Transcriptional Profiling of Whisker Follicles and of the Striatum in Methamphetamine Self-Administered Rats

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
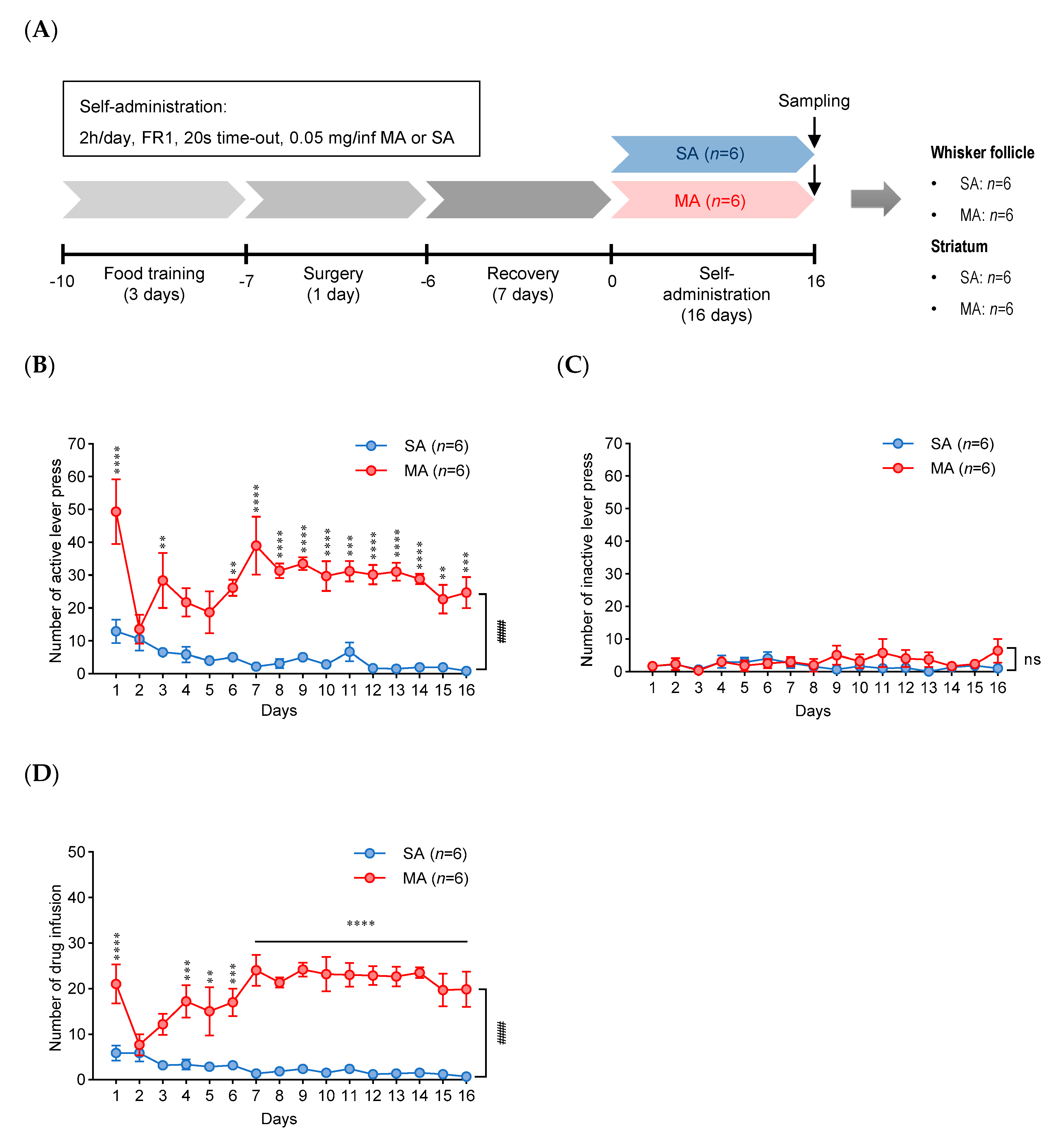
2.1. Methamphetamine Self-Administration
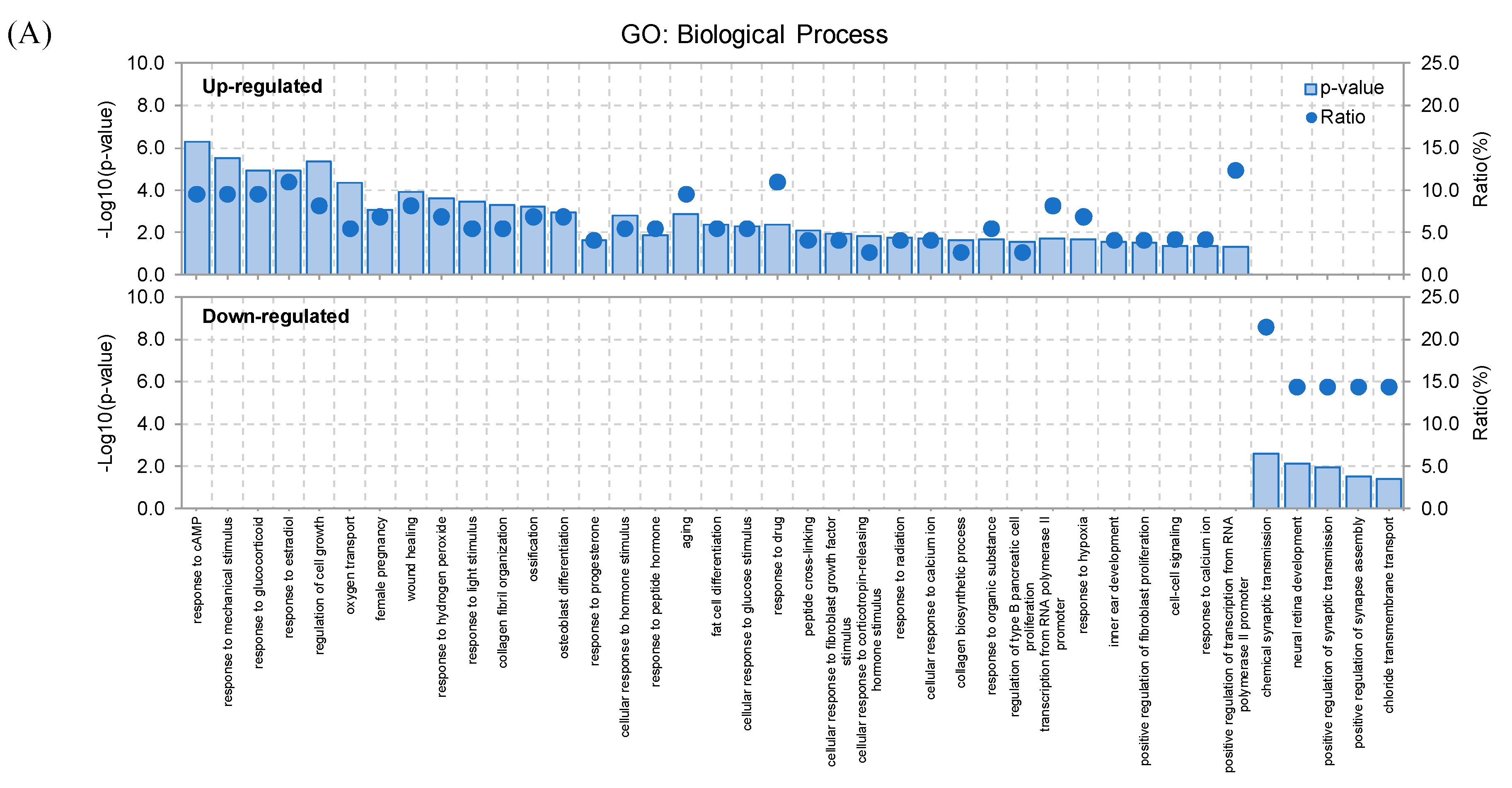
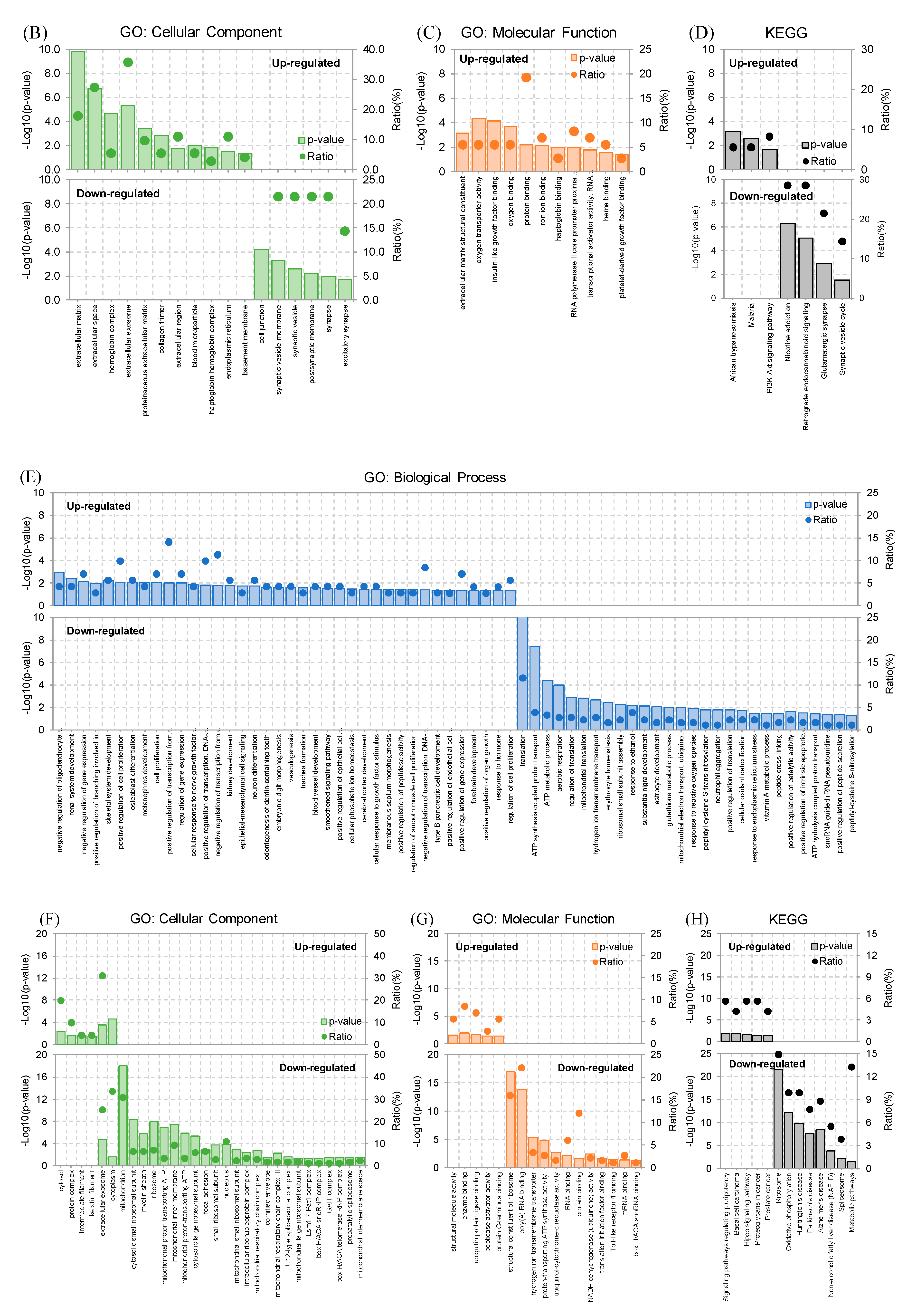
2.2. RNA-Seq Analysis and Gene Expression Profiling
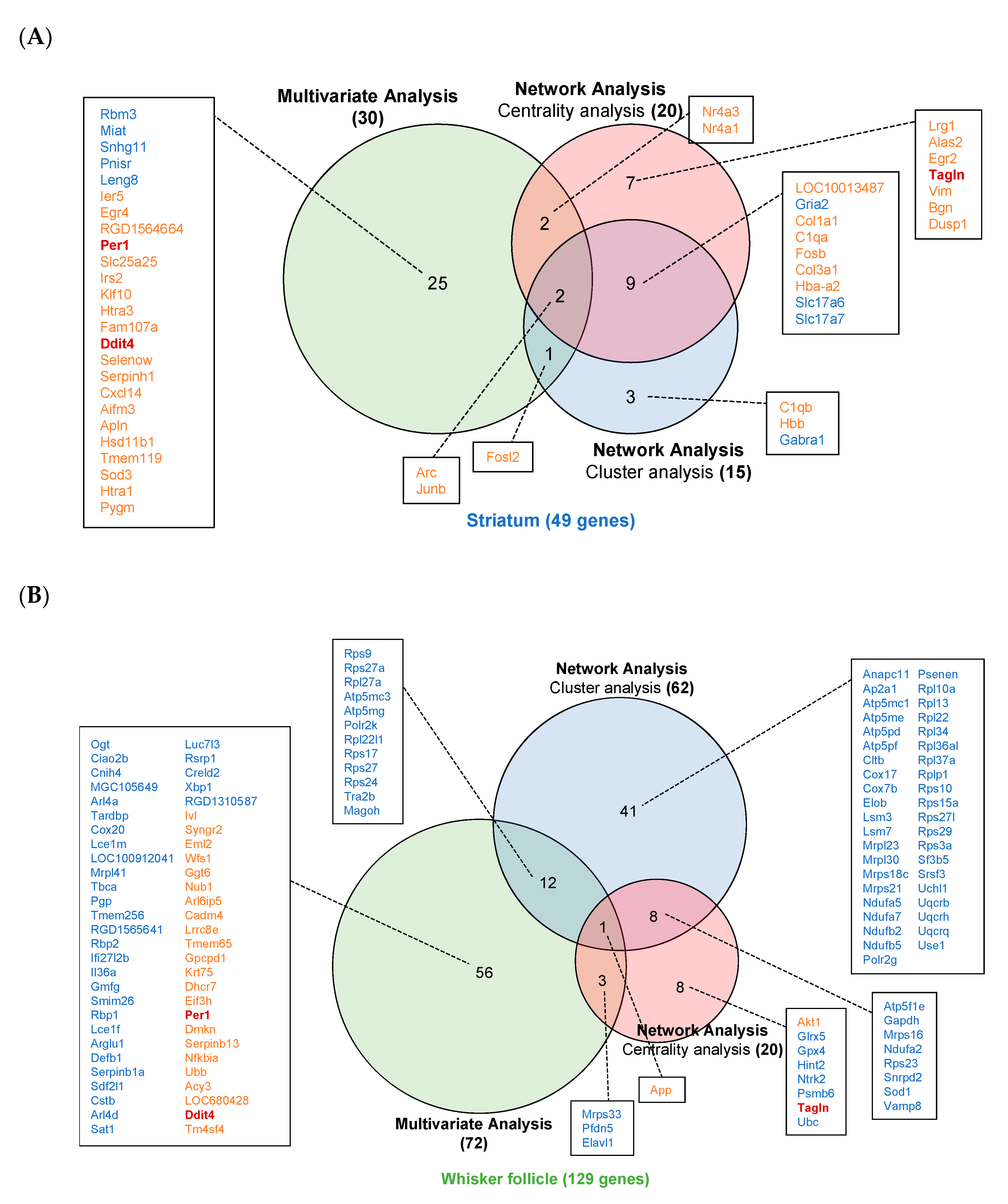
