RAGE Signaling in Melanoma Tumors



Abstract
:1. Melanoma
1.1. Driver Mutations in Melanomagenesis
1.2. Cutaneous and Non-Cutaneous Melanoma
1.3. Staging of Melanoma and Patient Survival
1.4. Melanoma Biomarkers
1.5. Treatment of Cutaneous Melanoma
2. RAGE
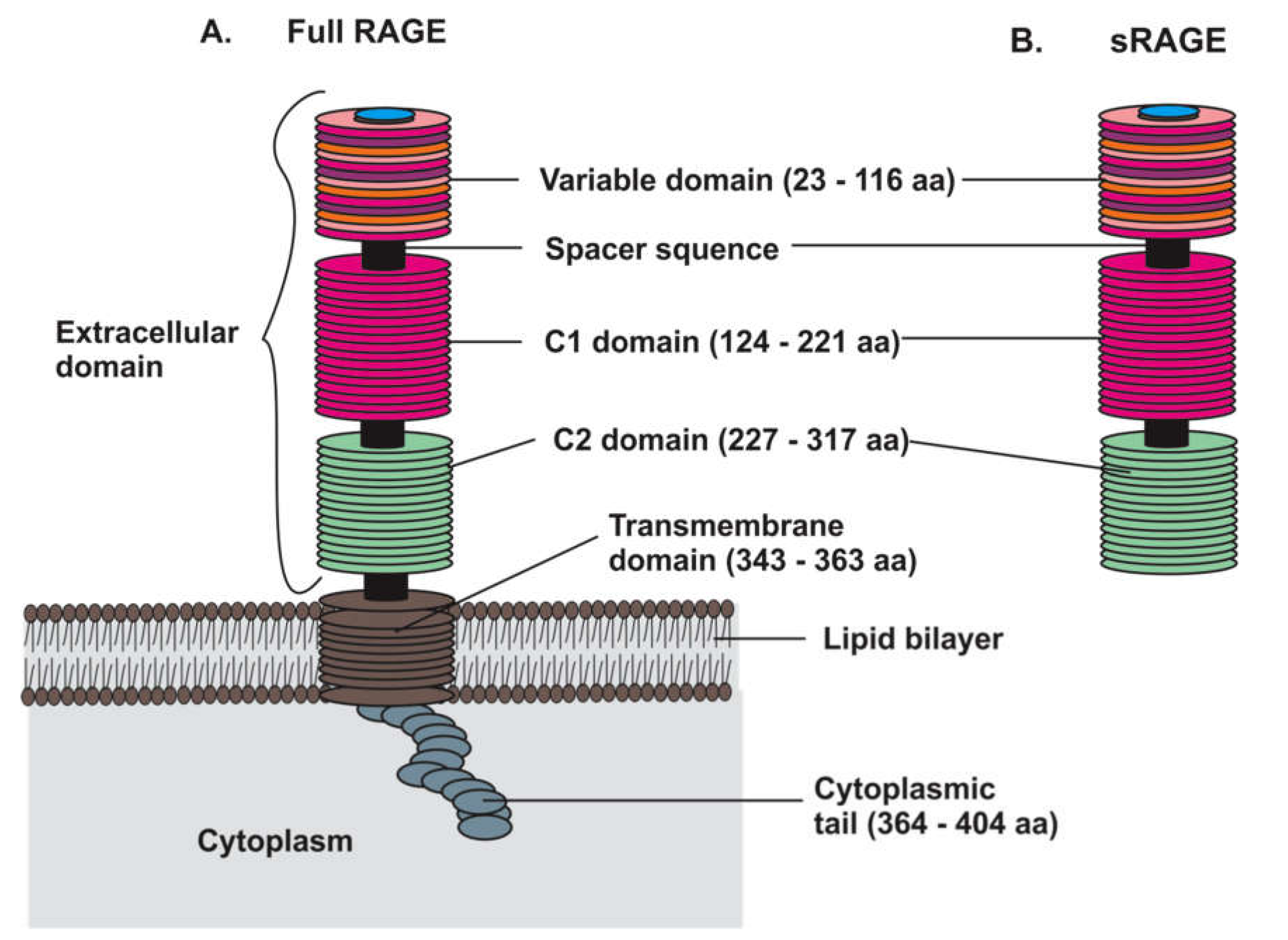
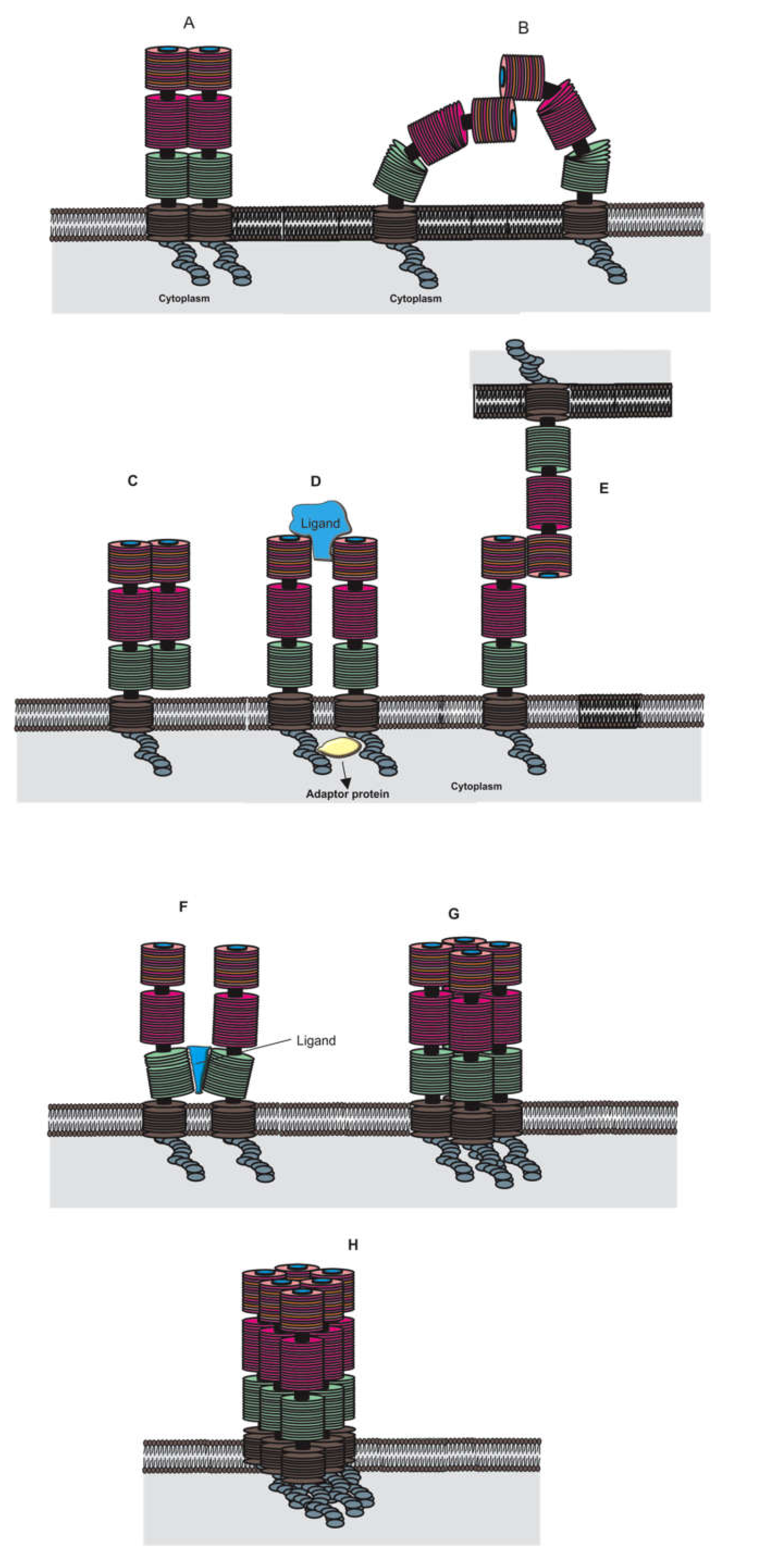
2.1. RAGE Structure and Isoforms
2.2. RAGE Ligands
2.3. S100 Proteins Family
2.3.1. S100B
2.3.2. S100A1
2.3.3. S100A2
2.3.4. S100A4
2.3.5. S100A6
2.3.6. S100A8/A9
2.3.7. S100A13
2.3.8. S100P
2.4. HMGB1
2.5. Advanced Glycation End Products
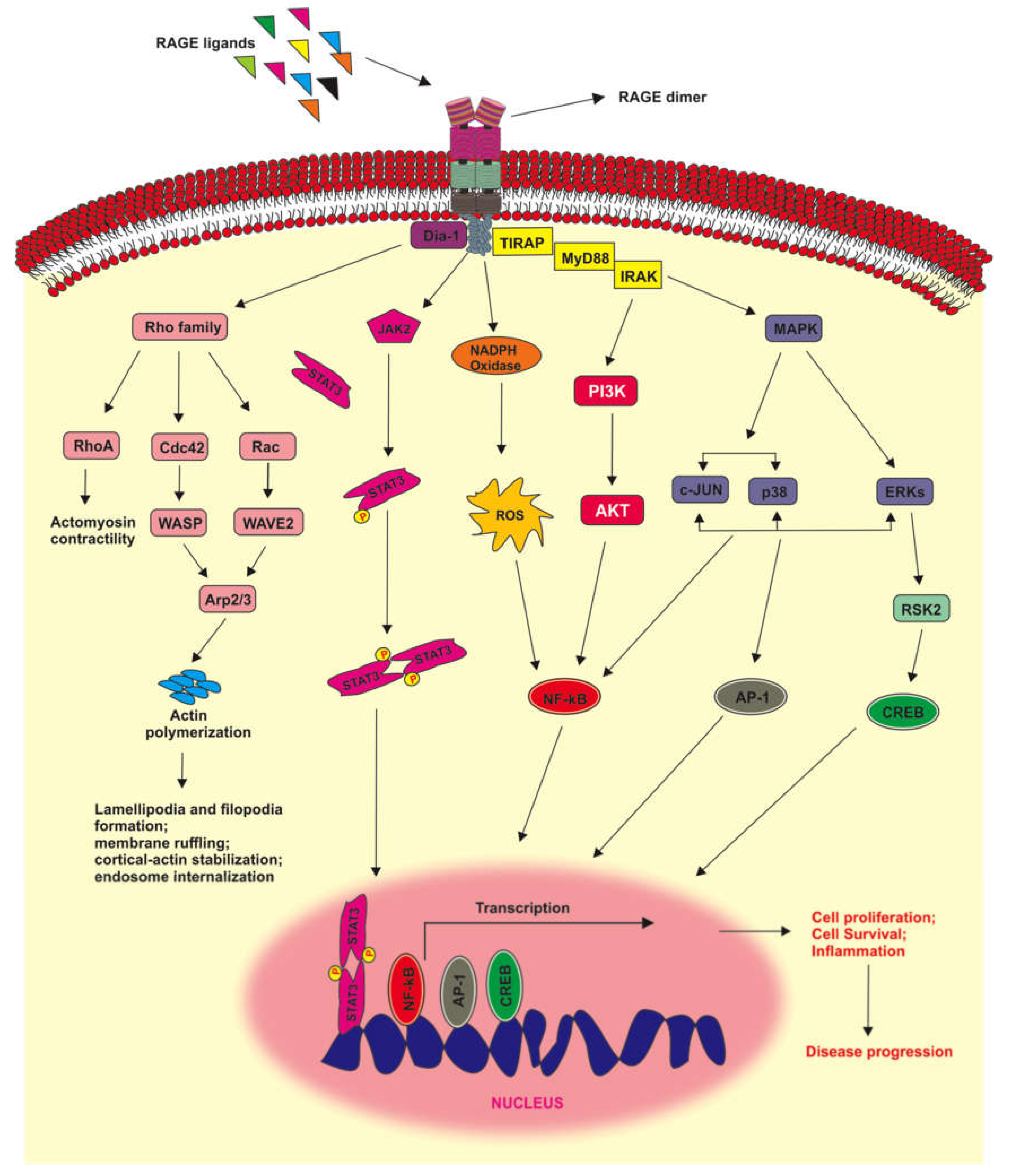
2.6. RAGE Signaling Pathways
3. RAGE Signaling in Melanoma Tumors
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RAGE | Receptor for advanced glycation end product |
| HMGB1 | High Mobility Group Box 1 |
| MSH | Melanocyte stimulating hormone |
| MC1-R | Melanocortin 1 receptor |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| MAPK | Mitogen-activated protein kinase |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| BRAF | V-raf murine sarcoma viral oncogene homolog B1 |
| PI3K | Phosphatidylinositol-3 kinase |
| mTOR | Mammalian target of rapamycin |
| NF1 | Neurofibromatosis type1 |
| ARID2 | AT-rich interactive domain-containing protein 2 |
| TP53 | Tumor protein p53 |
| PPP6C | Serine/threonine-protein phosphatase 6 catalytic subunit |
| DDX3X | DEAD-box helicase 3 X-linked |
| PTEN | Phosphatase and tension homology |
| RAC1 | ras-related C3 botulinum toxin substrate 1 |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| SNX31 | sorting nexin 31 |
| PREX2 | phosphatidylinositol-3,4,5- trisphosphate-dependent rac exchange factor 2 |
| TACC1 | transforming acidic coiled-coil-containing protein 1 |
| KIT | KIT tyrosine-protein kinase |
| IDH1 | isocitrate dehydrogenase1 |
| RB1 | retinoblastoma protein 1 |
| SF3B1 | splicing factor 3b subunit 1 |
| CTNNB1 | catenin (cadherin-associate protein) beta 1 |
| PIK3CA | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| CDK4 | cyclin-dependent kinase 4 |
| RASA2 | ras p21 protein activator 2 |
