Failed Neuroprotection of Combined Inhibition of L-Type and ASIC1a Calcium Channels with Nimodipine and Amiloride

 , and
, and
Abstract
:1. Introduction
2. Results
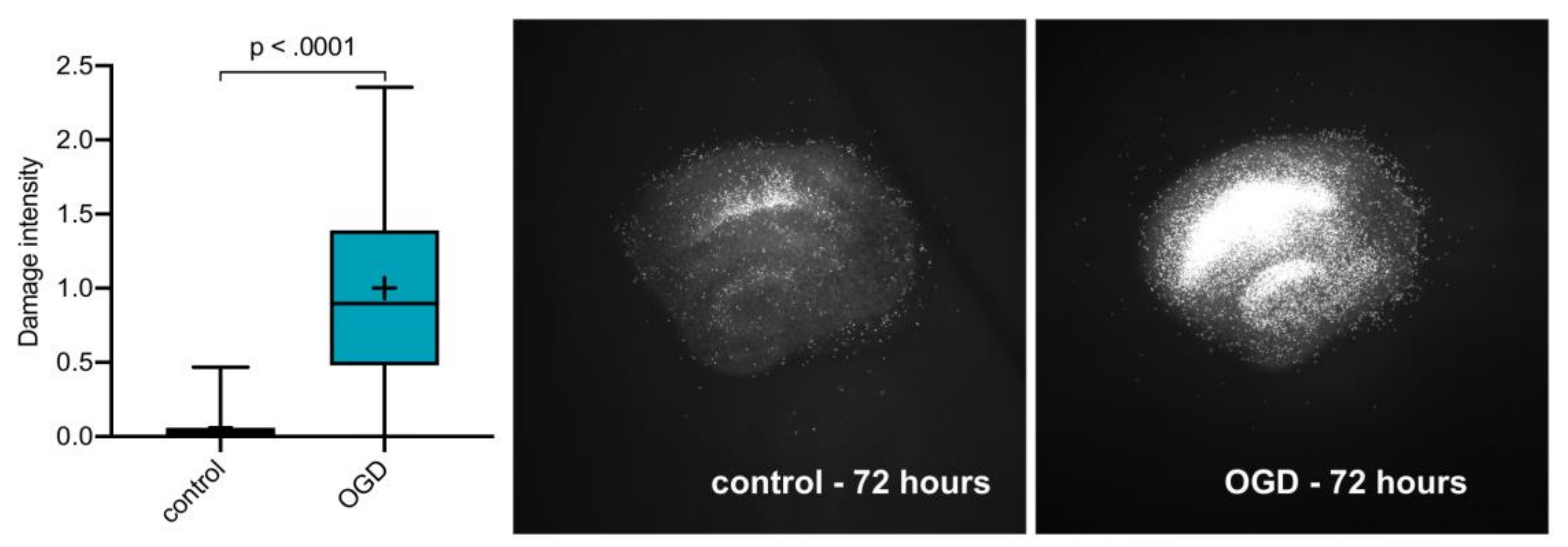
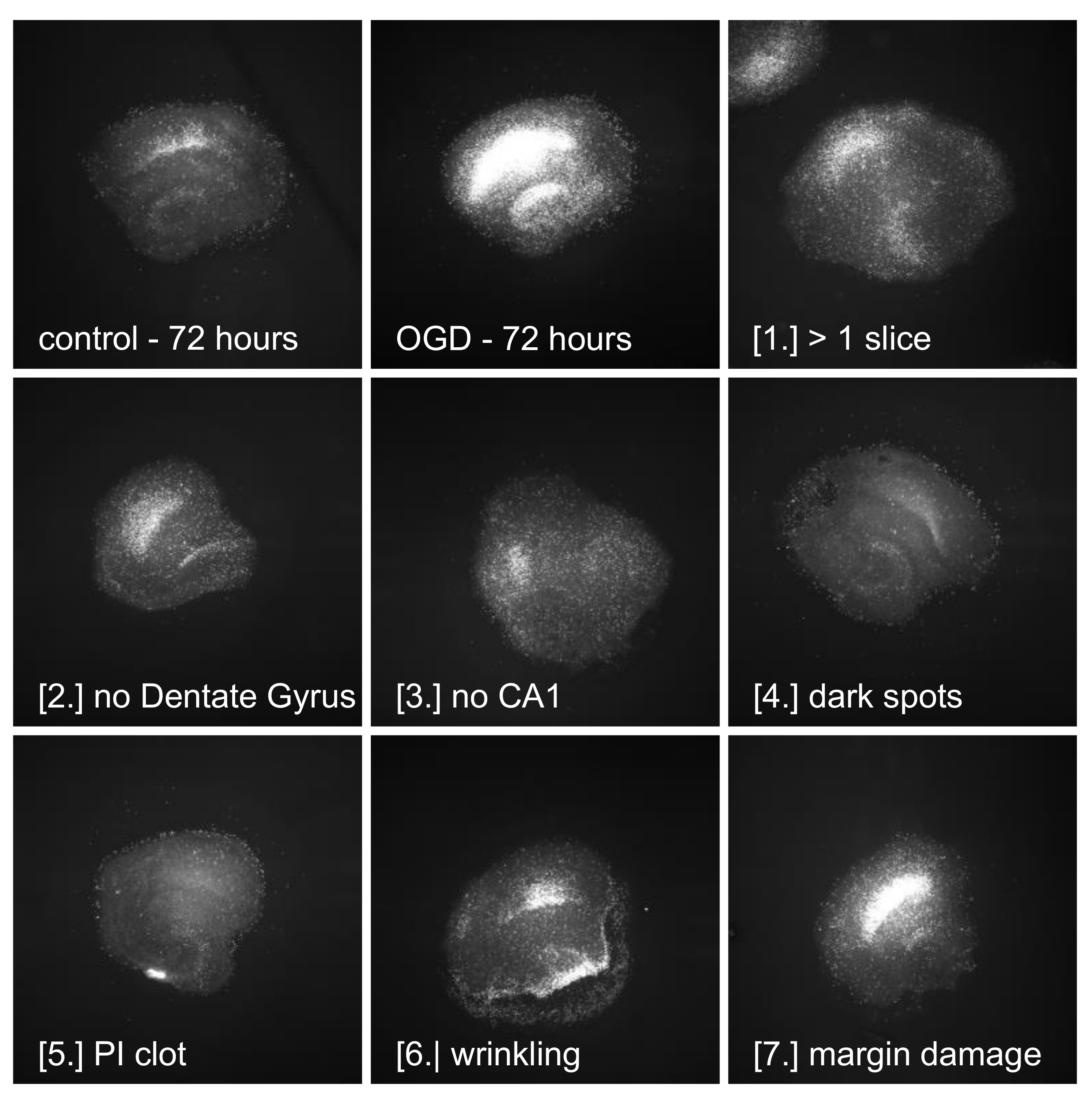
2.1. OGD Damage
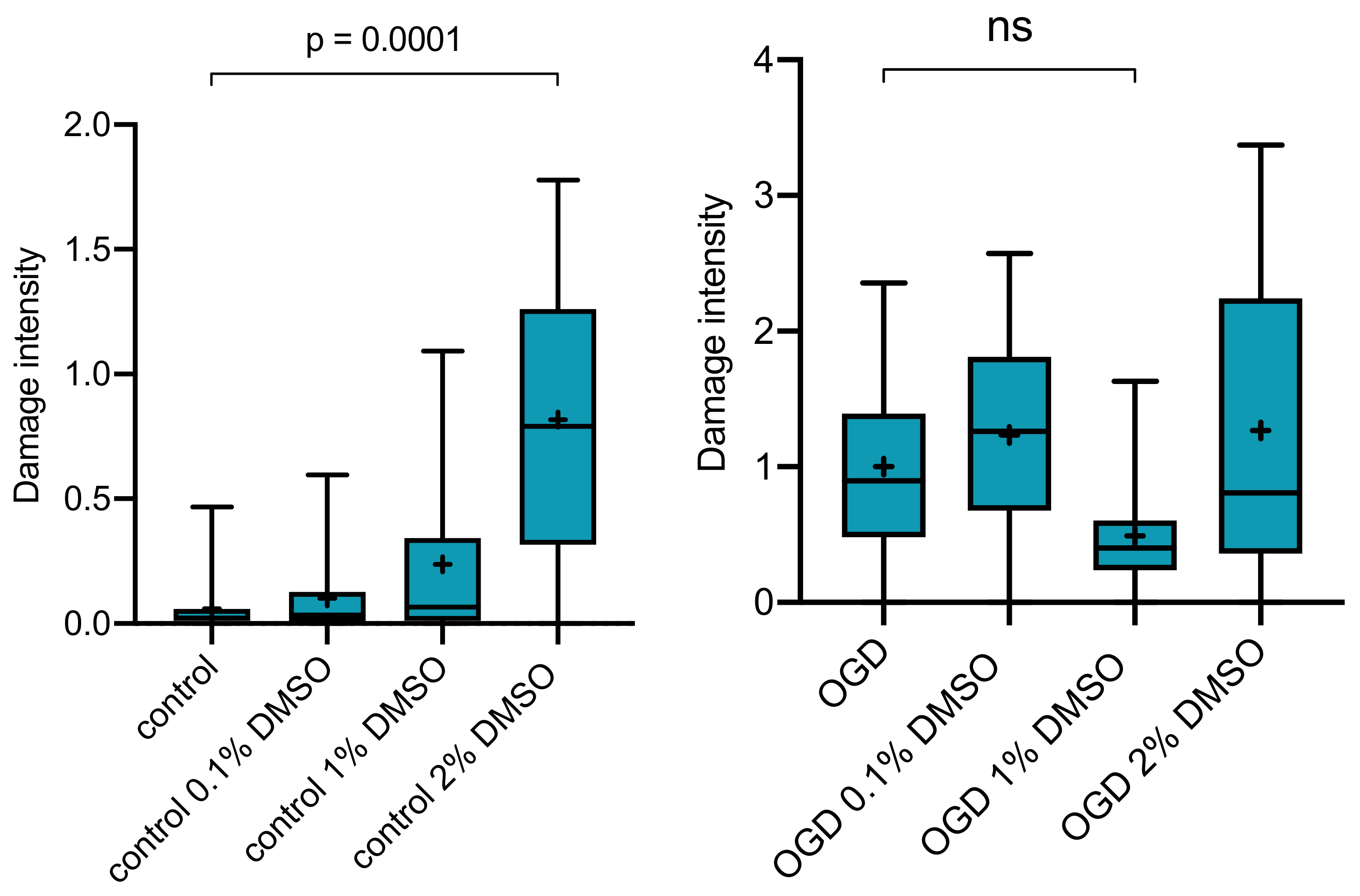
2.2. Effect of DMSO as Vehicle on OGD-Induced Damage
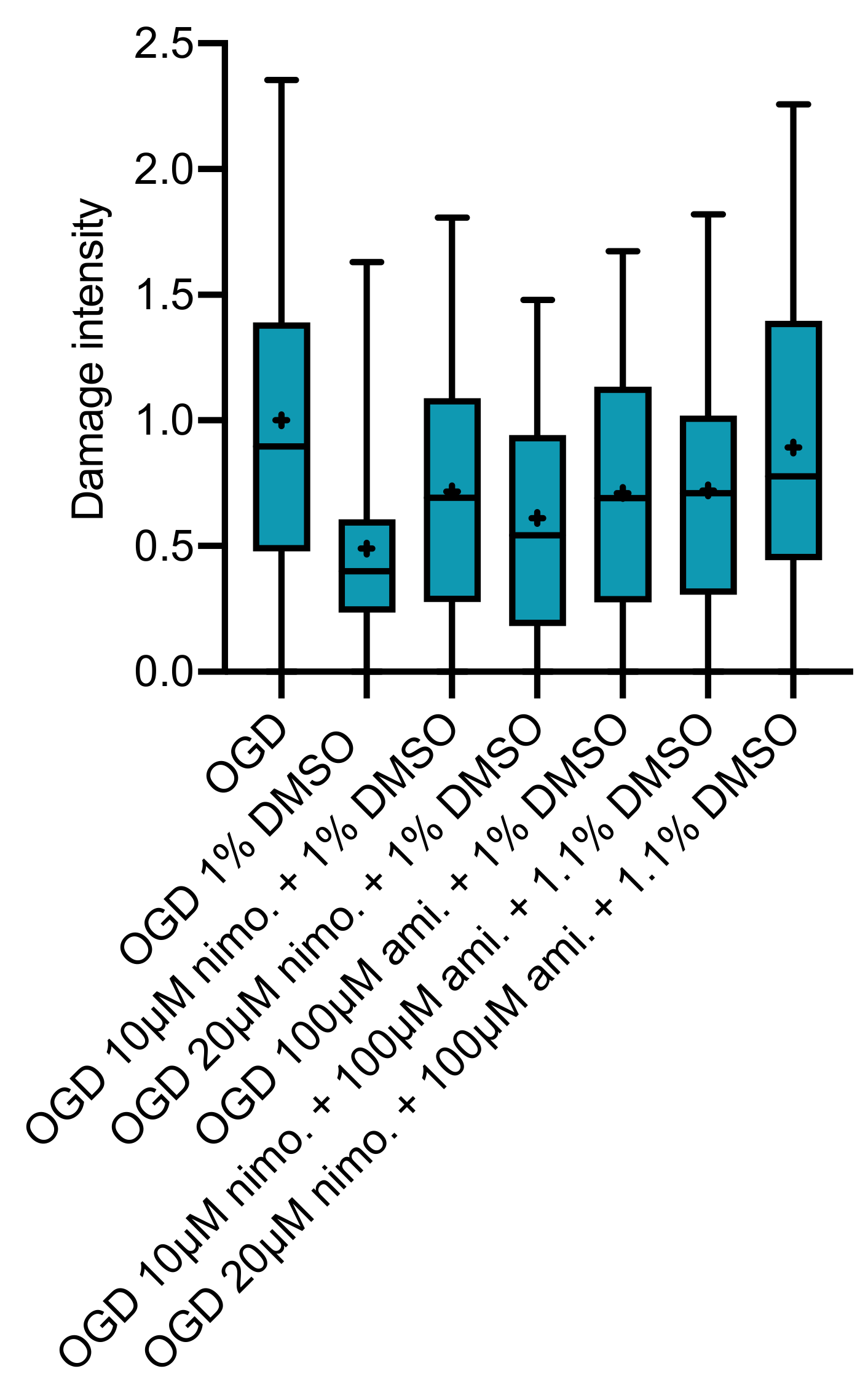
2.3. Effect of Nimodipine at 10 or 20 µM Dissolved in Varying DMSO Concentrations
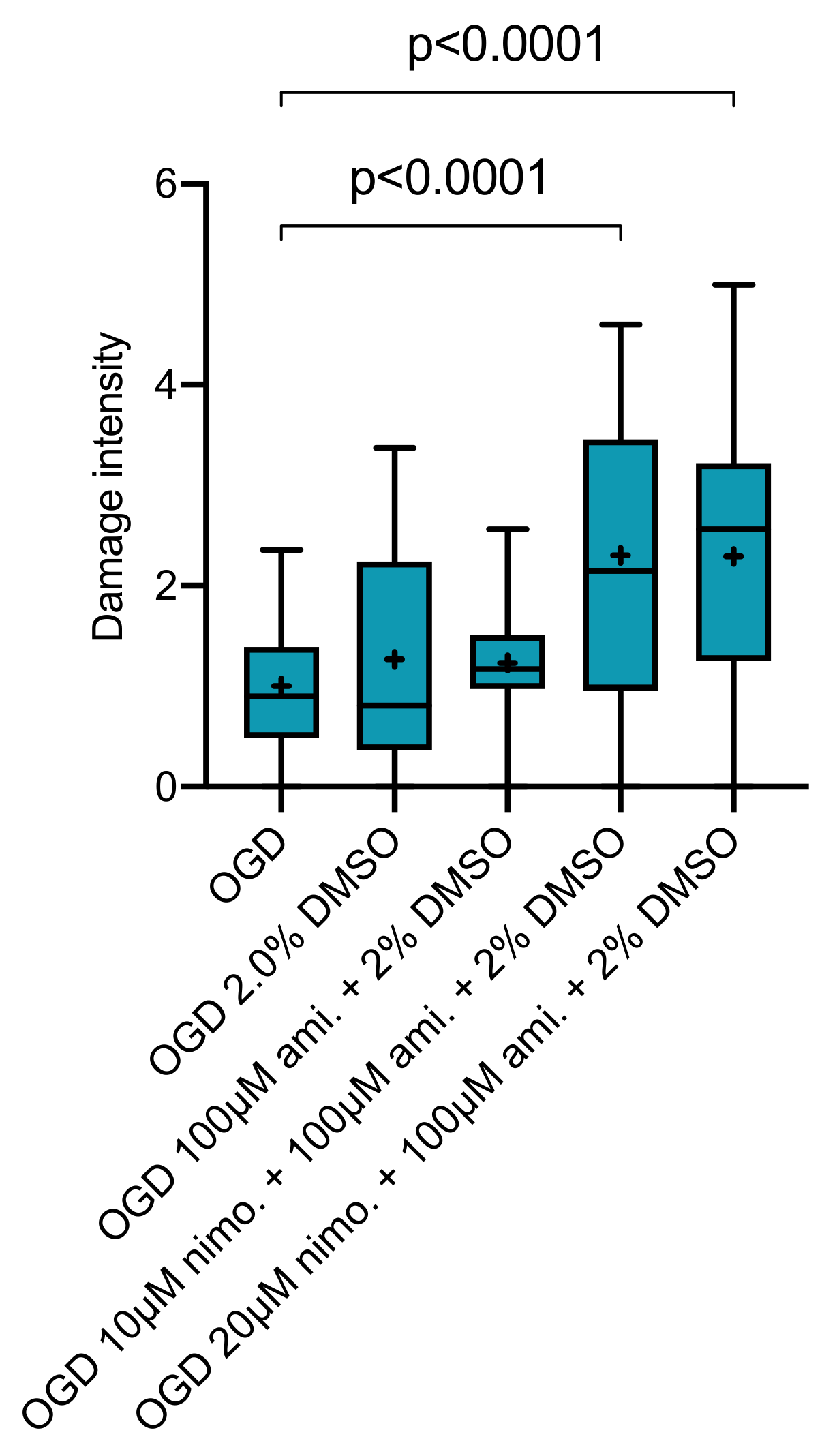
2.4. Effect of Nimodipine at 10 or 20 µM in Combination with 100 µM Amiloride Dissolved in Varying DMSO Concentrations
3. Discussion
3.1. The Role of Calcium for Cell Death and Failure to Provide Neuroprotection
3.2. Amiloride and Combined L-Type Calcium Channel and ASICs Inhibition
3.3. Nimodipine—Most Important Effect on the Vasculature
3.4. DMSO as Solvent
3.5. Quality of Slices—Need for Defined Exclusion Criteria
3.6. Limitations
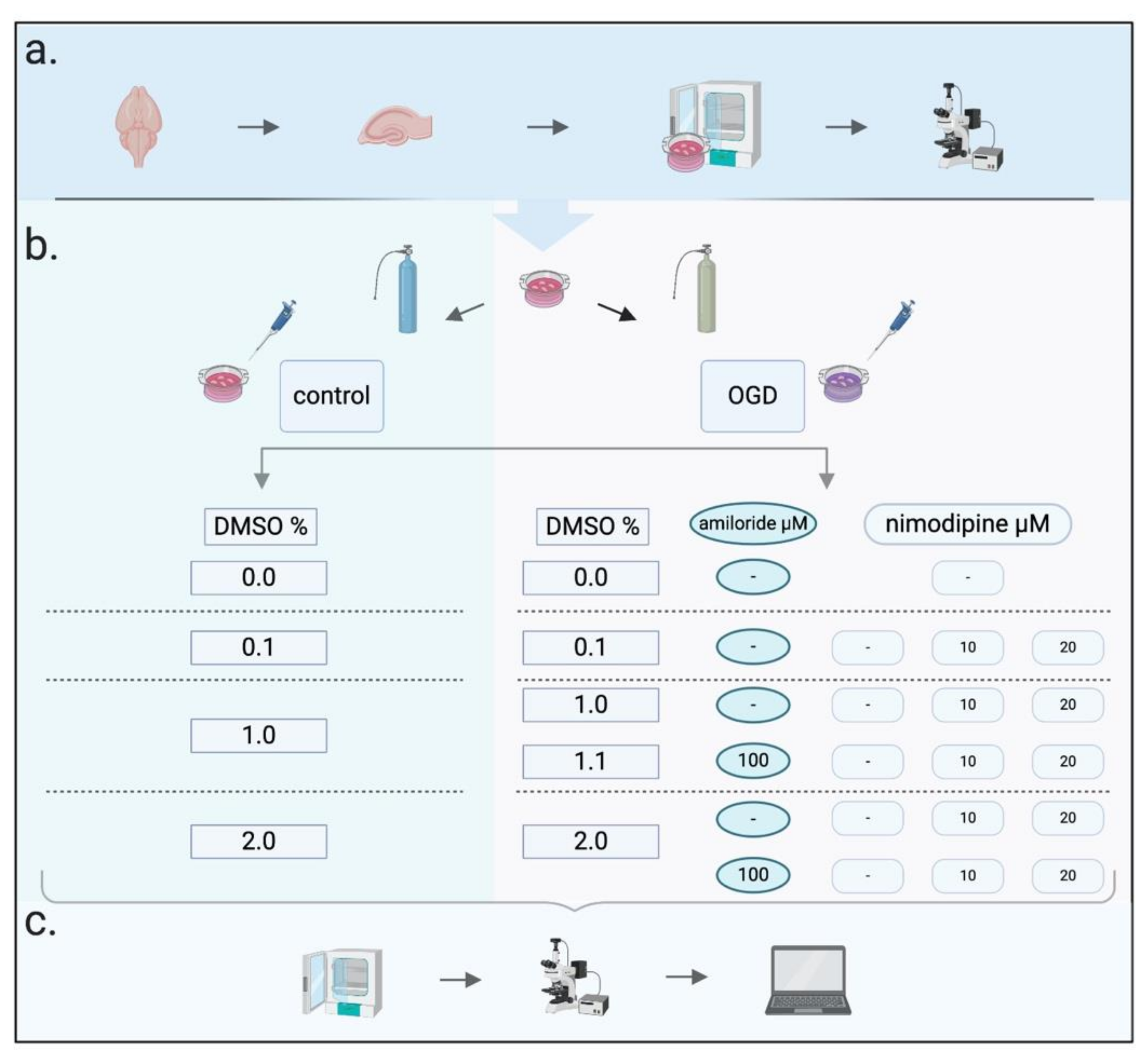
4. Materials and Methods
4.1. Mediums
4.2. Slice Preparation and Cultivation
4.3. Imaging
4.4. Oxygen–Glucose Deprivation (OGD)
4.5. Neuroprotective Protocols
4.6. Cell Death Assessment
4.7. Pre-Statistical Image Processing
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASIC | Acid-sensing ion channel |
| DCI | Delayed cerebral ischaemia |
| DMSO | Dimethyl-sulfoxide |
| OGD | Oxygen–glucose deprivation |
| PI | Propidium iodide |
| SAH | Subarachnoid haemorrhage |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OGD/Control | Vol.% DMSO | Nimodipine Concentration in µM | Amiloride Concentration in µM | n Slices | Median | 95% CI (Lower–Upper) |
|---|---|---|---|---|---|---|
| Control | - | - | - | 101 | 0.02308 | 0.01774–0.03227 |
| Control | 0.1 | - | - | 25 | 0.03409 | 0.01500–0.05189 |
| Control | 1.0 | - | - | 12 | 0.06632 | 0.01006–0.3592 |
| Control | 1.0 | - | 100 | 63 | 0.1470 | 0.09480–0.1877 |
| Control | 2.0 | - | - | 20 | 0.7912 | 0.3633–1.253 |
| Control | 2.0 | - | 100 | 6 | 0.02141 | 0.000–0.1145 |
| OGD | - | - | - | 96 | 0.8978 | 0.8099–1.118 |
| OGD | 0.1 | - | - | 41 | 1.261 | 0.8406–1.697 |
| OGD | 0.1 | 10 | - | 77 | 0.7449 | 0.4920–1.027 |
| OGD | 0.1 | 20 | - | 64 | 1.152 | 0.9478–1.471 |