2.3. Multivariate Analysis
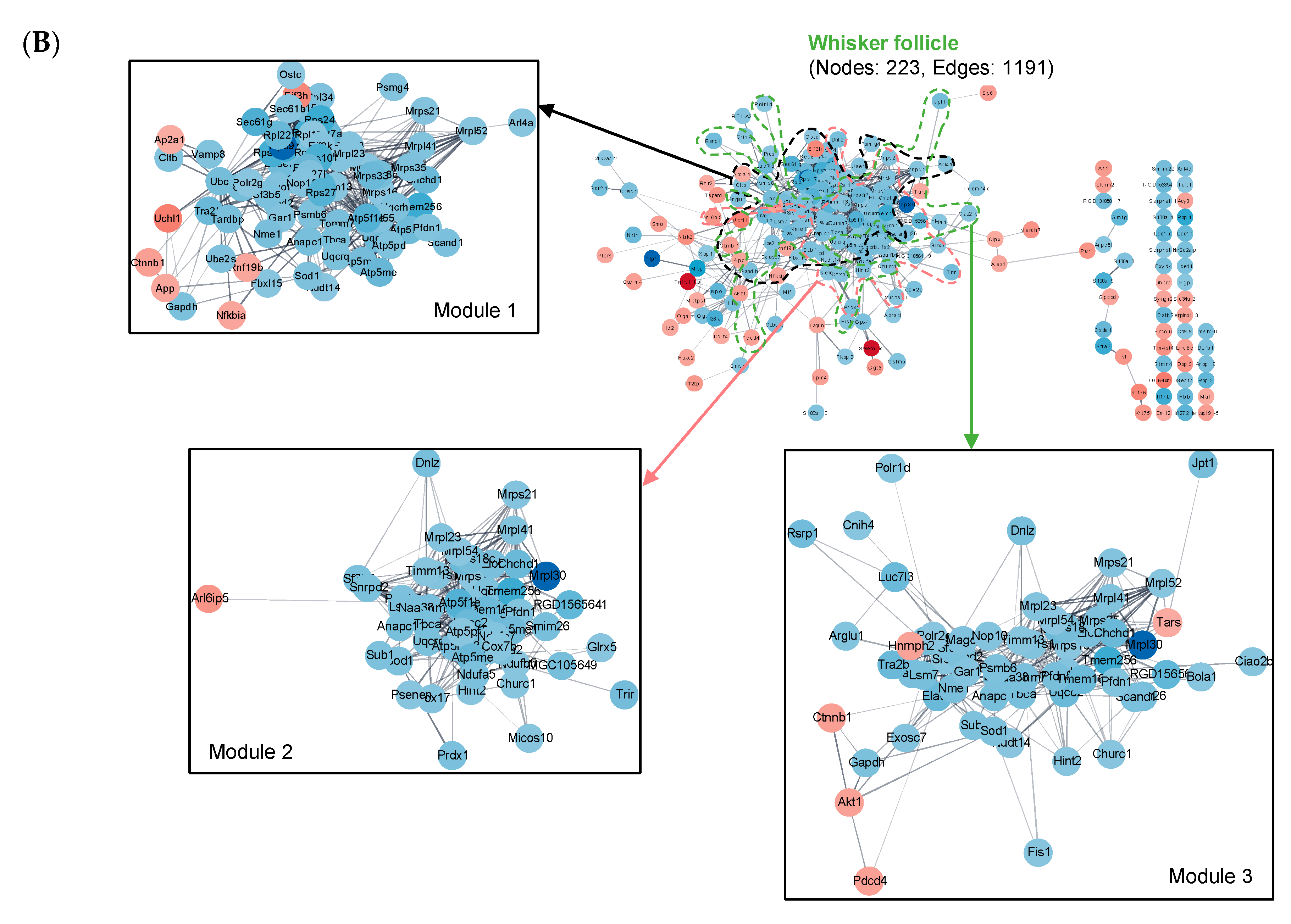
2.4. Construction of Protein–Protein Interaction (PPI) Networks
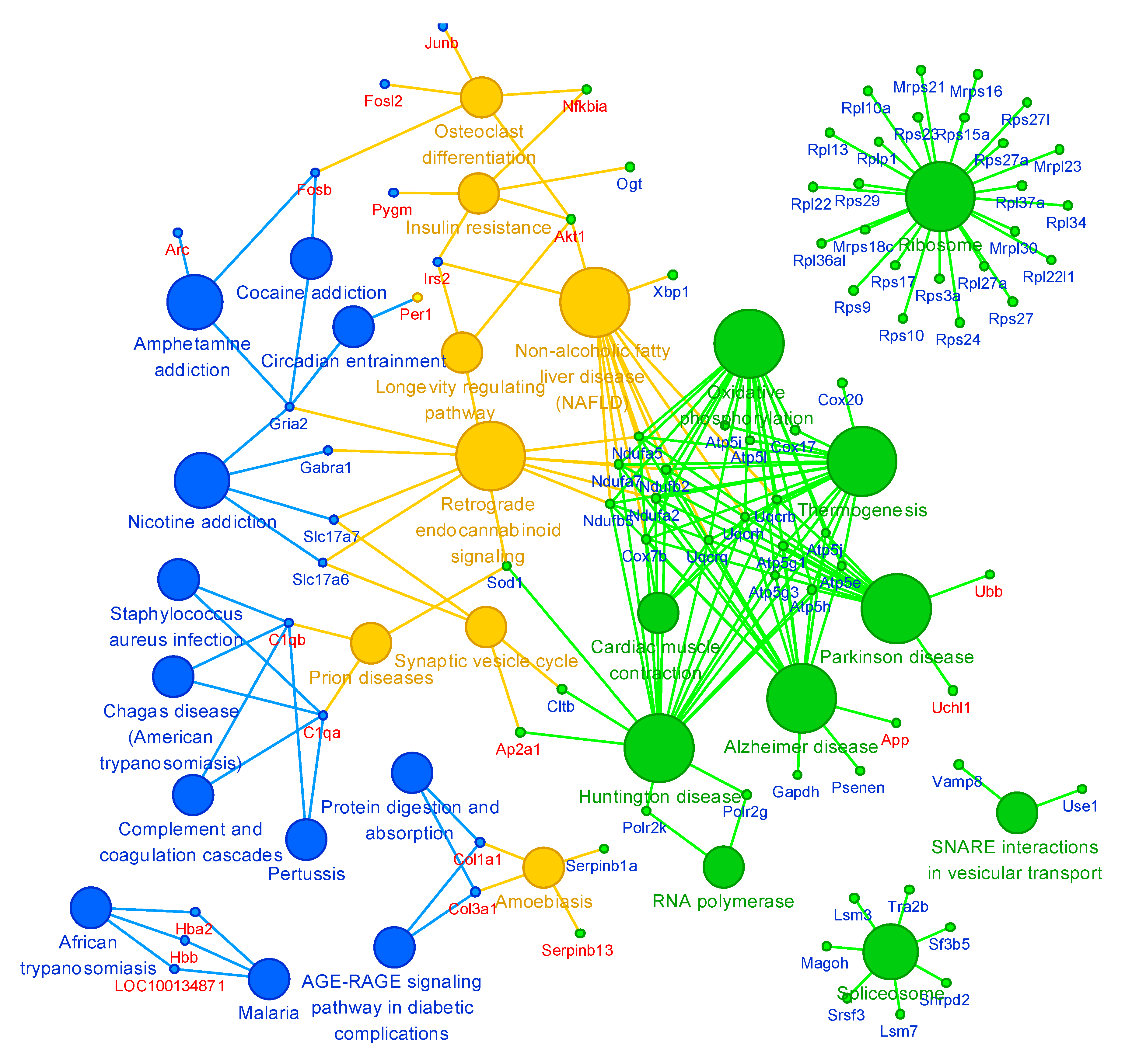
2.5. Cluster Analysis
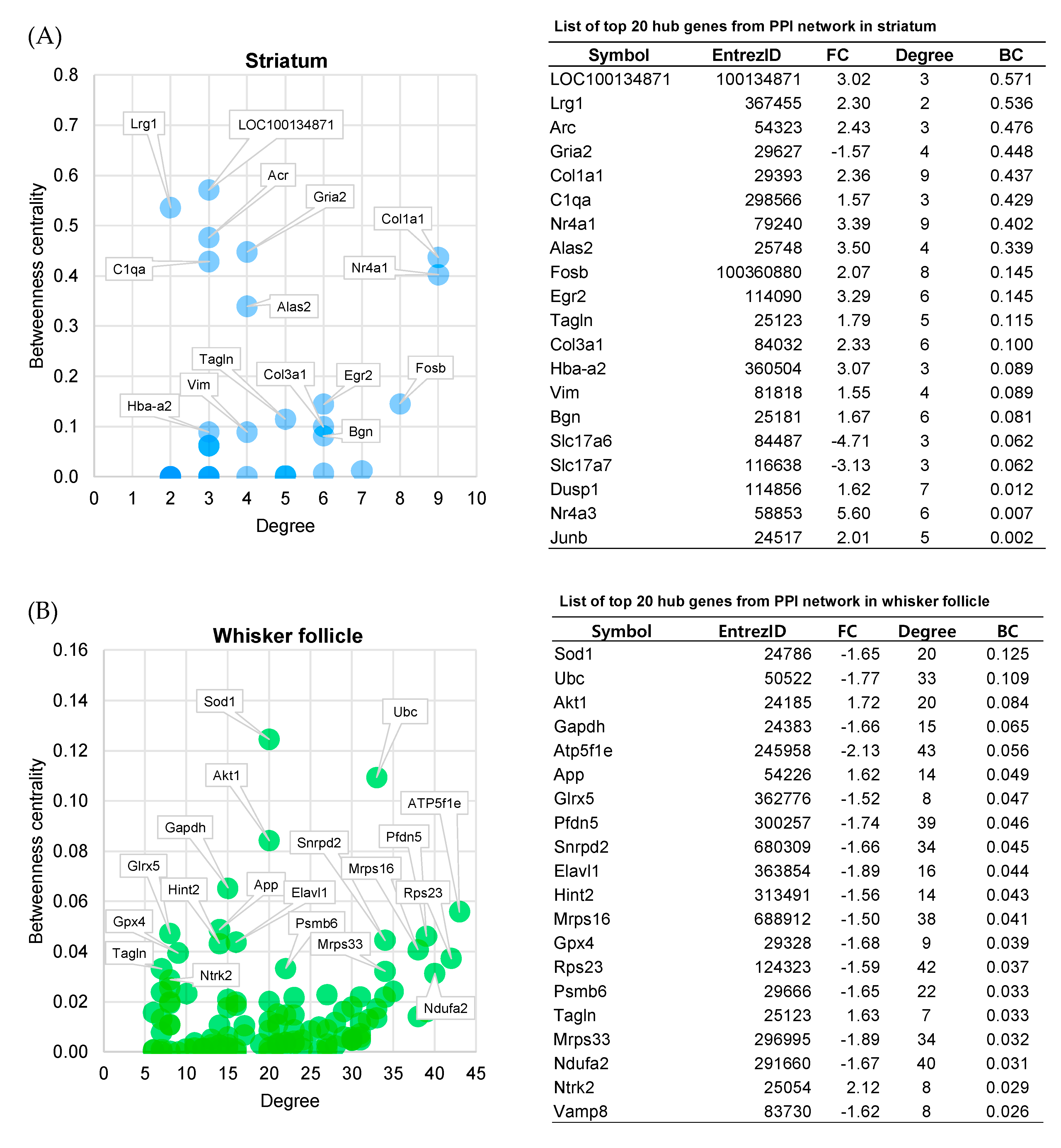
2.6. Centrality Analysis
2.7. Identification of Potential Diagnostic Markers
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Methamphetamine Self-Administration and Sample Collection
