The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer


Abstract
:1. Introduction
2. The Role of cGAS, STING, and Type I Interferons in Antitumor Immunity
2.1. A Primer on Immune-Mediated Destruction of Cancer
2.2. Nucleic Acid Sensing and Sources of Cytosolic DNA
2.3. cGAS, STING, and Type I Interferon Signaling Transduction
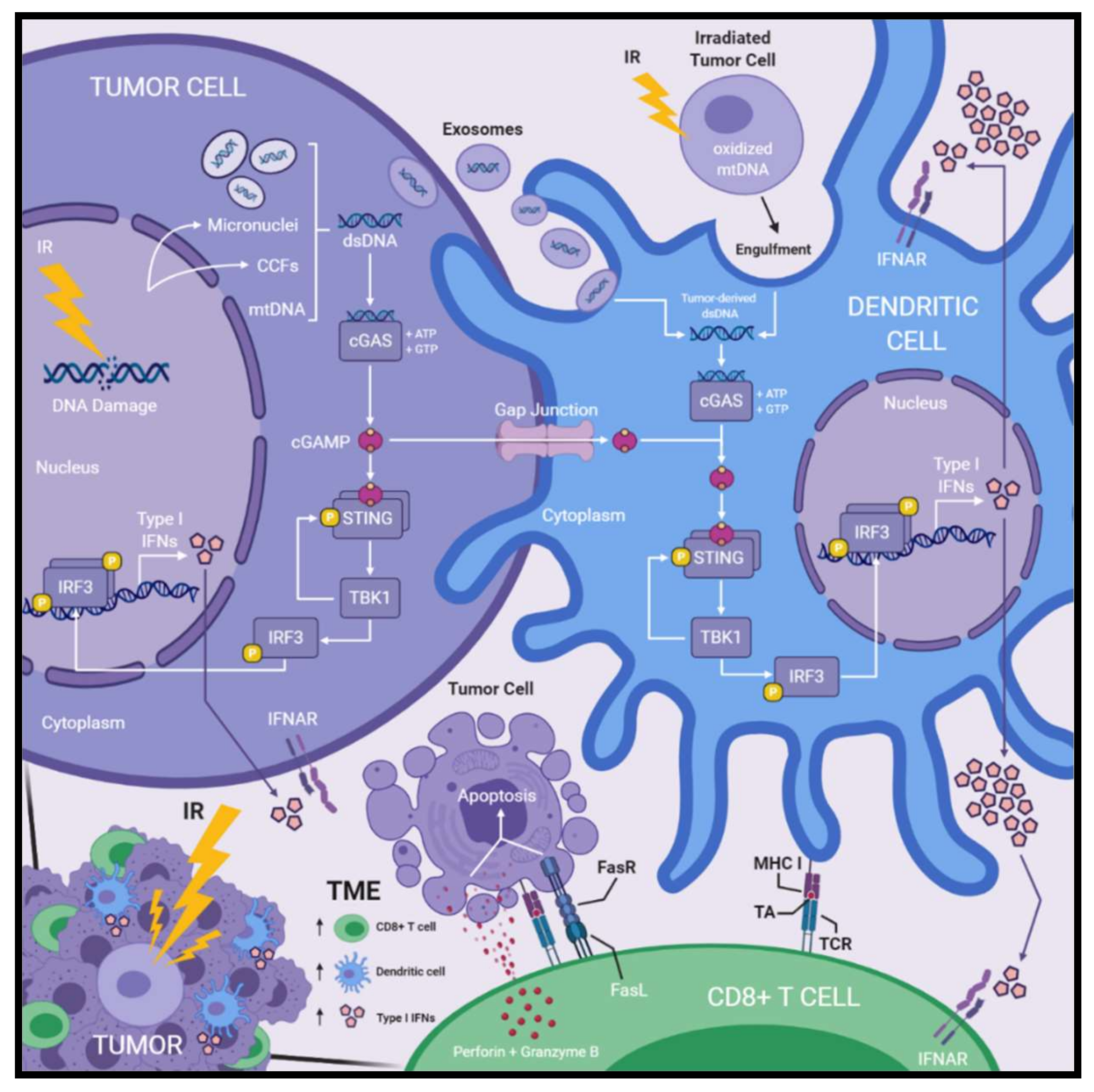
3. The Connection between Radiation-Induced DNA Damage and Innate Immune Activation
4. Radiation-Induced Antitumor Responses Involving cGas, STING, or Type I Interferons
4.1. Radiation-Induced Immunological Contributions to Antitumor Immunity
4.2. Activation of cGAS–STING and Type I Interferon Production Following Ionizing Radiation Treatment
4.3. Sources Activating cGAS–STING Signaling in Dendritic Cells
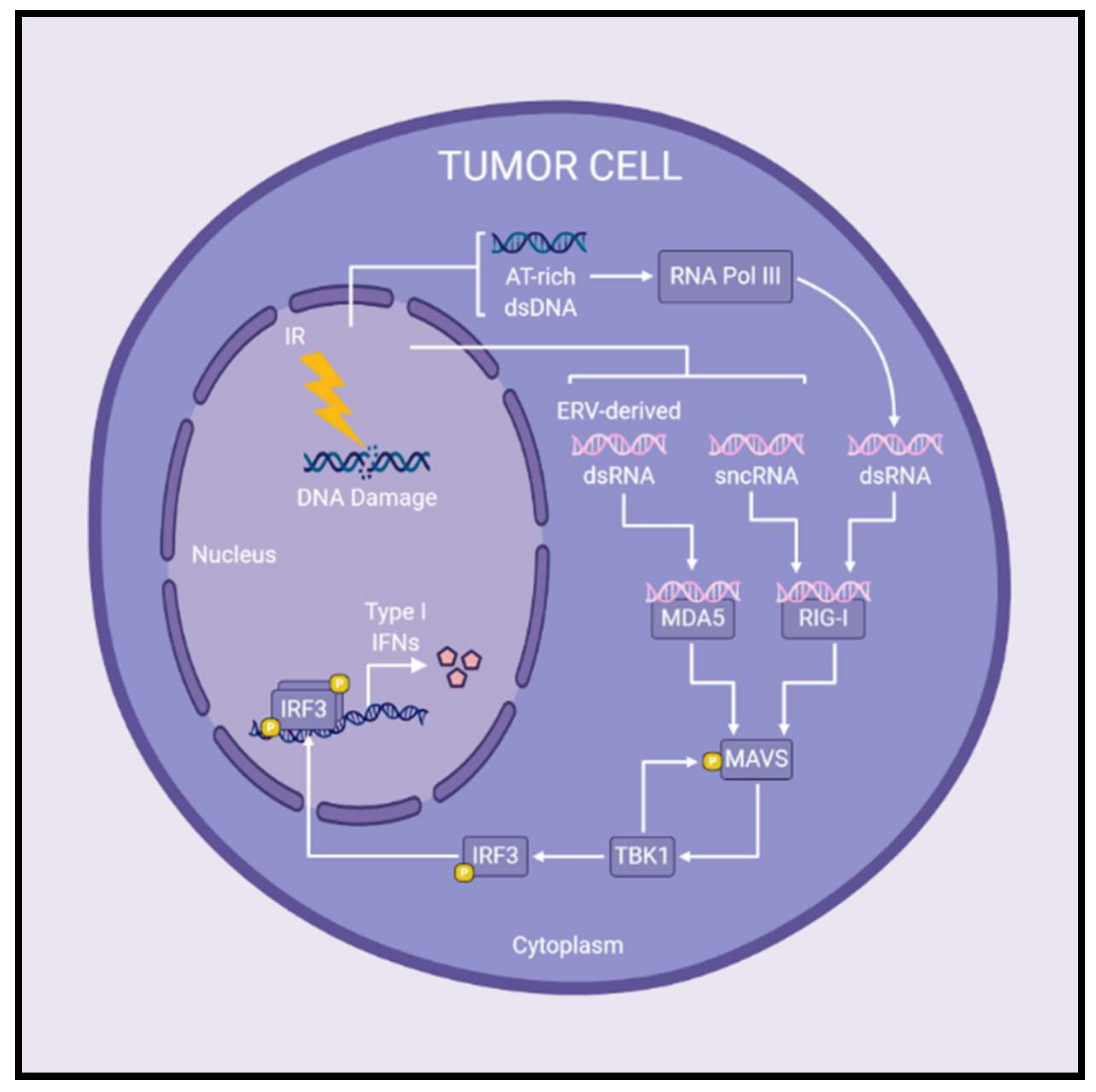
5. STING-Independent Activation of Type I Interferons Following Radiation
6. Pathways Hindering Radiation-Induced cGAS-STING-Type I Interferon Signaling and Subsequent Antitumor Immunity
7. Detrimental Effects of cGAS, STING, and Type I Interferons Following Radiation
8. Augmenting STING Signaling to Enhance Radiation-Induced Antitumor Immunity
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cDC1s | conventional dendritic cells type 1 |
| CDN | cyclic dinucleotide |
| cGAMP | cyclic GMP-AMP |
| cGAS | cGAMP synthase |
| CCFs | cytoplasmic chromatin fragments |
| DAMPs | damage-associated molecular patterns |
| DCs | dendritic cells |
| dsDNA | double-stranded DNA |
| IDO1 | indoleamine 2,3 dioxygenase 1 |
| IFN | interferon |
| IFNAR | IFNα/β receptor |
| IRF3 | interferon regulatory factor 3 |
| IR | ionizing radiation |
| MHC I | major histocompatibility complex class I molecules |
| MDA5 | melanoma differentiation-associated protein 5 |
| MAVS | mitochondrial antiviral-signaling protein |
| mtDNA | mitochondria DNA |
| MDSCs | myeloid derived suppressor cells |
| RIG-I | retinoic acid-inducible gene I |
| STING | stimulator of interferon genes |
| TBK1 | TANK-binding kinase 1 |
| TCR | T cell receptor |
| TME | Tumor microenvironment |
References
- Liauw, S.L.; Connell, P.P.; Weichselbaum, R.R. New Paradigms and Future Challenges in Radiation Oncology: An Update of Biological Targets and Technology. Sci. Transl. Med. 2013, 5, 173sr2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philchenkov, A. Radiation-Induced Cell Death: Signaling and Pharmacological Modulation. Crit. Rev. Oncog. 2018, 23, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Martinez Monge, R.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation Effects on Antitumor Immune Responses: Current Perspectives and Challenges. Ther. Adv. Med. Oncol. 2018, 10, 1–27. [Google Scholar] [CrossRef]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of Tumor Cells Up-Regulates Fas and Enhances CTL Lytic Activity and CTL Adoptive Immunotherapy. J. Immunol. 2003, 170, 6338. [Google Scholar] [CrossRef] [Green Version]
- Garnett, C.T.; Palena, C.; Chakarborty, M.; Tsang, K.-Y.; Schlom, J.; Hodge, J.W. Sublethal Irradiation of Human Tumor Cells Modulates Phenotype Resulting in Enhanced Killing by Cytotoxic T Lymphocytes. Cancer Res. 2004, 64, 7985. [Google Scholar] [CrossRef]
