L.; Witzig, T.E. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012, 26, 1356–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottamal, M.; Zheng, S.L.; Huang, T.L.; Wang, G.D. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. Biomed. Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.T.; Chen, Z.J.; Jiang, G.M.; Wu, Y.M.; Liu, T.; Yi, Y.M.; Zeng, J.; Du, J.; Wang, H.S. Histone deacetylase inhibitors suppress mutant p53 transcription via HDAC8/YY1 signals in triple negative breast cancer cells. Cell. Signal. 2016, 28, 506–515. [Google Scholar] [CrossRef]
- Qiu, X.Y.; Xiao, X.; Li, N.; Li, Y.M. Histone deacetylases inhibitors (HDACis) as novel therapeutic application in various clinical diseases. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 72, 60–72. [Google Scholar] [CrossRef]
- Sarkar, R.; Banerjee, S.; Amin, S.A.; Adhikari, N.; Jha, T. Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review. Eur. J. Med. Chem. 2020, 192, 49. [Google Scholar] [CrossRef]
- Banerjee, S.; Adhikari, N.; Amin, S.A.; Jha, T. Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview. Eur. J. Med. Chem. 2019, 164, 214–240. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIB multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [Green Version]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.L.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma, (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; O’Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef]
- Blumenschein, G.R., Jr.; Kies, M.S.; Papadimitrakopoulou, V.A.; Lu, C.; Kumar, A.J.; Ricker, J.L.; Chiao, J.H.; Chen, C.; Frankel, S.R. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza (TM), suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Investig. New Drugs 2008, 26, 81–87. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S.; et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 2006, 24, 166–173. [Google Scholar] [CrossRef]
- Ueda, H.; Manda, T.; Matsumoto, S.; Mukumoto, S.; Nishigaki, F.; Kawamura, I.; Shimomura, K. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968: III. Antitumor activities on experimental tumors in mice. J. Antibiot. (Tokyo) 1994, 47, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II Multi-Institutional Trial of the Histone Deacetylase Inhibitor Romidepsin as Monotherapy for Patients with Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.J.; Demierre, M.-F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final Results from a Multicenter, International, Pivotal Study of Romidepsin in Refractory Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Reddy, S.A. Romidepsin for the treatment of relapsed/refractory cutaneous T-cell lymphoma (mycosis fungoides/Sezary syndrome): Use in a community setting. Crit. Rev. Oncol. Hematol. 2016, 106, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [Green Version]
- Ning, Z.Q.; Li, Z.B.; Newman, M.J.; Shan, S.; Wang, X.H.; Pan, D.S.; Zhang, J.; Dong, M.; Du, X.; Lu, X.P. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901–909. [Google Scholar] [CrossRef]
- Chan, T.S.; Tse, E.; Kwong, Y.L. Chidamide in the treatment of peripheral T-cell lymphoma. Onco Targets Ther. 2017, 10, 347–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.F.; Li, W.; Hu, X.C.; Zhang, Q.Y.; Sun, T.; Cui, S.D.; Wang, S.S.; Ouyang, Q.C.; Yin, Y.M.; Geng, C.Z.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Shen, Y.L.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492. [Google Scholar] [CrossRef] [Green Version]
- San-Miguel, J.F.; Hungria, V.T.M.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef] [Green Version]