4.3. RNA Extraction
4.4. Construction of Transcriptome Libraries
4.5. Processing of the RNA-Seq Data
4.6. Statistical Analysis of Differentially Expressed Genes (DEGs)
4.7. Principal Component Analysis (PCA) and Hierarchical Clustering
4.8. Correlation Analysis
4.9. Centrality Analysis
4.10. Functional and Pathway Enrichment Analyses
4.11. Cluster and Functional Annotation Analyses of PPI Networks
4.12. Identification of Potential Markers among the DEGs of Whisker Follicles or of the Striatum
4.13. Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DEGs | Differentially expressed genes |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery |
| GO | Gene Ontology |
| BP | Biological process |
| CC | Cellular component |
| MF | Molecular function |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PCA | Principal component analysis |
| PPI | Protein–protein interaction |
| STRING | Search Tool for the Retrieval of Interacting Genes/Proteins |
| MCODE | Molecular complex detection |
| BC | Betweenness centrality |
| MA | Methamphetamine |
| SA | Saline |
| FR1 | Fixed ratio 1 |
| FDR | False discovery rate |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| FC | Fold change |
| FPKM | Fragments per kilobase of transcript per million mapped reads |
References
- Centers for Disease Control and Prevention. Acute public health consequences of methamphetamine laboratories—16 states, January 2000-June 2004. MMWR Morb. Mortal. Wkly. Rep. 2005, 54, 356–359. Available online: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5414a3.htm (accessed on 14 April 2005).