| WT1 | Wilms’ tumor suppressor gene 1 |
| EZH2 | enhancer of zeste homolog 2 |
| STK19 | serine/threonine-protein kinase 19 |
| SSM | superficial spreading melanoma |
| AJCC | American joint commission in cancer |
| OS | Overall survival |
| DSS | Disease-specific survival |
| LDH | Lactate dehydrogenase |
| PD1L1 | Programmed cell death receptor 1 ligand 1 |
| PD-1 | Programmed cell death receptor 1 |
| IL-2 | Interleukin 2 |
| IFN-α | Interferon alpha |
| FDA | Food and drug administration |
| CTLA-4 | Cytotoxic T lymphocyte antigen 4 |
| MHC | Major histocompatibility complex |
| TM | Transmembrane |
| V | Variable |
| C1 | Constant domain 1 |
| C2 | Constant domain 2 |
| DAMPS | Damage-associated molecular patterns |
| AGE | Advanced glycation end products |
| TRPM-1 | Transient receptor potential melastatin-1 |
| SSSRs | S100 soil sensor receptor |
| EMMPRIN | Extracellular matrix metalloproteinase inducer |
| ALCAM | Activated leukocyte cell adhesion molecule |
| TLR-4 | Toll-like receptor 4 |
| NPTNβ | Neuroplastin β |
| MCAM | Melanoma cells adhesion molecule |
| NF-κB | Nuclear factor kappa beta |
| ROS | Reactive oxygen species |
| TPL2 | Tumor progression locus 2 |
| ETV4 | ETS translocation variant 4 |
| MMP-25 | Matrix metalloproteinase 25 |
| VEGF | Vascular endothelial growth factor |
| FGF | Fibroblast growth factor |
| CMM | Cutaneous malignant melanoma |
| MCP-1 | Monocyte chemoattractant protein 1 |
| Dia-1 | Diaphanous-1 |
| cdc 42 | Cell division control protein 42 |
| TIRAP | Toll-like receptor 2/4 adaptors |
| MyD88 | Myeloid differentiation primary response 88 |
| ERK | Extracellular signal-related kinase |
| JNK | C-Jun N-terminal kinase |
| TXNIP | Thioredoxin interacting protein |
| NLRP3 | NLR family pyrin domain containing 3 |
| JAK | Janus kinase |
| STAT | Signal transducer and activator of transcription |
| CREB | Cyclin AMP response element-binding protein |
| RSK2 | Ribosomal S6 kinase 2 |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| ICAM-1 | Intercellular adhesion molecule 1 |
| TNF-α | Tumor necrosis factor alpha |
| NO | Nitric oxide |