- Gameiro, S.R.; Ardiani, A.; Kwilas, A.; Hodge, J.W. Radiation-Induced Survival Responses Promote Immunogenic Modulation to Enhance Immunotherapy in Combinatorial Regimens. OncoImmunology 2014, 3, e28643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.R.; Malamas, A.S.; Bernstein, M.B.; Tsang, K.Y.; Vassantachart, A.; Sahoo, N.; Tailor, R.; Pidikiti, R.; Guha, C.P.; Hahn, S.M.; et al. Tumor Cells Surviving Exposure to Proton or Photon Radiation Share a Common Immunogenic Modulation Signature, Rendering Them More Sensitive to T Cell–Mediated Killing. Part. Ther. Spec. Ed. 2016, 95, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, S.R.; Jammeh, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-Induced Immunogenic Modulation of Tumor Enhances Antigen Processing and Calreticulin Exposure, Resulting in Enhanced T-Cell Killing. Oncotarget 2013, 5, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; De Ru, A.H.; Neijssen, J.; et al. Radiation Modulates the Peptide Repertoire, Enhances MHC Class I Expression, and Induces Successful Antitumor Immunotherapy. J. Exp. Med. 2006, 203, 1259. [Google Scholar] [CrossRef]
- Golden, E.B.; Frances, D.; Pellicciotta, I.; Demaria, S.; Helen Barcellos-Hoff, M.; Formenti, S.C. Radiation Fosters Dose-Dependent and Chemotherapy-Induced Immunogenic Cell Death. OncoImmunology 2014, 3, e28518. [Google Scholar] [CrossRef] [Green Version]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local Radiation Therapy of B16 Melanoma Tumors Increases the Generation of Tumor Antigen-Specific Effector Cells That Traffic to the Tumor. J. Immunol. 2005, 174, 7516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic Effects of Ablative Radiation on Local Tumor Require CD8+ T Cells: Changing Strategies for Cancer Treatment. Blood 2009, 114, 589. [Google Scholar] [CrossRef] [PubMed]
- Filatenkov, A.; Baker, J.; Mueller, A.M.S.; Kenkel, J.; Ahn, G.-O.; Dutt, S.; Zhang, N.; Kohrt, H.; Jensen, K.; Dejbakhsh-Jones, S.; et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin. Cancer Res. 2015, 21, 3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Li, R.; Yin, L.-M.; Deng, L.; Gui, J.; Chen, B.-Q.; Zhou, L.; Meng, M.-B.; Huang, Q.-R.; Mo, X.-M.; et al. Targeting Myeloid-Derived Suppressor Cells and Programmed Death Ligand 1 Confers Therapeutic Advantage of Ablative Hypofractionated Radiation Therapy Compared With Conventional Fractionated Radiation Therapy. Int. J. Radiat. Oncol. 2018, 101, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Dar, T.B.; Henson, R.M.; Shiao, S.L. Targeting Innate Immunity to Enhance the Efficacy of Radiation Therapy. Front. Immunol. 2019, 9, 3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiontov, S.I.; Pitroda, S.P.; Chmura, S.J.; Arina, A.; Weichselbaum, R.R. Cytoreduction and the Optimization of Immune Checkpoint Inhibition with Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Deloch, L.; Derer, A.; Hartmann, J.; Frey, B.; Fietkau, R.; Gaipl, U.S. Modern Radiotherapy Concepts and the Impact of Radiation on Immune Activation. Front. Oncol. 2016, 6, 141. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Liang, H.; Fu, S.; Weichselbaum, R.R.; Fu, Y.-X. From DNA Damage to Nucleic Acid Sensing: A Strategy to Enhance Radiation Therapy. Clin. Cancer Res. 2016, 22, 20. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. CGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic Progression Following DNA Damage Enables Pattern Recognition within Micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through CGAS Promotes Senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, Z.; Zhang, Y.; Shen, A.; Dong, C.; Zhang, A.; Moore, C.; Ren, Z.; Lu, C.; Cao, X.; et al. Tumor Cells Suppress Radiation-Induced Immunity by Hijacking Caspase 9 Signaling. Nat. Immunol. 2020, 21, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Mo, F.; Liu, L.; Du, J.; Luo, M.; Men, K.; Na, F.; Wang, W.; Yang, H.; Wei, X. Oxidized Mitochondrial DNA Sensing by STING Signaling Promotes the Antitumor Effect of an Irradiated Immunogenic Cancer Cell Vaccine. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.-P.; Chapman, J.; Ueberheide, B.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory DsDNA from Irradiated Cancer Cells to Dendritic Cells. Cancer Immunol. Res. 2018. canimm.0581.2017. [Google Scholar] [CrossRef] [Green Version]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic CGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248.e7. [Google Scholar] [CrossRef] [Green Version]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.M.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Carozza, J.A.; Böhnert, V.; Nguyen, K.C.; Skariah, G.; Shaw, K.E.; Brown, J.A.; Rafat, M.; Von Eyben, R.; Graves, E.E.; Glenn, J.S.; et al. Extracellular CGAMP Is a Cancer-Cell-Produced Immunotransmitter Involved in Radiation-Induced Anticancer Immunity. Nat. Cancer 2020, 1, 184–196. [Google Scholar] [CrossRef]
- Turvey, S.E.; Broide, D.H. Innate Immunity. J. Allergy Clin. Immunol. 2010, 125, S24–S32. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a Systems Understanding of MHC Class I and MHC Class II Antigen Presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T Cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatner, R.E.; Janssen, E.M. STING, DCs and the Link between Innate and Adaptive Tumor Immunity. Tumor Microenviron. Mol. Regul. Innate Immune Cells 2019, 110, 13–23. [Google Scholar] [CrossRef]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat. Rev. Immunol. 2015, 15, 405. [Google Scholar] [CrossRef]
- Medrano, R.F.V.; Hunger, A.; Mendonça, S.A.; Barbuto, J.A.M.; Strauss, B.E. Immunomodulatory and Antitumor Effects of Type I Interferons and Their Application in Cancer Therapy. Oncotarget 2017, 8, 71249–71284. [Google Scholar] [CrossRef] [Green Version]
- Platanias, L.C. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Le Bon, A.; Tough, D.F. Links between Innate and Adaptive Immunity via Type I Interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I Interferon Is Selectively Required by Dendritic Cells for Immune Rejection of Tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.-R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host Type I IFN Signals Are Required for Antitumor CD8+ T Cell Responses through CD8α+ Dendritic Cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarquist, J.; Hennies, C.M.; Lehn, M.A.; Reboulet, R.A.; Feau, S.; Janssen, E.M. STING-Mediated DNA Sensing Promotes Antitumor and Autoimmune Responses to Dying Cells. J. Immunol. 2014, 193, 6124. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Li, Y.; Chen, L.; Chen, H.; You, F.; Zhou, X.; Zhou, Y.; Zhai, Z.; Chen, D.; Jiang, Z. ERIS, an Endoplasmic Reticulum IFN Stimulator, Activates Innate Immune Signaling through Dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 8653. [Google Scholar] [CrossRef] [Green Version]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING Is a Direct Innate Immune Sensor of Cyclic Di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-ΚB Activation through TBK1. J. Virol. 