- Kwon, N.J.; Han, E. A commentary on the effects of methamphetamine and the status of methamphetamine abuse among youths in South Korea, Japan, and China. Forensic. Sci. Int. 2018, 286, 81–85. [Google Scholar] [CrossRef]
- Kalechstein, A.D.; Newton, T.F.; Green, M. Methamphetamine dependence is associated with neurocognitive impairment in the initial phases of abstinence. J. Neuropsychiatry Clin. Neurosci. 2003, 15, 215–220. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Taylor, J.R. Impulsivity resulting from frontostriatal dysfunction in drug abuse: Implications for the control of behavior by reward-related stimuli. Psychopharmacology 1999, 146, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef] [Green Version]
- Luscher, C.; Malenka, R.C. Drug-evoked synaptic plasticity in addiction: From molecular changes to circuit remodeling. Neuron 2011, 69, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Robison, A.J.; Nestler, E.J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 2011, 12, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Yamada, K.; Toyoshima, M.; Ohnishi, T.; Iwayama, Y.; Shimamoto, C.; Toyota, T.; Nozaki, Y.; Balan, S.; Matsuzaki, H.; et al. Utility of Scalp Hair Follicles as a Novel Source of Biomarker Genes for Psychiatric Illnesses. Biol. Psychiatry 2015, 78, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.H.; Jang, W.J.; Hwang, J.; Park, B.; Jang, J.H.; Seo, Y.H.; Yang, C.H.; Lee, S.; Jeong, C.H. Transcriptome profiling of whisker follicles in methamphetamine self-administered rats. Sci. Rep. 2018, 8, 11420. [Google Scholar] [CrossRef]
- O’Connor, E.C.; Chapman, K.; Butler, P.; Mead, A.N. The predictive validity of the rat self-administration model for abuse liability. Neurosci. Biobehav. Rev. 2011, 35, 912–938. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Fan, P.; Arolfo, M.; Jiang, Z.; Olive, M.F.; Zablocki, J.; Sun, H.L.; Chu, N.; Lee, J.; Kim, H.Y.; et al. Inhibition of aldehyde dehydrogenase-2 suppresses cocaine seeking by generating THP, a cocaine use-dependent inhibitor of dopamine synthesis. Nat. Med. 2010, 16, 1024–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnova, I.N.; Chiflikyan, M.; Justinova, Z.; McCoy, M.T.; Ladenheim, B.; Jayanthi, S.; Quintero, C.; Brannock, C.; Barnes, C.; Adair, J.E.; et al. CREB phosphorylation regulates striatal transcriptional responses in the self-administration model of methamphetamine addiction in the rat. Neurobiol. Dis. 2013, 58, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Miryala, S.K.; Anbarasu, A.; Ramaiah, S. Discerning molecular interactions: A comprehensive review on biomolecular interaction databases and network analysis tools. Gene 2018, 642, 84–94. [Google Scholar] [CrossRef]
- Gursoy, A.; Keskin, O.; Nussinov, R. Topological properties of protein interaction networks from a structural perspective. Biochem. Soc. Trans. 2008, 36, 1398–1403. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007, 3, e59. [Google Scholar] [CrossRef]
- Cami, J.; Farre, M. Drug addiction. N. Engl. J. Med. 2003, 349, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, R.Z.; Volkow, N.D. Drug addiction and its underlying neurobiological basis: Neuroimaging evidence for the involvement of the frontal cortex. Am. J. Psychiatry 2002, 159, 1642–1652. [Google Scholar] [CrossRef]
- Olkin, I.; Sampson, A.R. Multivariate Analysis: Overview. In International Encyclopedia of the Social & Behavioral Sciences, 1st ed.; Smelser, N.J., Baltes, P.B., Eds.; Pergamon: Oxford, UK, 2001; pp. 10240–10247. [Google Scholar] [CrossRef]
- Marcello Manfredi, E.R. Biomarkers Discovery through Multivariate Statistical Methods: A Review of Recently Developed Methods and Applications in Proteomics. J. Proteom. Bioinform. 2013, s3. [Google Scholar] [CrossRef] [Green Version]
- Vella, D.; Marini, S.; Vitali, F.; Di Silvestre, D.; Mauri, G.; Bellazzi, R. MTGO: PPI Network Analysis Via Topological and Functional Module Identification. Sci. Rep. 2018, 8, 5499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Mohanty, P.; Bhatnagar, S. Integrative analysis of ocular complications in atherosclerosis unveils pathway convergence and crosstalk. J. Recept. Signal. Transduct. Res. 2015, 35, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Jayanthi, S.; McCoy, M.T.; Vawter, M.; Ladenheim, B. Temporal profiling of methamphetamine-induced changes in gene expression in the mouse brain: Evidence from cDNA array. Synapse 2001, 41, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Jayanthi, S.; McCoy, M.T.; Beauvais, G.; Ladenheim, B.; Gilmore, K.; Wood, W., 3rd; Becker, K.; Cadet, J.L. Methamphetamine induces dopamine D1 receptor-dependent endoplasmic reticulum stress-related molecular events in the rat striatum. PLoS ONE 2009, 4, e6092. [Google Scholar] [CrossRef] [Green Version]