| Tregs | Regulatory T cells |
References
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Rockville, MD, USA, 2020. [Google Scholar]
- Bertolotto, C. Melanoma: From Melanocyte to Genetic Alterations and Clinical Options. Scientifica 2013, 2013, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordlund, J.J. The Melanocyte and the Epidermal Melanin Unit: An Expanded Concept. Dermatol. Clin. 2007, 25, 271–281. [Google Scholar] [CrossRef]
- Riley, P. Melanin. Int. J. Biochem. Cell Biol. 1997, 29, 1235–1239. [Google Scholar] [CrossRef]
- Breathnach, A.S. Extra-Cutaneous Melanin. Pigment. Cell Res. 1988, 1, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Italian Melanoma Intergroup for the Italian Melanoma Intergroup (IMI); Colombino, M.; Casula, M.; Manca, A.; Mandalà, M.; Cossu, A. Molecular Pathways in Melanomagenesis: What We Learned from Next-Generation Sequencing Approaches. Curr. Oncol. Rep. 2018, 20, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, G.; Eombra, M.; Ecolombino, M.; Ecasula, M.; Esini, M.; Emanca, A.; Epaliogiannis, P.; Eascierto, P.A.; Ecossu, A. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front. Oncol. 2015, 5, 183. [Google Scholar] [CrossRef]
- Bertrand, J.; Steingrimsson, E.; Jouenne, F.; Paillerets, B.; LaRue, L. Melanoma Risk and Melanocyte Biology. Acta Derm. Venereol. 2020, 100, adv00139. [Google Scholar] [CrossRef]
- Curioni-Fontecedro, A.; Pitocco, R.; Schoenewolf, N.L.; Holzmann, D.; Soldini, D.; Dummer, R.; Calvieri, S.; Moch, H.; Fitsche, A.; Mihic-Probst, D. Erratum to “intratumoral heterogeneity of MAGE-C1/CT7 and MAGE-C2/CT10 expression in mucosal melanoma”. BioMed Res. Int. 2019, 2019, 5256364. [Google Scholar] [CrossRef] [Green Version]
- Iga, N.; Otsuka, A.; Hirata, M.; Kataoka, T.; Irie, H.; Nakashima, C.; Matsushita, S.; Uchi, H.; Yamamoto, Y.; Funakoshi, T.; et al. Variable indoleamine 2,3-dioxygenase expression in acral/mucosal melanoma and its possible link to immunotherapy. Cancer Sci. 2019, 110, 3434–3441. [Google Scholar] [CrossRef]