2014, 88, 5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranzusch, P.J.; Wilson, S.C.; Lee, A.S.Y.; Berger, J.M.; Doudna, J.A.; Vance, R.E. Ancient Origin of CGAS-STING Reveals Mechanism of Universal 2′,3′ CGAMP Signaling. Mol. Cell 2015, 59, 891–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Barber, G.N. STING Signaling and Host Defense against Microbial Infection. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Li, T.; Li, X.-D.; Chen, X.; Li, Q.-Z.; Wight-Carter, M.; Chen, Z.J. Activation of Cyclic GMP-AMP Synthase by Self-DNA Causes Autoimmune Diseases. Proc. Natl. Acad. Sci. USA 2015, 112, E5699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA Sensing by the CGAS–STING Pathway in Health and Disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The Cytosolic DNA Sensor CGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2′,5′)PA(3′,5′)p] Is the Metazoan Second Messenger Produced by DNA-Activated Cyclic GMP-AMP Synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. CGAS Produces a 2′-5′-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The Innate Immune DNA Sensor CGAS Produces a Noncanonical Cyclic Dinucleotide That Activates Human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Hengge, R.; Gründling, A.; Jenal, U.; Ryan, R.; Yildiz, F. Bacterial Signal Transduction by Cyclic Di-GMP and Other Nucleotide Second Messengers. J. Bacteriol. 2016, 198, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilchanka, O.; Mekalanos, J.J. Cyclic Dinucleotides and the Innate Immune Response. Cell 2013, 154, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is an Endogenous High-Affinity Ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell Intrinsic Immunity Spreads to Bystander Cells via the Intercellular Transfer of CGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Gentili, M.; Kowal, J.; Tkach, M.; Satoh, T.; Lahaye, X.; Conrad, C.; Boyron, M.; Lombard, B.; Durand, S.; Kroemer, G.; et al. Transmission of Innate Immune Signaling by Packaging of CGAMP in Viral Particles. Science 2015, 349, 1232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, B.; Zhang, L.; Lei, C.; Li, Y.; Mao, A.-P.; Yang, Y.; Wang, Y.-Y.; Zhang, X.-L.; Shu, H.-B. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity 2009, 30, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonugunta, V.K.; Sakai, T.; Pokatayev, V.; Yang, K.; Wu, J.; Dobbs, N.; Yan, N. Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-Tumor Response. Cell Rep. 2017, 21, 3234–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelige, R.; Searles, S.; Bui, J.D. Mechanisms Regulating Immune Surveillance of Cellular Stress in Cancer. Cell. Mol. Life Sci. 2018, 75, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing CGAS–STING Pathway in Cancer. Cancer Discov. 2020, 10, 26. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Kim, T.Y.; Song, Y.H.; Min, I.M.; Yim, J.; Kim, T.K. Activation of Interferon Regulatory Factor 3 in Response to DNA-Damaging Agents. J. Biol. Chem. 1999, 274, 30686–30689. [Google Scholar] [CrossRef] [Green Version]