- Novotny-Diermayr, V.; Sangthongpitag, K.; Hu, C.Y.; Wu, X.; Sausgruber, N.; Yeo, P.; Greicius, G.; Pettersson, S.; Liang, A.L.; Loh, Y.K.; et al. SB939, a Novel Potent and Orally Active Histone Deacetylase Inhibitor with High Tumor Exposure and Efficacy in Mouse Models of Colorectal Cancer. Mol. Cancer Ther. 2010, 9, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Manero, G.; Abaza, Y.; Takahashi, K.; Medeiros, B.C.; Arellano, M.; Khaled, S.K.; Patnaik, M.; Odenike, O.; Sayar, H.; Tummala, M.; et al. Pracinostat plus azacitidine in older patients with newly diagnosed acute myeloid leukemia: Results of a phase 2 study. Blood Adv. 2019, 3, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Zhang, L.; Jiang, Q.; Song, W.; Yan, F.; Zhang, L. Histone deacetylase inhibitor based prodrugs. Eur. J. Med. Chem. 2020, 203, 112628. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.R.; Tan, C.P.; Chen, B.C.; Li, R.T.; Mao, Z.W. Zinc-Containing Metalloenzymes: Inhibition by Metal-Based Anticancer Agents. Front. Chem. 2020, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.A.; Pinheiro, P.D.M.; Sagrillo, F.S.; Bolognesi, M.L.; Fraga, C.A.M. Histone deacetylases as targets for the treatment of neurodegenerative disorders: Challenges and future opportunities. Med. Res. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kelly, J.; Yu, W.; Clausen, D.; Yu, Y.; Kim, H.; Duffy, J.L.; Chung, C.C.; Myers, R.W.; Carroll, S.; et al. Selective Class I HDAC Inhibitors Based on Aryl Ketone Zinc Binding Induce HIV-1 Protein for Clearance. ACS Med. Chem. Lett. 2020, 11, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Micelli, C.; Rastelli, G. Histone deacetylases: Structural determinants of inhibitor selectivity. Drug Discov. Today 2015, 20, 718–735. [Google Scholar] [CrossRef]
- Kandasamy, S.; Subramani, P.; Srinivasan, K.; Jayaraj, J.M.; Prasanth, G.; Muthusamy, K.; Rajakannan, V.; Vilwanathan, R. Design and synthesis of imidazole based zinc binding groups as novel small molecule inhibitors targeting Histone deacetylase enzymes in lung cancer. J. Mol. Struct. 2020, 1214, 13. [Google Scholar] [CrossRef]
- Sixto-Lopez, Y.; Bello, M.; Correa-Basurto, J. Insights into structural features of HDAC1 and its selectivity inhibition elucidated by Molecular dynamic simulation and Molecular Docking. J. Biomol. Struct. Dyn. 2019, 37, 584–610. [Google Scholar] [CrossRef]
- Bieliauskas, A.V.; Pflum, M.K.H. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.F.; Wiest, O.; Helquist, P.; Lan-Hargest, H.Y.; Wiech, N.L. On the function of the 14 angstrom long internal cavity of histone deacetylase-like protein: Implications for the design of histone deacetylase inhibitors. J. Med. Chem. 2004, 47, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, L.; Dobler, M.R.; Radetich, B.; Zhu, Y.Y.; Atadja, P.W.; Claiborne, T.; Grob, J.E.; McRiner, A.; Pancost, M.R.; Patnaik, A.; et al. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Biorg. Med. Chem. 2011, 19, 4626–4634. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, N.J.; Christianson, D.W. Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr. Opin. Struct. Biol. 2019, 59, 9–18. [Google Scholar] [CrossRef]
- Lopez, J.E.; Sullivan, E.D.; Fierke, C.A. Metal-dependent Deacetylases: Cancer and Epigenetic Regulators. Acs Chem. Biol. 2016, 11, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, R.; Luo, H.B. Inhibition mechanism of SAHA in HDAC: A revisit. Phys. Chem. Chem. Phys. 2015, 17, 29483–29488. [Google Scholar] [CrossRef]
- Lee, H.; Rezai-Zadeh, N.; Seto, E. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol. Cell. Biol. 2004, 24, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers (Basel) 2020, 12, 1664. [Google Scholar] [CrossRef]