| OGD | 1.0 | - | - | 19 | 0.4013 | 0.2374–0.6065 |
| OGD | 1.0 | 10 | - | 30 | 0.6925 | 0.3331–1.012 |
| OGD | 1.0 | 20 | - | 25 | 0.5422 | 0.2462–0.8696 |
| OGD | 1.0 | - | 100 | 50 | 0.6901 | 0.4543–0.8942 |
| OGD | 1.1 | 10 | 100 | 56 | 0.7106 | 0.4539–0.8876 |
| OGD | 1.1 | 20 | 100 | 59 | 0.7777 | 0.5228–1.001 |
| OGD | 2.0 | - | - | 45 | 0.8082 | 0.5730–1.515 |
| OGD | 2.0 | - | 100 | 43 | 1.172 | 1.067–1.353 |
| OGD | 2.0 | 10 | 100 | 47 | 2.147 | 1.775–3.085 |
| OGD | 2.0 | 20 | 100 | 46 | 2.563 | 1.526–2.939 |
Appendix B

References
- De Rooij, N.K.; Linn, F.H.; van der Plas, J.A.; Algra, A.; Rinkel, G.J. Incidence of subarachnoid haemorrhage: A systematic review with emphasis on region, age, gender and time trends. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. 2015, 157, 1683–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanne, F.A.; Kane, A.B.; Young, E.E.; Farber, J.L. Calcium dependence of toxic cell death: A final common pathway. Science 1979, 206, 700–702. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K. Calcium and cell death. Magnesium 1989, 8, 223–237. [Google Scholar] [PubMed]
- Siesjo, B.K.; Bengtsson, F.; Grampp, W.; Theander, S. Calcium, excitotoxins, and neuronal death in the brain. Ann. N. Y. Acad. Sci. 1989, 568, 234–251. [Google Scholar] [CrossRef]
- Dubinsky, J.M. Examination of the role of calcium in neuronal death. Ann. N. Y. Acad. Sci. 1993, 679, 34–42. [Google Scholar] [CrossRef]
- Dubinsky, J.M. Intracellular calcium levels during the period of delayed excitotoxicity. J. Neurosci. 1993, 13, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Wahlgren, N.G.; Ahmed, N. Neuroprotection in cerebral ischaemia: Facts and fancies--the need for new approaches. Cerebrovasc. Dis. 2004, 153–166. [Google Scholar] [CrossRef]
- O’Bryant, Z.; Vann, K.T.; Xiong, Z.G. Translational strategies for neuroprotection in ischemic stroke--focusing on acid-sensing ion channel 1a. Transl. Stroke Res. 2014, 5, 59–68. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, S. Aneurysmal Subarachnoid Hemorrhage. J. Neurosurg. Anesthesiol. 2015, 27, 222–240. [Google Scholar] [CrossRef] [Green Version]