- Karpova, A.Y.; Trost, M.; Murray, J.M.; Cantley, L.C.; Howley, P.M. Interferon Regulatory Factor-3 Is an in Vivo Target of DNA-PK. Proc. Natl. Acad. Sci. USA 2002, 99, 2818. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular Mechanisms of Micronucleus, Nucleoplasmic Bridge and Nuclear Bud Formation in Mammalian and Human Cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.-H.; Karagounis, I.V.; Thomas, C.; François, N.B.; Facciabene, A.; Koumenis, C.; Maity, A. Combination of CHEK1/2 Inhibition and Ionizing Radiation Results in Abscopal Tumor Response through Increased Micronuclei Formation. Oncogene 2020, 39, 4344–4357. [Google Scholar] [CrossRef]
- Mirzayans, R.; Scott, A.; Cameron, M.; Murray, D. Induction of Accelerated Senescence by γ Radiation in Human Solid Tumor-Derived Cell Lines Expressing Wild-Type TP53. Radiat. Res. 2005, 163, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy Road to Therapy-Induced Cellular Senescence and Escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S. Three Steps to the Immortality of Cancer Cells: Senescence, Polyploidy and Self-Renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; Van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic Caspases Suppress MtDNA-Induced STING-Mediated Type I IFN Production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.-X.; Auh, S.L. The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon–Dependent Innate and Adaptive Immunity. Cancer Res. 2011, 71, 2488. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Probst, H.C.; Vuong, V.; Landshammer, A.; Muth, S.; Yagita, H.; Schwendener, R.; Pruschy, M.; Knuth, A.; Van den Broek, M. Radiotherapy Promotes Tumor-Specific Effector CD8+ T Cells via Dendritic Cell Activation. J. Immunol. 2012, 189, 558. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.H.; Gerber, S.A.; Murphy, S.P.; Lord, E.M. Type I Interferons Induced by Radiation Therapy Mediate Recruitment and Effector Function of CD8+ T Cells. Cancer Immunol. Immunother. 2014, 63, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Blair, T.C.; Bambina, S.; Alice, A.F.; Kramer, G.F.; Medler, T.R.; Baird, J.R.; Broz, M.L.; Tormoen, G.W.; Troesch, V.; Crittenden, M.R.; et al. Dendritic Cell Maturation Defines Immunological Responsiveness of Tumors to Radiation Therapy. J. Immunol. 2020, 204, 3416. [Google Scholar] [CrossRef]
- Liu, Y.; Crowe, W.N.; Wang, L.; Lu, Y.; Petty, W.J.; Habib, A.A.; Zhao, D. An Inhalable Nanoparticulate STING Agonist Synergizes with Radiotherapy to Confer Long-Term Control of Lung Metastases. Nat. Commun. 2019, 10, 5108. [Google Scholar] [CrossRef] [Green Version]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING Activation of Tumor Endothelial Cells Initiates Spontaneous and Therapeutic Antitumor Immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, A.-K.; Giebel, B. Exosomes: Small Vesicles Participating in Intercellular Communication. Int. J. Biochem. Cell Biol. 2012, 44, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Kitai, Y.; Kawasaki, T.; Sueyoshi, T.; Kobiyama, K.; Ishii, K.J.; Zou, J.; Akira, S.; Matsuda, T.; Kawai, T. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J. Immunol. 2017, 198, 1649. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-Derived CGAMP Triggers a STING-Mediated Interferon Response in Non-Tumor Cells to Activate the NK Cell Response. Immunity 2018, 49, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Ding, T.; Zuo, Z.; Xu, Z.; Deng, J.; Wei, Z. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front. Immunol. 2020, 11, 1030. [Google Scholar] [CrossRef] [PubMed]