- Kodama, M.; Akiyama, K.; Ujike, H.; Shimizu, Y.; Tanaka, Y.; Kuroda, S. A robust increase in expression of arc gene, an effector immediate early gene, in the rat brain after acute and chronic methamphetamine administration. Brain Res. 1998, 796, 273–283. [Google Scholar] [CrossRef]
- El-Boustani, S.; Ip, J.P.K.; Breton-Provencher, V.; Knott, G.W.; Okuno, H.; Bito, H.; Sur, M. Locally coordinated synaptic plasticity of visual cortex neurons in vivo. Science 2018, 360, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, J.D.; Bear, M.F. New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 2011, 14, 279–284. [Google Scholar] [CrossRef]
- Gao, M.; Sossa, K.; Song, L.; Errington, L.; Cummings, L.; Hwang, H.; Kuhl, D.; Worley, P.; Lee, H.K. A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. J. Neurosci. 2010, 30, 7168–7178. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, J.D.; Rumbaugh, G.; Wu, J.; Chowdhury, S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 2006, 52, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Petralia, R.S.; Kurushima, H.; Patel, H.; Jung, M.Y.; Volk, L.; Chowdhury, S.; Shepherd, J.D.; Dehoff, M.; Li, Y.; et al. Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell 2011, 147, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Francois, M.; Leifert, W.; Martins, R.; Thomas, P.; Fenech, M. Biomarkers of Alzheimer’s disease risk in peripheral tissues; focus on buccal cells. Curr. Alzheimer Res. 2014, 11, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Falcon, E.; McClung, C.A. A role for the circadian genes in drug addiction. Neuropharmacology 2009, 56 (Suppl. 1), 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibley, H.L.; Malcolm, R.J.; Veatch, L.M. Adolescents with insomnia and substance abuse: Consequences and comorbidities. J. Psychiatr. Pract. 2008, 14, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, T.; Akiyama, M.; Moriya, T.; Shibata, S. Sensitized increase of period gene expression in the mouse caudate/putamen caused by repeated injection of methamphetamine. Mol. Pharmacol. 2001, 59, 894–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Hida, A.; Kitamura, S.; Enomoto, M.; Ohsawa, Y.; Katayose, Y.; Nozaki, K.; Moriguchi, Y.; Aritake, S.; Higuchi, S.; et al. Rhythmic expression of circadian clock genes in human leukocytes and beard hair follicle cells. Biochem. Biophys. Res. Commun. 2012, 425, 902–907. [Google Scholar] [CrossRef]
- Yoon, S.S.; Kim, H.; Choi, K.H.; Lee, B.H.; Lee, Y.K.; Lim, S.C.; Choi, S.H.; Hwang, M.; Kim, K.J.; Yang, C.H. Acupuncture suppresses morphine self-administration through the GABA receptors. Brain Res. Bull. 2010, 81, 625–630. [Google Scholar] [CrossRef]
- Yoon, S.S.; Yang, E.J.; Lee, B.H.; Jang, E.Y.; Kim, H.Y.; Choi, S.M.; Steffensen, S.C.; Yang, C.H. Effects of acupuncture on stress-induced relapse to cocaine-seeking in rats. Psychopharmacology 2012, 222, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.A.; Kiss Von Soly, S.; Ratnayake, U.; Klug, M.; Binder, M.D.; Hannan, A.J.; van den Buuse, M. Long-term effects of combined neonatal and adolescent stress on brain-derived neurotrophic factor and dopamine receptor expression in the rat forebrain. Biochim. Biophys. Acta 2014, 1842, 2126–2135. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, W.-J.; Son, T.; Song, S.-H.; Ryu, I.S.; Lee, S.; Jeong, C.-H. Transcriptional Profiling of Whisker Follicles and of the Striatum in Methamphetamine Self-Administered Rats. Int. J. Mol. Sci. 2020, 21, 8856. https://doi.org/10.3390/ijms21228856
Jang W-J, Son T, Song S-H, Ryu IS, Lee S, Jeong C-H. Transcriptional Profiling of Whisker Follicles and of the Striatum in Methamphetamine Self-Administered Rats. International Journal of Molecular Sciences. 2020; 21(22):8856. https://doi.org/10.3390/ijms21228856
Chicago/Turabian StyleJang, Won-Jun, Taekwon Son, Sang-Hoon Song, In Soo Ryu, Sooyeun Lee, and Chul-Ho Jeong. 2020. "Transcriptional Profiling of Whisker Follicles and of the Striatum in Methamphetamine Self-Administered Rats" International Journal of Molecular Sciences 21, no. 22: 8856. https://doi.org/10.3390/ijms21228856
APA StyleJang, W. -J., Son, T., Song, S. -H., Ryu, I. S., Lee, S., & Jeong, C. -H. (2020). Transcriptional Profiling of Whisker Follicles and of the Striatum in Methamphetamine Self-Administered Rats. International Journal of Molecular Sciences, 21(22), 8856. https://doi.org/10.3390/ijms21228856