- Zhang, Y.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Bird, A.; Reinberg, D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999, 13, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.F.; Wade, P.A.; Kutateladze, T.G. The NuRD architecture. Cell. Mol. Life Sci. 2013, 70, 3513–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.Y.; Wade, P.A. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat. Rev. Cancer 2011, 11, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and function insights into the NuRD chromatin remodeling complex. Cell. Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Varma, N.; Saleh, A.; Morris, K.; Watson, P.J.; Bottrill, A.R.; Fairall, L.; Smith, C.J.; Schwabe, J.W. The structure of the core NuRD repression complex provides insights into its interaction with chromatin. eLife 2016, 5, e13941. [Google Scholar] [CrossRef]
- Leighton, G.; Williams, D.C. The Methyl-CpG-Binding Domain 2 and 3 Proteins and Formation of the Nucleosome Remodeling and Deacetylase Complex. J. Mol. Biol. 2020, 432, 1624–1639. [Google Scholar] [CrossRef]
- Hayakawa, T.; Nakayama, J. Physiological Roles of Class I HDAC Complex and Histone Demethylase. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Bansal, N.; David, G.; Farias, E.; Waxman, S. Emerging Roles of Epigenetic Regulator Sin3 in Cancer. Adv. Cancer Res. 2016, 130, 113–135. [Google Scholar] [CrossRef]
- Banks, C.A.S.; Thornton, J.L.; Eubanks, C.G.; Adams, M.K.; Miah, S.; Boanca, G.; Liu, X.Y.; Katt, M.L.; Parmely, T.J.; Florens, L.; et al. A Structured Workflow for Mapping Human Sin3 Histone Deacetylase Complex Interactions Using Halo-MudPIT Affinity-Purification Mass Spectrometry. Mol. Cell. Proteom. 2018, 17, 1432–1447. [Google Scholar] [CrossRef] [Green Version]
- Banks, C.A.S.; Zhang, Y.; Miah, S.; Hao, Y.; Adams, M.K.; Wen, Z.H.; Thornton, J.L.; Florens, L.; Washburn, M.P. Integrative Modeling of a Sin3/HDAC Complex Sub-structure. Cell Rep. 2020, 31, 14. [Google Scholar] [CrossRef]
- Silverstein, R.A.; Ekwall, K. Sin3: A flexible regulator of global gene expression and genome stability. Curr. Genet. 2005, 47, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Andres, M.E.; Burger, C.; Peral-Rubio, M.J.; Battaglioli, E.; Anderson, M.E.; Grimes, J.; Dallman, J.; Ballas, N.; Mandel, G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 9873–9878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalin, J.H.; Wu, M.Z.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.X.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.J.; Gocke, C.B.; Luo, X.L.; Borek, D.; Tomchick, D.R.; Machius, M.; Otwinowski, Z.; Yu, H.T. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef]
- Kim, S.A.; Zhu, J.; Yennawar, N.; Eek, P.; Tan, S. Crystal Structure of the LSD1/CoREST Histone Demethylase Bound to Its Nucleosome Substrate. Mol. Cell 2020, 78, 903–914.e4. [Google Scholar] [CrossRef]
- Song, Y.; Dagil, L.; Fairall, L.; Robertson, N.; Wu, M.X.; Ragan, T.J.; Savva, C.G.; Saleh, A.; Morone, N.; Kunze, M.B.A.; et al. Mechanism of Crosstalk between the LSD1 Demethylase and HDAC1 Deacetylase in the CoREST Complex. Cell Rep. 2020, 30, 2699–2711. [Google Scholar] [CrossRef] [Green Version]
- Mondal, B.; Jin, H.J.; Kallappagoudar, S.; Sedkov, Y.; Martinez, T.; Sentmanat, M.F.; Poet, G.J.; Li, C.L.; Fan, Y.P.; Pruett-Miller, S.M.; et al. The histone deacetylase complex MiDAC regulates a neurodevelopmental gene expression program to control neurite outgrowth. eLife 2020, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, R.E.; Fairall, L.; Saleh, A.; Kelsall, E.; Morris, K.L.; Ragan, T.J.; Savva, C.G.; Chandru, A.; Millard, C.J.; Makarova, O.V.; et al. The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure. Nat. Commun. 2020, 11, 15. [Google Scholar] [CrossRef]