- Petrella, T.M.; Fletcher, G.G.; Knight, G.; McWhirter, E.; Rajagopal, S.; Song, X.; Baetz, T.D. Systemic adjuvant therapy for adult patients at high risk for recurrent cutaneous or mucosal melanoma: An Ontario Health (Cancer Care Ontario) clinical practice guideline. Curr. Oncol. 2020, 27, e43–e52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donizy, P.; Wu, C.-L.; Mull, J.; Fujimoto, M.; Chłopik, A.; Peng, Y.; Shalin, S.C.; Selim, M.A.; Puig, S.; Fernández-Figueras, M.T.; et al. Up-Regulation of PARP1 Expression Significantly Correlated with Poor Survival in Mucosal Melanomas. Cells 2020, 9, 1135. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Ohuchi, K.; Amagai, R.; Sato, Y.; Tanita, K.; Hashimoto, A.; Aiba, S. Successful Treatment of a Patient with anti-PD1 Antibody-Resistant Advanced Mucosal Melanoma with Nivolumab, Ipilimumab plus Denosumab Combination Therapy. Case Rep. Oncol. 2020, 13, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, A.T.; Nagamine, M.K.; Da Fonseca, I.I.M.; Miraldo, A.C.; Scattone, N.V.; Guerra, J.L.; Xavier, J.G.; Santos, M.; Gomes, C.O.M.D.S.; Ward, J.M.; et al. Inhibitory Effects of a Reengineered Anthrax Toxin on Canine Oral Mucosal Melanomas. Toxins 2020, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Seban, R.-D.; Moya-Plana, A.; Antonios, L.; Yeh, R.; Marabelle, A.; Deutsch, E.; Schwartz, L.H.; Gómez, R.G.H.; Saenger, Y.; Robert, C.; et al. Prognostic 18F-FDG PET biomarkers in metastatic mucosal and cutaneous melanoma treated with immune checkpoint inhibitors targeting PD-1 and CTLA-4. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Palsgrove, D.; Kurian, E.; Yan, S.; Oliai, B.R.; Bishop, J.A. Variable Expression of S100 Protein in Sinonasal Malignant Mucosal Melanoma: A Potential Diagnostic Pitfall. Head Neck Pathol. 2020, 14, 929–935. [Google Scholar] [CrossRef]
- Kashyap, S.; Meel, R.; Singh, L.; Singh, M. Uveal melanoma. Semin. Diagn. Pathol. 2016, 33, 141–147. [Google Scholar] [CrossRef]
- Mitre, V.; Heym, K.; Clark, G.D.; Venkatramani, R. Neurocutaneous Melanocytosis and Leptomeningeal Melanoma. J. Pediatr. Hematol. 2019. [Google Scholar] [CrossRef]