- Pala, A.; Schick, J.; Klein, M.; Mayer, B.; Schmitz, B.; Wirtz, C.R.; Konig, R.; Kapapa, T. The influence of nimodipine and vasopressors on outcome in patients with delayed cerebral ischemia after spontaneous subarachnoid hemorrhage. J. Neurosurg. 2019. [Google Scholar] [CrossRef]
- Lawton, M.T.; Vates, G.E. Subarachnoid Hemorrhage. N. Eng. J. Med. 2017, 377, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Dorhout Mees, S.M.; Rinkel, G.J.; Feigin, V.L.; Algra, A.; van den Bergh, W.M.; Vermeulen, M.; van Gijn, J. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.D.; Murray, G.D.; Illingworth, R.; Shaw, M.D.; Teasdale, G.M.; Foy, P.M.; Humphrey, P.R.; Lang, D.A.; Nelson, R.; Richards, P.; et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989, 298, 636–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohman, J.; Heiskanen, O. Effect of nimodipine on the outcome of patients after aneurysmal subarachnoid hemorrhage and surgery. J. Neurosurg. 1988, 69, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Towart, R.; Kazda, S. The cellular mechanism of action of nimodipine (BAY e 9736), a new calcium antagonist. Br. J. Pharmacol. 1979, 67, 409P–410P. [Google Scholar] [CrossRef] [PubMed]
- Petruk, K.C.; West, M.; Mohr, G.; Weir, B.K.; Benoit, B.G.; Gentili, F.; Disney, L.B.; Khan, M.I.; Grace, M.; Holness, R.O.; et al. Nimodipine treatment in poor-grade aneurysm patients. Results of a multicenter double-blind placebo-controlled trial. J. Neurosurg. 1988, 68, 505–517. [Google Scholar] [CrossRef]
- Uematsu, D.; Greenberg, J.H.; Hickey, W.F.; Reivich, M. Nimodipine attenuates both increase in cytosolic free calcium and histologic damage following focal cerebral ischemia and reperfusion in cats. Stroke 1989, 20, 1531–1537. [Google Scholar] [CrossRef] [Green Version]
- Greiner, C.; Schmidinger, A.; Hulsmann, S.; Moskopp, D.; Wolfer, J.; Kohling, R.; Speckmann, E.J.; Wassmann, H. Acute protective effect of nimodipine and dimethyl sulfoxide against hypoxic and ischemic damage in brain slices. Brain Res. 2000, 887, 316–322. [Google Scholar] [CrossRef]
- Krieglstein, J.; Lippert, K.; Poch, G. Apparent independent action of nimodipine and glutamate antagonists to protect cultured neurons against glutamate-induced damage. Neuropharmacology 1996, 35, 1737–1742. [Google Scholar] [CrossRef]