- Itoh, T.; Fairall, L.; Muskett, F.W.; Milano, C.P.; Watson, P.J.; Arnaudo, N.; Saleh, A.; Millard, C.J.; El-Mezgueldi, M.; Martino, F.; et al. Structural and functional characterization of a cell cycle associated HDAC1/2 complex reveals the structural basis for complex assembly and nucleosome targeting. Nucleic Acids Res. 2015, 43, 2033–2044. [Google Scholar] [CrossRef] [Green Version]
- Perissi, V.; Jepsen, K.; Glass, C.K.; Rosenfeld, M.G. Deconstructing repression: Evolving models of co-repressor action. Nat. Rev. Genet. 2010, 11, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell. Endocrinol. 2012, 348, 440–449. [Google Scholar] [CrossRef]
- Oberoi, J.; Fairall, L.; Watson, P.J.; Yang, J.C.; Czimmerer, Z.; Kampmann, T.; Goult, B.T.; Greenwood, J.A.; Gooch, J.T.; Kallenberger, B.C.; et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 2011, 18, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W.R. Class I HDACs Share a Common Mechanism of Regulation by Inositol Phosphates. Mol. Cell 2013, 51, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Huang, Y.; Cheng, C.; Wang, K.; Wu, R. Intrinsic Dynamics of the Binding Rail and Its Allosteric Effect in the Class I Histone Deacetylases. J. Chem. Inf. Model. 2017, 57, 2309–2320. [Google Scholar] [CrossRef]
- McClure, J.J.; Zhang, C.; Inks, E.S.; Peterson, Y.K.; Li, J.Y.; Chou, C.J. Development of Allosteric Hydrazide-Containing Class I Histone Deacetylase Inhibitors for Use in Acute Myeloid Leukemia. J. Med. Chem. 2016, 59, 9942–9959. [Google Scholar] [CrossRef] [Green Version]
- Werbeck, N.D.; Shukla, V.K.; Kunze, M.B.A.; Yalinca, H.; Pritchard, R.B.; Siemons, L.; Mondal, S.; Greenwood, S.O.R.; Kirkpatrick, J.; Marson, C.M.; et al. A distal regulatory region of a class I human histone deacetylase. Nat. Commun. 2020, 11, 9. [Google Scholar] [CrossRef]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2020, 27, 2449–2493. [Google Scholar] [CrossRef]
- Trapani, D.; Esposito, A.; Criscitiello, C.; Mazzarella, L.; Locatelli, M.; Minchella, I.; Minucci, S.; Curigliano, G. Entinostat for the treatment of breast cancer. Expert Opin. Investig. Drug. 2017, 26, 965–971. [Google Scholar] [CrossRef]
- Greenwood, S.O.R.; Chan, A.W.E.; Hansen, D.F.; Marson, C.M. Potent non-hydroxamate inhibitors of histone deacetylase-8: Role and scope of an isoindolin-2-yl linker with an alpha-amino amide as the zinc-binding unit. Bioorg. Med. Chem. Lett. 2020, 30, 126926. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Adhikari, N.; Amin, S.A.; Bobde, Y.; Ganesh, R.; Jha, T.; Ghosh, B. Design, synthesis, biological evaluation and molecular docking study of arylcarboxamido piperidine and piperazine-based hydroxamates as potential HDAC8 inhibitors with promising anticancer activity. Eur. J. Pharm. Sci. 2019, 138, 105046. [Google Scholar] [CrossRef] [PubMed]
- Estiu, G.; West, N.; Mazitschek, R.; Greenberg, E.; Bradner, J.E.; Wiest, O. On the inhibition of histone deacetylase 8. Bioorg. Med. Chem. 2010, 18, 4103–4110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, J.; Jiang, Q.X.; Song, W.G. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Hailu, G.S.; Robaa, D.; Forgione, M.; Sippl, W.; Rotili, D.; Mai, A. Lysine Deacetylase Inhibitors in Parasites: Past, Present, and Future Perspectives. J. Med. Chem. 2017, 60, 4780–4804. [Google Scholar] [CrossRef]