- Ranoa, D.R.E.; Parekh, A.D.; Pitroda, S.P.; Huang, X.; Darga, T.; Wong, A.C.; Huang, L.; Andrade, J.; Staley, J.P.; Satoh, T.; et al. Cancer Therapies Activate RIG-I-like Receptor Pathway through Endogenous Non-Coding RNAs. Oncotarget 2016, 7, 26496–26515. [Google Scholar] [CrossRef]
- Feng, X.; Tubbs, A.; Zhang, C.; Tang, M.; Sridharan, S.; Wang, C.; Jiang, D.; Su, D.; Zhang, H.; Chen, Z.; et al. ATR Inhibition Potentiates Ionizing Radiation-Induced Interferon Response via Cytosolic Nucleic Acid-Sensing Pathways. EMBO J. 2020, 39, e104036. [Google Scholar] [CrossRef]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-Dependent Sensing of Poly(DA:DT) through the Induction of an RNA Polymerase III–Transcribed RNA Intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.-H.; MacMillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.K.; Pan, D.; Bao, X.; Hu, M.; Li, F.; Li, C.-Y. Endogenous Retrovirus Activation as a Key Mechanism of Anti-Tumor Immune Response in Radiotherapy. Radiat. Res. 2020, 193, 305–317. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Buqué, A.; Hensler, M.; Chen, J.; Bloy, N.; Petroni, G.; Sato, A.; Yamazaki, T.; Fucikova, J.; Galluzzi, L. Apoptotic Caspases Inhibit Abscopal Responses to Radiation and Identify a New Prognostic Biomarker for Breast Cancer Patients. Oncoimmunology 2019, 8, e1655964. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liang, H.; Rao, E.; Zheng, W.; Huang, X.; Deng, L.; Zhang, Y.; Yu, X.; Xu, M.; Mauceri, H.; et al. Non-Canonical NF-ΚB Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 2018, 49, 490–503.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.-C. Non-Canonical NF-ΚB Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.F.; Muschel, R.J. Type I IFN Protects Cancer Cells from CD8+ T Cell–Mediated Cytotoxicity after Radiation. J. Clin. Investig. 2019, 129, 4224–4238. [Google Scholar] [CrossRef]
- Chen, B.; Alvarado, D.M.; Iticovici, M.; Kau, N.S.; Park, H.; Parikh, P.J.; Thotala, D.; Ciorba, M.A. Interferon-Induced IDO1 Mediates Radiation Resistance and Is a Therapeutic Target in Colorectal Cancer. Cancer Immunol. Res. 2020, 8, 451. [Google Scholar] [CrossRef] [Green Version]
- Jacquelot, N.; Yamazaki, T.; Roberti, M.P.; Duong, C.P.M.; Andrews, M.C.; Verlingue, L.; Ferrere, G.; Becharef, S.; Vétizou, M.; Daillère, R.; et al. Sustained Type I Interferon Signaling as a Mechanism of Resistance to PD-1 Blockade. Cell Res. 2019, 29, 846–861. [Google Scholar] [CrossRef]
- Bazhin, A.V.; Von Ahn, K.; Fritz, J.; Werner, J.; Karakhanova, S. Interferon-α Up-Regulates the Expression of PD-L1 Molecules on Immune Cells Through STAT3 and P38 Signaling. Front. Immunol. 2018, 9, 2129. [Google Scholar] [CrossRef]
- Shevtsov, M.; Sato, H.; Multhoff, G.; Shibata, A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front. Oncol. 2019, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Okonogi, N.; Nakano, T. Rationale of Combination of Anti-PD-1/PD-L1 Antibody Therapy and Radiotherapy for Cancer Treatment. Int. J. Clin. Oncol. 2020, 25, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Park, S.S.; Dong, H.; Liu, X.; Harrington, S.M.; Krco, C.J.; Grams, M.P.; Mansfield, A.S.; Furutani, K.M.; Olivier, K.R.; Kwon, E.D. PD-1 Restrains Radiotherapy-Induced Abscopal Effect. Cancer Immunol. Res. 2015, 3, 610. [Google Scholar] [CrossRef] [Green Version]