- Pisani, A.; Calabresi, P.; Tozzi, A.; D’Angelo, V.; Bernardi, G. L-type Ca2+ channel blockers attenuate electrical changes and Ca2+ rise induced by oxygen/glucose deprivation in cortical neurons. Stroke 1998, 29, 196–201; discussion 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, I.S.; Cottrell, J.E.; Chambers, G. Magnesium and cobalt, not nimodipine, protect neurons against anoxic damage in the rat hippocampal slice. Anesthesiology 1988, 69, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.P.; Choi, D.W. Combined oxygen and glucose deprivation in cortical cell culture: Calcium-dependent and calcium-independent mechanisms of neuronal injury. J. Neurosci 1993, 13, 3510–3524. [Google Scholar] [CrossRef] [PubMed]
- Small, D.L.; Monette, R.; Buchan, A.M.; Morley, P. Identification of calcium channels involved in neuronal injury in rat hippocampal slices subjected to oxygen and glucose deprivation. Brain Res. 1997, 753, 209–218. [Google Scholar] [CrossRef]
- Martinez-Sanchez, M.; Striggow, F.; Schroder, U.H.; Kahlert, S.; Reymann, K.G.; Reiser, G. Na(+) and Ca(2+) homeostasis pathways, cell death and protection after oxygen-glucose-deprivation in organotypic hippocampal slice cultures. Neuroscience 2004, 128, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.G.; Chu, X.P.; Simon, R.P. Ca2+ -permeable acid-sensing ion channels and ischemic brain injury. J. Membr. Biol 2006, 209, 59–68. [Google Scholar] [CrossRef]
- Yermolaieva, O.; Leonard, A.S.; Schnizler, M.K.; Abboud, F.M.; Welsh, M.J. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2004, 101, 6752–6757. [Google Scholar] [CrossRef] [Green Version]
- Pignataro, G.; Simon, R.P.; Xiong, Z.G. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain 2007, 130, 151–158. [Google Scholar] [CrossRef]
- Osmakov, D.I.; Andreev, Y.A.; Kozlov, S.A. Acid-sensing ion channels and their modulators. Biochemistry 2014, 79, 1528–1545. [Google Scholar] [CrossRef] [PubMed]
- Paukert, M.; Babini, E.; Pusch, M.; Grunder, S. Identification of the Ca2+ blocking site of acid-sensing ion channel (ASIC) 1: Implications for channel gating. J. Gen. Physiol. 