- Beckers, T.; Burkhardt, C.; Wieland, H.; Gimmnich, P.; Ciossek, T.; Maier, T.; Sanders, K. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int. J. Cancer 2007, 121, 1138–1148. [Google Scholar] [CrossRef]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [Green Version]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef]
- Zagni, C.; Floresta, G.; Monciino, G.; Rescifina, A. The Search for Potent, Small-Molecule HDACIs in Cancer Treatment: A Decade after Vorinostat. Med. Res. Rev. 2017, 37, 1373–1428. [Google Scholar] [CrossRef]
- Zhan, P.; Wang, X.S.; Liu, X.Y.; Suzuki, T. Medicinal Chemistry Insights into Novel HDAC Inhibitors: An Updated Patent Review (2012–2016). Recent Pat. Anticancer Drug Discov. 2017, 12, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Uba, A.I.; Yelekçi, K. Identification of potential isoform-selective histone deacetylase inhibitors for cancer therapy: A combined approach of structure-based virtual screening, ADMET prediction and molecular dynamics simulation assay. J. Biomol. Struct. Dyn. 2018, 36, 3231–3245. [Google Scholar] [CrossRef] [PubMed]
- Halder, A.K.; Mallick, S.; Shikha, D.; Saha, A.; Saha, K.D.; Jha, T. Design of dual MMP-2/HDAC-8 inhibitors by pharmacophore mapping, molecular docking, synthesis and biological activity. RSC Adv. 2015, 5, 72373–72386. [Google Scholar] [CrossRef]
- Noh, H.; Seo, H. Age-dependent effects of valproic acid in alzheimer’s disease (ad) mice are associated with nerve growth factor (Ngf) regulation. Neuroscience 2014, 266, 255–265. [Google Scholar] [CrossRef]
- Su, J.M.; Li, X.-N.; Thompson, P.; Ou, C.-N.; Ingle, A.M.; Russell, H.; Lau, C.C.; Adamson, P.C.; Blaney, S.M. Phase 1 Study of Valproic Acid in Pediatric Patients with Refractory Solid or CNS Tumors: A Children’s Oncology Group Report. Clin. Cancer Res. 2011, 17, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Gore, S.D.; Weng, L.J.; Figg, W.D.; Zhai, S.P.; Donehower, R.C.; Dover, G.; Grever, M.R.; Griffin, C.; Grochow, L.B.; Hawkins, A.; et al. Impact of prolonged infusions of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin. Cancer Res. 2002, 8, 963–970. [Google Scholar]
- Zhang, M.; Ying, J.B.; Wang, S.S.; He, D.; Zhu, H.; Zhang, C.; Tang, L.; Lin, R.; Zhang, Y. Exploring the binding mechanism of HDAC8 selective inhibitors: Lessons from the modification of Cap group. J. Cell. Biochem. 2020, 121, 3162–3172. [Google Scholar] [CrossRef]
- Wagner, F.F.; Weiwer, M.; Steinbacher, S.; Schomburg, A.; Reinemer, P.; Gale, J.P.; Campbell, A.J.; Fisher, S.L.; Zhao, W.N.; Reis, S.A.; et al. Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1, 2) inhibitors. Biorg. Med. Chem. 2016, 24, 4008–4015. [Google Scholar] [CrossRef]
- Suzuki, T.; Ota, Y.; Ri, M.; Bando, M.; Gotoh, A.; Itoh, Y.; Tsumoto, H.; Tatum, P.R.; Mizukami, T.; Nakagawa, H.; et al. Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. J. Med. Chem. 2012, 55, 9562–9575. [Google Scholar] [CrossRef]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Ramos-Morales, E.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef]
- Suzuki, T.; Muto, N.; Bando, M.; Itoh, Y.; Masaki, A.; Ri, M.; Ota, Y.; Nakagawa, H.; Iida, S.; Shirahige, K.; et al. Design, synthesis, and biological activity of NCC149 derivatives as histone deacetylase 8-selective inhibitors. ChemMedChem 2014, 9, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Luo, T.; Greenberg, E.F.; Bradner, J.E.; Schreiber, S.L. Discovery of histone deacetylase 8 selective inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2601–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.J.; Wang, Y.C.; Chao, S.W.; Yang, C.Y.; Chen, L.C.; Lin, M.H.; Hou, W.C.; Chen, M.Y.; Lee, T.L.; Yang, P.; et al. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. ChemMedChem 2012, 7, 1815–1824. [Google Scholar] [CrossRef]