- Besra, K.; Panda, S.; Dash, S.; Samantaray, S.; Pathy, P.C.; Rout, N. Clinicopathological study of malignant melanoma in a regional cancer center. Indian J. Cancer 2018, 55, 292. [Google Scholar] [CrossRef]
- Yde, S.S.; Sjoegren, P.; Heje, M.; Stolle, L.B. Mucosal Melanoma: A Literature Review. Curr. Oncol. Rep. 2018, 20, 28. [Google Scholar] [CrossRef]
- Seetharamu, N.; Ott, P.A.; Pavlick, A.C. Mucosal Melanomas: A Case-Based Review of the Literature. Oncologist 2010, 15, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Spencer, K.R.; Mehnert, J.M. Mucosal Melanoma: Epidemiology, Biology and Treatment. Cancer Treat. Res. 2015, 167, 295–320. [Google Scholar] [CrossRef]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef] [Green Version]
- Patrick, R.J.; Fenske, N.A.; Messina, J.L. Primary mucosal melanoma. J. Am. Acad. Dermatol. 2007, 56, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Laver, N.V.; McLaughlin, M.E.; Duker, J.S. Ocular melanoma. Arch. Pathol. Lab. Med. 2010, 134, 1778–1784. [Google Scholar] [PubMed]
- Char, D.H. Ocular melanoma. Surg. Clin. N. Am. 2003, 83, 253–274. [Google Scholar] [CrossRef]
- Da Costa, N.F.; Fernandes, N.C.; Borges, M.R.M.M. Study of the histopathological types of cutaneous melanoma in Palmas-TO from 2001 to 2011. An. Bras. de Dermatol. 2015, 90, 638–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Guy, G.P.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [Green Version]
- Hahn, H.M.; Lee, K.G.; Choi, W.; Cheong, S.H.; Myung, K.B. An updated review of mucosal melanoma: Survival meta-analysis. Mol. Clin. Oncol. 2019, 11, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Han, A.Y.; Dhanjani, S.; Pettijohn, K.; Patel, P.B.; John, M.A.S. Optimal resection margin for head and neck cutaneous melanoma. Laryngoscope 2019, 129, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Cayrefourcq, L.; De Roeck, A.; Garcia, C.; Stoebner, P.-E.; Fichel, F.; Garima, F.; Perriard, F.; Daures, J.-P.; Meunier, L.; Alix-Panabières, C. S100-EPISPOT: A New Tool to Detect Viable Circulating Melanoma Cells. Cells 2019, 8, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Tandler, N.; Mosch, B.; Pietzsch, J. Protein and non-protein biomarkers in melanoma: A critical update. Amino Acids 2012, 43, 2203–2230. [Google Scholar] [CrossRef] [PubMed]