2004, 124, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babini, E.; Paukert, M.; Geisler, H.S.; Grunder, S. Alternative splicing and interaction with di- and polyvalent cations control the dynamic range of acid-sensing ion channel 1 (ASIC1). J. Biol. Chem. 2002, 277, 41597–41603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.J.; Symon, L.; Branston, N.M.; Bayhan, M. Changes in extracellular calcium activity in cerebral ischaemia. J. Cereb. Blood Flow Metab. 1981, 1, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loetscher, P.D.; Rossaint, J.; Rossaint, R.; Weis, J.; Fries, M.; Fahlenkamp, A.; Ryang, Y.M.; Grottke, O.; Coburn, M. Argon: Neuroprotection in in vitro models of cerebral ischemia and traumatic brain injury. Crit. Care 2009, 13, R206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Ogawa, S.; Kitao, Y.; Hori, O. Ischemia-induced neuronal cell death and stress response. Antioxid. Redox. Signal. 2007, 9, 573–587. [Google Scholar] [CrossRef]
- Gelmers, H.J.; Gorter, K.; de Weerdt, C.J.; Wiezer, H.J. A controlled trial of nimodipine in acute ischemic stroke. N. Engl. J. Med. 1988, 318, 203–207. [Google Scholar] [CrossRef]
- Lee, K.S.; Lee, K.C.; Choi, J.U. Clinical trial of a calcium channel blocker in patients with aneurysmal subarachnoid hemorrhage--prevention of delayed ischemic deficits. Yonsei Med. J. 1987, 28, 126–130. [Google Scholar] [CrossRef]
- Rowland, M.J.; Hadjipavlou, G.; Kelly, M.; Westbrook, J.; Pattinson, K.T. Delayed cerebral ischaemia after subarachnoid haemorrhage: Looking beyond vasospasm. Br. J. Anaesth. 2012, 109, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Won, S.J.; Kim, D.Y.; Gwag, B.J. Cellular and molecular pathways of ischemic neuronal death. J. Biochem. Mol. Biol. 2002, 35, 67–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, I.S.; Lipton, P. Calcium and long-term transmission damage following anoxia in dentate gyrus and CA1 regions of the rat hippocampal slice. J. Physiol. 1986, 378, 313–334. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, H.; Jorgensen, M.B.; Diemer, N.H.; Hansen, A.J. Calcium accumulation by glutamate receptor activation is involved in hippocampal cell damage after ischemia. Acta Neurol. Scand. 1988, 78, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Mattson, M.P. Dimethyl sulfoxide suppresses NMDA- and AMPA-induced ion currents and calcium influx and protects against excitotoxic death in hippocampal neurons. Exp. Neurol. 