- Krennhrubec, K.; Marshall, B.L.; Hedglin, M.; Verdin, E.; Ulrich, S.M. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Tabackman, A.A.; Frankson, R.; Marsan, E.S.; Perry, K.; Cole, K.E. Structure of ‘linkerless’ hydroxamic acid inhibitor-HDAC8 complex confirms the formation of an isoform-specific subpocket. J. Struct. Biol. 2016, 195, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, N.; Amin, S.A.; Jha, T. Selective and nonselective HDAC8 inhibitors: A therapeutic patent review. Pharm. Pat. Anal. 2018, 7, 259–276. [Google Scholar] [CrossRef]
- Taha, T.Y.; Aboukhatwa, S.M.; Knopp, R.C.; Ikegaki, N.; Abdelkarim, H.; Neerasa, J.; Lu, Y.; Neelarapu, R.; Hanigan, T.W.; Thatcher, G.R.J.; et al. Design, Synthesis, and Biological Evaluation of Tetrahydroisoquinoline-Based Histone Deacetylase 8 Selective Inhibitors. Acs Med. Chem. Lett. 2017, 8, 824–829. [Google Scholar] [CrossRef]
- Ononye, S.N.; VanHeyst, M.D.; Oblak, E.Z.; Zhou, W.; Ammar, M.; Anderson, A.C.; Wright, D.L. Tropolones as Lead-Like Natural Products: The Development of Potent and Selective Histone Deacetylase Inhibitors. ACS Med. Chem. Lett. 2013, 4, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Tilekar, K.; Upadhyay, N.; Jansch, N.; Schweipert, M.; Mrowka, P.; Meyer-Almes, F.J.; Ramaa, C.S. Discovery of 5-naphthylidene-2,4-thiazolidinedione derivatives as selective HDAC8 inhibitors and evaluation of their cytotoxic effects in leukemic cell lines. Bioorg. Chem. 2020, 95, 103522. [Google Scholar] [CrossRef]
- Galletti, P.; Quintavalla, A.; Ventrici, C.; Giannini, G.; Cabri, W.; Penco, S.; Gallo, G.; Vincenti, S.; Giacomini, D. Azetidinones as zinc-binding groups to design selective HDAC8 inhibitors. ChemMedChem 2009, 4, 1991–2001. [Google Scholar] [CrossRef]
- Ravichandiran, P.; Jegan, A.; Premnath, D.; Periasamy, V.S.; Muthusubramanian, S.; Vasanthkumar, S. Synthesis, molecular docking and cytotoxicity evaluation of novel 2-(4-amino-benzosulfonyl)-5H-benzo[b]carbazole-6,11-dione derivatives as histone deacetylase (HDAC8) inhibitors. Bioorg. Chem. 2014, 53, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P.; et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1: 2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, M.; Chen, N.; Wang, S.; Luo, H.B.; Zhang, Y.; Wu, R. Computational design of a time-dependent histone deacetylase 2 selective inhibitor. ACS Chem. Biol. 2015, 10, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; De Jong, R.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef] [PubMed]
- Weerasinghe, S.V.W.; Estiu, G.; Wiest, O.; Pflum, M.K.H. Residues in the 11 A channel of histone deacetylase 1 promote catalytic activity: Implications for designing isoform-selective histone deacetylase inhibitors. J. Med. Chem. 2008, 51, 5542–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, F.Y.; Zwinderman, M.R.H.; Dekker, F.J. The Process and Strategy for Developing Selective Histone Deacetylase 3 Inhibitors. Molecules 2018, 23, 551. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.Y.; Yang, F.; Jiang, H.; Fan, L. RGFP966 Suppresses Tumor Growth and Migration through Inhibition of EGFR Expression in Hepatocellular Carcinoma Cells in vitro. Drug Des. Devel. Ther. 2020, 14, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, F.F.; Lundh, M.; Kaya, T.; McCarren, P.; Zhang, Y.L.; Chattopadhyay, S.; Gale, J.P.; Galbo, T.; Fisher, S.L.; Meier, B.C.; et al. An Isochemogenic Set of Inhibitors to Define the Therapeutic Potential of Histone Deacetylases in beta-Cell Protection. ACS Chem. Biol. 2016, 11, 363–374. [Google Scholar] [CrossRef]