- Pfannenstiel, L.W.; McNeilly, C.; Xiang, C.; Kang, K.; Diaz-Montero, C.M.; Yu, J.S.; Gastman, B.R. Combination PD-1 Blockade and Irradiation of Brain Metastasis Induces an Effective Abscopal Effect in Melanoma. Oncoimmunology 2019, 8, e1507669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondini, M.; Levy, A.; Meziani, L.; Milliat, F.; Deutsch, E. Radiotherapy–Immunotherapy Combinations – Perspectives and Challenges. Mol. Oncol. 2020, 14, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-Dependent MDSC Mobilization Drives Extrinsic Radiation Resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Jia, S.; Shao, C.; Shi, Y. Irradiation Induces Cancer Lung Metastasis through Activation of the CGAS–STING–CCL5 Pathway in Mesenchymal Stromal Cells. Cell Death Dis. 2020, 11, 326. [Google Scholar] [CrossRef]
- Du, S.; Chen, G.; Yuan, B.; Hu, Y.; Yang, P.; Chen, Y.; Zhao, Q.; Zhou, J.; Fan, J.; Zeng, Z. DNA Sensing and Associated Type 1 Interferon Signaling Contributes to Progression of Radiation-Induced Liver Injury. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. CGAS Is Essential for Cellular Senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, D.; Mandal, M. Senescence in Polyploid Giant Cancer Cells: A Road That Leads to Chemoresistance. Cytokine Growth Factor Rev. 2020, 52, 68–75. [Google Scholar] [CrossRef]
- Luo, M.; Liu, Z.; Zhang, X.; Han, C.; Samandi, L.Z.; Dong, C.; Sumer, B.D.; Lea, J.; Fu, Y.-X.; Gao, J. Synergistic STING Activation by PC7A Nanovaccine and Ionizing Radiation Improves Cancer Immunotherapy. J. Control. Release 2019, 300, 154–160. [Google Scholar] [CrossRef]
- Baird, J.R.; Friedman, D.; Cottam, B.; Dubensky, T.W.; Kanne, D.B.; Bambina, S.; Bahjat, K.; Crittenden, M.R.; Gough, M.J. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016, 76, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marill, J.; Mohamed Anesary, N.; Paris, S. DNA Damage Enhancement by Radiotherapy-Activated Hafnium Oxide Nanoparticles Improves CGAS-STING Pathway Activation in Human Colorectal Cancer Cells. Radiother. Oncol. 2019, 141, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Huang, Y.; Xiao, Y.; Zhu, Z.; Shen, M.; Zhou, P.; Guo, Z.; Wang, J.; Wang, H.; Dai, W.; et al. ATR Inhibitor AZD6738 Enhances the Antitumor Activity of Radiotherapy and Immune Checkpoint Inhibitors by Potentiating the Tumor Immune Microenvironment in Hepatocellular Carcinoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Ref | Radiation Dose | Cells/Model | Cell/Tissue Type | Response(s) |
|---|---|---|---|---|
| [19] | 1 Gy, 5 Gy b | MEFs in vitro | mouse embryonic fibroblasts | micronuclei formation |
| [20] | 20 Gy c | MCF10A in vitro | human breast epithelial cells | micronuclei formation |
| [21] | 12 Gy b | MEFs in vitro; WI-38 in vitro | mouse embryonic fibroblasts; human lung fibroblasts | CCF a production |
| [22] | 40 Gy | MC38 in vitro | mouse colon carcinoma | mtDNA a release into cytosol |
| [23] | 75 Gy b in vitro pre-treatment | EG7 in vivo (s.c.) a | mouse lymphoma | oxidized mtDNA |
| [24] | 4 Gy whole mouse | C57Bl/6 | liver | ↓ d inflammatory factors with host STING-deficiency |