2001, 170, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Gao, J.Y.; Guo, J.C.; Bai, L.; Marshall, C.; Cai, Z.Y.; Wang, L.M.; Xiao, M. Dimethyl Sulfoxide Damages Mitochondrial Integrity and Membrane Potential in Cultured Astrocytes. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.G.; Pignataro, G.; Li, M.; Chang, S.Y.; Simon, R.P. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr. Opin. Pharmacol. 2008, 8, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, R.L.; Kassell, N.F.; Mayer, S.; Ruefenacht, D.; Schmiedek, P.; Weidauer, S.; Frey, A.; Roux, S.; Pasqualin, A.; CONSCIOUS-1 Investigators. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): Randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke 2008, 39, 3015–3021. [Google Scholar] [CrossRef] [Green Version]
- Vergouwen, M.D.; Vermeulen, M.; Coert, B.A.; Stroes, E.S.; Roos, Y.B. Microthrombosis after aneurysmal subarachnoid hemorrhage: An additional explanation for delayed cerebral ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 1761–1770. [Google Scholar] [CrossRef]
- Suzuki, S.; Kimura, M.; Souma, M.; Ohkima, H.; Shimizu, T.; Iwabuchi, T. Cerebral microthrombosis in symptomatic cerebral vasospasm--a quantitative histological study in autopsy cases. Neurol. Med. Chir. 1990, 30, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, B.; Muller, F.; Feiler, S.; Scholler, K.; Plesnila, N. Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: An in-vivo microscopy study. J. Cereb. Blood Flow Metab. 2012, 32, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Wellman, G.C.; Koide, M. Impact of subarachnoid hemorrhage on parenchymal arteriolar function. Acta Neurochir. Suppl. 2013, 115, 173–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budohoski, K.P.; Guilfoyle, M.; Helmy, A.; Huuskonen, T.; Czosnyka, M.; Kirollos, R.; Menon, D.K.; Pickard, J.D.; Kirkpatrick, P.J. The pathophysiology and treatment of delayed cerebral ischaemia following subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Logsdon, A.F.; Manoranjan, B.; Turner, R.C.; McConnell, E.; Vates, G.E.; Huber, J.D.; Rosen, C.L.; Simard, J.M. Aneurysmal Subarachnoid Hemorrhage and Neuroinflammation: A Comprehensive Review. Int. J. Mol. Sci. 2016, 17, 497. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Manoel, A.L.; Macdonald, R.L. Neuroinflammation as a Target for Intervention in Subarachnoid Hemorrhage. Front. Neurol. 2018, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P.; Major, S.; Manning, A.; Woitzik, J.; Drenckhahn, C.; Steinbrink, J.; Tolias, C.; Oliveira-Ferreira, A.I.; Fabricius, M.; Hartings, J.A.; et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009, 132, 1866–1881. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, A.M.; Hou, Y.; Zhao, X.; Zhang, B.; Lyeth, B.G.; Russell, M.J. Dimethyl sulfoxide provides neuroprotection in a traumatic brain injury model. Restor. Neurol. Neurosci. 2008, 26, 501–507. [Google Scholar]
- Shimizu, S.; Simon, R.P.; Graham, S.H. Dimethylsulfoxide (DMSO) treatment reduces infarction volume after permanent focal cerebral ischemia in rats. Neurosci. Lett. 1997, 239, 125–127. [Google Scholar] [CrossRef]
- Phillis, J.W.; Estevez, A.Y.; O’Regan, M.H. Protective effects of the free radical scavengers, dimethyl sulfoxide and ethanol, in cerebral ischemia in gerbils. Neurosci. Lett. 1998, 244, 109–111. [Google Scholar] [CrossRef]
- Stoppini, L.; Buchs, P.-A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Galvao, J.; Davis, B.; Tilley, M.; Normando, E.; Duchen, M.R.; Cordeiro, M.F. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014, 28, 1317–1330. [Google Scholar] [CrossRef]
- Zhang, C.; Deng, Y.; Dai, H.; Zhou, W.; Tian, J.; Bing, G.; Zhao, L. Effects of dimethyl sulfoxide on the morphology and viability of primary cultured neurons and astrocytes. Brain Res. Bull. 2017, 128, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, F.A.; Garner, B.; Ball, G.E.; Rae, C. Modulation of brain metabolism by very low concentrations of the commonly used drug delivery vehicle dimethyl sulfoxide (DMSO). J. Neurosci. Res. 2008, 86, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Grüßer, L.; Blaumeiser-Debarry, R.; Krings, M.; Kremer, B.; Höllig, A.; Rossaint, R.; Coburn, M. Argon attenuates the emergence of secondary injury after traumatic brain injury within a 2-hour incubation period compared to desflurane: An in vitro study. Med. Gas. Res. 2017, 7, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macklis, J.D.; Madison, R.D. Progressive incorporation of propidium iodide in cultured mouse neurons correlates with declining electrophysiological status: A fluorescence scale of membrane integrity. J. Neurosci. Methods 1990, 31, 43–46. [Google Scholar] [CrossRef]