- Marson, C.M.; Matthews, C.J.; Yiannaki, E.; Atkinson, S.J.; Soden, P.E.; Shukla, L.; Lamadema, N.; Thomas, N.S.B. Discovery of Potent, Isoform-Selective Inhibitors of Histone Deacetylase Containing Chiral Heterocyclic Capping Groups and a N-(2-Aminophenyl)benzamide Binding Unit. J. Med. Chem. 2013, 56, 6156–6174. [Google Scholar] [CrossRef]
- Suzuki, T.; Kasuya, Y.; Itoh, Y.; Ota, Y.; Zhan, P.; Asamitsu, K.; Nakagawa, H.; Okamoto, T.; Miyata, N. Identification of Highly Selective and Potent Histone Deacetylase 3 Inhibitors Using Click Chemistry-Based Combinatorial Fragment Assembly. PLoS ONE 2013, 8, e68669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; He, R.; Chen, Y.; D’Annibale, M.A.; Langley, B.; Kozikowski, A.P. Studies of Benzamide- and Thiol-Based Histone Deacetylase Inhibitors in Models of Oxidative-Stress-Induced Neuronal Death: Identification of Some HDAC3-Selective Inhibitors. Chemmedchem 2009, 4, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.Y.; Chuang, H.C.; Shen, F.H.; Detroja, K.; Hsin, L.W.; Chen, C.S. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur. J. Med. Chem. 2017, 140, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.; Soragni, E.; Chou, C.J.; Barnes, G.; Jones, S.; Rusche, J.R.; Gottesfeld, J.M.; Pandolfo, M. Two New Pimelic Diphenylamide HDAC Inhibitors Induce Sustained Frataxin Upregulation in Cells from Friedreich’s Ataxia Patients and in a Mouse Model. PLoS ONE 2010, 5, e8825. [Google Scholar] [CrossRef]
- Chou, C.J.; Herman, D.; Gottesfeld, J.M. Pimelic Diphenylamide 106 Is a Slow, Tight-binding Inhibitor of Class I Histone Deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef] [Green Version]
- Zwinderman, M.R.H.; de Weerd, S.; Dekker, F.J. Targeting HDAC Complexes in Asthma and COPD. Epigenomes 2019, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Becher, I.; Dittmann, A.; Savitski, M.M.; Hopf, C.; Drewes, G.; Bantscheff, M. Chemoproteomics Reveals Time-Dependent Binding of Histone Deacetylase Inhibitors to Endogenous Repressor Complexes. ACS Chem. Biol. 2014, 9, 1736–1746. [Google Scholar] [CrossRef]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Fuller, N.O.; Pirone, A.; Lynch, B.A.; Hewitt, M.C.; Quinton, M.S.; McKee, T.D.; Ivarsson, M. CoREST Complex-Selective Histone Deacetylase Inhibitors Show Prosynaptic’ Effects and an Improved Safety Profile to Enable Treatment of Synaptopathies. ACS Chem. Neurosci. 2019, 10, 1729–1743. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Wang, S.; Min, H.; Li, J.; Qin, L.-S.; Li, D. Pre-clinical characterization of 4SC-202, a novel class I HDAC inhibitor, against colorectal cancer cells. Tumor Biol. 2016, 37, 10257–10267. [Google Scholar] [CrossRef]
- Von Tresckow, B.; Sayehli, C.; Aulitzky, W.E.; Goebeler, M.-E.; Schwab, M.; Braz, E.; Krauss, B.; Krauss, R.; Hermann, F.; Bartz, R.; et al. Phase I study of domatinostat (4SC-202), a class I histone deacetylase inhibitor in patients with advanced hematological malignancies. Eur. J. Haematol. 2019, 102, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Petrie, K.; Christova, R.; Chung, C.-Y.; Leibovitch, B.A.; Howell, L.; Gil, V.; Sbirkov, Y.; Lee, E.; Wexler, J.; et al. Targeting the SIN3A-PF1 interaction inhibits epithelial to mesenchymal transition and maintenance of a stem cell phenotype in triple negative breast cancer. Oncotarget 2015, 6, 34087–34105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.