| [25] | 8 Gy b × 3 a | TSA and 4T1 in vitro MDA-MB-231 in vitro MCA38 in vitro | mouse mammary carcinomas human breast adenocarcinoma mouse colorectal carcinoma | ↓ type I IFN a with cGAS- or STING-deficiency |
| [26] | 20 Gy 40 Gy | MC38 in vivo (s.c.) MC38-SIY in vitro | mouse colon adenocarcinoma mouse colon adenocarcinoma (expressing tumor-antigen) | ↓ efficacy with host STING-deficiency ↓ DC a activation + T cell stimulation with cGAS- or STING-deficient DCs |
| [27] | 8 Gy b × 3 | TSA in vitro | mouse mammary carcinoma | ↓ DC activation with STING-deficient DCs |
| [28] | 20 Gy b | CT26 in vivo (s.c.) | mouse colorectal carcinoma | ↓ efficacy with host cGAS-deficiency |
| [29] | 1 Gy, 5 Gy b | BMDMs in vitro | bone-marrow-derived macrophages | ↓ type I IFN with STING-deficiency |
| [30] | 8 Gy b | E0771 in vivo (orthotopic) | mouse breast carcinoma | ↓ efficacy with cGAMP depletion or host STING-deficiency |
| Ref | Radiation Dose | Cells/Model | Cell Type | Responses |
|---|---|---|---|---|
| [96] | 3, 6, 9 Gy c 5 Gy × 6 a,b | MEFs in vitro; D54 in vitro; HCT116 in vitro D54 in vivo (s.c.) a; HCT116 in vivo (s.c.) | mouse embryonic fibroblasts; human glioblastoma; human colon carcinoma | ↓ d type I IFN a with MAVS- or RIG-I-deficiency ↓ efficacy with cellular MAVS- or RIG-I-deficiency |
| [97] | 20 Gy b | MCF10A in vitro; MC38 in vitro; MDA-MB-468 in vitro; PANC-1 in vitro; HCC1937 in vitro | human breast epithelial cells; mouse colon adenocarcinoma; human breast adenocarcinoma; human pancreas carcinoma; human breast carcinoma | ↓ type I IFN activation with MAVS-deficiency |
| [98] | 8 Gy b | A549 B16F10 | human lung carcinoma mouse melanoma | ↑ e cytosolic dsRNA a + ERV a activation ↑ ISGs a |
| Ref | Radiation Dose | Cells/Model | Cell Type | Responses |
|---|---|---|---|---|
| [101] | 20 Gy b 8 Gy b | TSA in vivo (s.c.) a TSA in vitro | mouse mammary carcinoma | ↑ e efficacy with cellular CASP3-deficiency ↑ type I IFN a with cellular CASP3-deficiency |
| [22] | 40 Gy 15 Gy | MC38 in vitro MC38 in vivo (s.c.) | mouse colon adenocarcinoma | ↑ type I IFN with CASP9-deficiency ↑ efficacy with CASP9-deficiency |
| [102] | 20 Gy | MC38 in vivo (s.c) | mouse colon adenocarcinoma | ↑ efficacy, ↑ type I IFN, and ↑ DC priming capacity with host non-canonical NF-κB-deficiency |
| [25] | 8 Gy b × 3 a + αCTLA4 8 Gy b × 3 | TSA in vivo (s.c.) TSA in vitro | mouse mammary carcinoma | ↓ d systemic (abscopal) efficacy with induction of cellular Trex1 ↓ type I IFN with induction of cellular Trex1 |
| [27] | 8 Gy b × 3 | TSA in vitro | mouse mammary carcinoma | ↓ dsDNA a cargo in exosomes with induction of cellular Trex1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Storozynsky, Q.; Hitt, M.M. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int. J. Mol. Sci. 2020, 21, 8877. https://doi.org/10.3390/ijms21228877
Storozynsky Q, Hitt MM. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. International Journal of Molecular Sciences. 2020; 21(22):8877. https://doi.org/10.3390/ijms21228877
Chicago/Turabian StyleStorozynsky, Quinn, and Mary M. Hitt. 2020. "The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer" International Journal of Molecular Sciences 21, no. 22: 8877. https://doi.org/10.3390/ijms21228877
APA StyleStorozynsky, Q., & Hitt, M. M. (2020). The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. International Journal of Molecular Sciences, 21(22), 8877. https://doi.org/10.3390/ijms21228877