- Brayton, C.F. Dimethyl sulfoxide (DMSO): A review. Cornell Vet. 1986, 76, 61–90. [Google Scholar] [PubMed]
- Krings, M.; Höllig, A.; Liu, J.; Grüßer, L.; Rossaint, R.; Coburn, M. Desflurane impairs outcome of organotypic hippocampal slices in an in vitro model of traumatic brain injury. Med. Gas. Res. 2016, 6, 3–9. [Google Scholar] [CrossRef]
- Schampel, A.; Volovitch, O.; Koeniger, T.; Scholz, C.J.; Jorg, S.; Linker, R.A.; Wischmeyer, E.; Wunsch, M.; Hell, J.W.; Ergun, S.; et al. Nimodipine fosters remyelination in a mouse model of multiple sclerosis and induces microglia-specific apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3295–E3304. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ort, J.; Kremer, B.; Grüßer, L.; Blaumeiser-Debarry, R.; Clusmann, H.; Coburn, M.; Höllig, A.; Lindauer, U. Failed Neuroprotection of Combined Inhibition of L-Type and ASIC1a Calcium Channels with Nimodipine and Amiloride. Int. J. Mol. Sci. 2020, 21, 8921. https://doi.org/10.3390/ijms21238921
Ort J, Kremer B, Grüßer L, Blaumeiser-Debarry R, Clusmann H, Coburn M, Höllig A, Lindauer U. Failed Neuroprotection of Combined Inhibition of L-Type and ASIC1a Calcium Channels with Nimodipine and Amiloride. International Journal of Molecular Sciences. 2020; 21(23):8921. https://doi.org/10.3390/ijms21238921
Chicago/Turabian StyleOrt, Jonas, Benedikt Kremer, Linda Grüßer, Romy Blaumeiser-Debarry, Hans Clusmann, Mark Coburn, Anke Höllig, and Ute Lindauer. 2020. "Failed Neuroprotection of Combined Inhibition of L-Type and ASIC1a Calcium Channels with Nimodipine and Amiloride" International Journal of Molecular Sciences 21, no. 23: 8921. https://doi.org/10.3390/ijms21238921
APA StyleOrt, J., Kremer, B., Grüßer, L., Blaumeiser-Debarry, R., Clusmann, H., Coburn, M., Höllig, A., & Lindauer, U. (2020). Failed Neuroprotection of Combined Inhibition of L-Type and ASIC1a Calcium Channels with Nimodipine and Amiloride. International Journal of Molecular Sciences, 21(23), 8921. https://doi.org/10.3390/ijms21238921