- Belter, B.; Haase-Kohn, C.; Pietzsch, J. Biomarkers in Malignant Melanoma: Recent Trends and Critical Perspective. In Cutaneous Melanoma: Etiology and Therapy; Codon Publications: Brisbane, Australia, 2017; pp. 39–56. [Google Scholar]
- Mandalà, M.; Merelli, B.; Massi, D. PD-L1 in melanoma: Facts and myths. Melanoma Manag. 2016, 3, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.K.; Zarzoso, I.; Daud, A. PD-1 and PD-L1 antibodies for melanoma. Hum. Vaccines Immunother. 2014, 10, 3111–3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finck, S.J.; Giuliano, A.E.; Morton, D.L. LDH and melanoma. Cancer 1983, 51, 840–843. [Google Scholar] [CrossRef]
- Petrelli, F.; Ardito, R.; Merelli, B.; Lonati, V.; Cabiddu, M.; Seghezzi, S.; Barni, S.; Ghidini, A. Prognostic and predictive role of elevated lactate dehydrogenase in patients with melanoma treated with immunotherapy and BRAF inhibitors: A systematic review and meta-analysis. Melanoma Res. 2019, 29, 1–12. [Google Scholar] [CrossRef]
- Gray, M.R.; Martin del Campo, S.; Zhang, X.; Zhang, H.; Souza, F.F.; Carson, W.E., III; Smith, A.D. Metastatic melanoma: Lactate dehydrogenase levels and CT imaging findings of tumor devascularization allow accurate prediction of survival in patients treated with bevacizumab. Radiology 2014, 270, 425–434. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Palmer, S.R.; Erickson, L.A.; Ichetovkin, I.; Knauer, D.J.; Markovic, S.N. Circulating Serologic and Molecular Biomarkers in Malignant Melanoma. Mayo Clin. Proc. 2011, 86, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegre, E.; Sammamed, M.; Fernández-Landázuri, S.; Zubiri, L.; González, Á. Circulating Biomarkers in Malignant Melanoma. Int. Rev. Cytol. 2015, 69, 47–89. [Google Scholar] [CrossRef]