-J.; Petrie, K.; Leibovitch, B.A.; Zeng, L.; Mezei, M.; Howell, L.; Gil, V.; Christova, R.; Bansal, N.; Yang, S.; et al. Selective Inhibition of SIN3 Corepressor with Avermectins as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2015, 14, 1824–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farias, E.F.; Petrie, K.; Leibovitch, B.; Murtagh, J.; Chornet, M.B.; Schenk, T.; Zelent, A.; Waxman, S. Interference with Sin3 function induces epigenetic reprogramming and differentiation in breast cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11811–11816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.J.; Leibovitch, B.A.; Bansal, N.; Pereira, L.; Chung, C.Y.; Ariztia, E.V.; Zelent, A.; Farias, E.F.; Waxman, S. Targeted interference of SIN3A-TGIF1 function by SID decoy treatment inhibits Wnt signaling and invasion in triple negative breast cancer cells. Oncotarget 2017, 8, 88421–88436. [Google Scholar] [CrossRef]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Leitner, A.; Faini, M.; Stengel, F.; Aebersold, R. Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochem. Sci. 2016, 41, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Vasilescu, J.; Guo, X.C.; Kast, J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics 2004, 4, 3845–3854. [Google Scholar] [CrossRef]
- Kostyukevich, Y.; Acter, T.; Zherebker, A.; Ahmed, A.; Kim, S.; Nikolaev, E. Hydrogen/deuterium exchange in mass spectrometry. Mass Spectrom. Rev. 2018, 37, 811–853. [Google Scholar] [CrossRef]
- Marcsisin, S.R.; Engen, J.R. Hydrogen exchange mass spectrometry: What is it and what can it tell us? Anal. Bioanal. Chem. 2010, 397, 967–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabjerg, E.; Nazari, Z.E.; Rand, K.D. Conformational analysis of complex protein states by hydrogen/deuterium exchange mass spectrometry (HDX-MS): Challenges and emerging solutions. Trends Analyt. Chem. 2018, 106, 125–138. [Google Scholar] [CrossRef]
- Li, H.; Sheng, Y.; McGee, W.; Cammarata, M.; Holden, D.; Loo, J.A. Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal. Chem. 2017, 89, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Heck, A.J.R. Native mass spectrometry: A bridge between interactomics and structural biology. Nat. Methods 2008, 5, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Nguyen, H.H.; Ogorzalek Loo, R.R.; Campuzano, I.D.G.; Loo, J.A. An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat. Chem. 2018, 10, 139–148. [Google Scholar] [CrossRef]
- Li, H.; Wongkongkathep, P.; Van Orden, S.L.; Ogorzalek Loo, R.R.; Loo, J.A. Revealing ligand binding sites and quantifying subunit variants of noncovalent protein complexes in a single native top-down FTICR MS experiment. J. Am. Soc. Mass Spectrom. 2014, 25, 2060–2068. [Google Scholar] [CrossRef] [Green Version]
- Lossl, P.; van de Waterbeemd, M.; Heck, A.J.R. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016, 35, 2634–2657. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Y.; Li, H. Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs). Int. J. Mol. Sci. 2020, 21, 8828. https://doi.org/10.3390/ijms21228828
Luo Y, Li H. Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs). International Journal of Molecular Sciences. 2020; 21(22):8828. https://doi.org/10.3390/ijms21228828
Chicago/Turabian StyleLuo, Yuxiang, and Huilin Li. 2020. "Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs)" International Journal of Molecular Sciences 21, no. 22: 8828. https://doi.org/10.3390/ijms21228828
APA StyleLuo, Y., & Li, H. (2020). Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs). International Journal of Molecular Sciences, 21(22), 8828. https://doi.org/10.3390/ijms21228828