- Petrelli, F.; Cabiddu, M.; Coinu, A.; Borgonovo, K.; Ghilardi, M.; Lonati, V.; Barni, S. Prognostic role of lactate dehydrogenase in solid tumors: A systematic review and meta-analysis of 76 studies. Acta Oncol. 2015, 54, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Kelderman, S.; Heemskerk, B.; Van Tinteren, H.; Brom, R.R.H.V.D.; Hospers, G.A.P.; Eertwegh, A.J.M.V.D.; Kapiteijn, E.W.; De Groot, J.W.B.; Soetekouw, P.; Jansen, R.L.; et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol. Immunother. 2014, 63, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef]
- Vereecken, P.; Cornélis, F.; Van Baren, N.; Vandersleyen, V.; Baurain, J.-F. A Synopsis of Serum Biomarkers in Cutaneous Melanoma Patients. Dermatol. Res. Pract. 2012, 2012, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Osella-Abate, S.; Savoia, P.; Quaglino, P.; Fierro, M.T.; Leporati, C.; Ortoncelli, M.; Bernengo, M.G. Tyrosinase expression in the peripheral blood of stage III melanoma patients is associated with a poor prognosis: A clinical follow-up study of 110 patients. Br. J. Cancer 2003, 89, 1457–1462. [Google Scholar] [CrossRef]
- Quaglino, P.; Osella-Abate, S.; Cappello, N.; Ortoncelli, M.; Nardò, T.; Fierro, M.T.; Cavallo, F.; Savoia, P.; Bernengo, M.G. Prognostic relevance of baseline and sequential peripheral blood tyrosinase expression in 200 consecutive advanced metastatic melanoma patients. Melanoma Res. 2007, 17, 75–82. [Google Scholar] [CrossRef]
- Karagiannis, P.; Fittall, M.W.; Karagiannis, S.N. Evaluating biomarkers in melanoma. Front. Oncol. 2014, 4, 383. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, T.R.; Taube, J.M. PD-L1 and Emerging Biomarkers in Immune Checkpoint Blockade Therapy. Cancer J. 2018, 24, 41–46. [Google Scholar] [CrossRef]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Merelli, B.; Massi, D.; Cattaneo, L.; Mandalà, M. Targeting the PD1/PD-L1 axis in melanoma: Biological rationale, clinical challenges and opportunities. Crit. Rev. Oncol. 2014, 89, 140–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, E.; Ascierto, P.A. Anti-PD-1 and PD-L1 antibodies in metastatic melanoma. Melanoma Manag. 2017, 4, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Madore, J.; Vilain, R.E.; Menzies, A.M.; Kakavand, H.; Wilmott, J.S.; Hyman, J.; Yearley, J.H.; Kefford, R.F.; Thompson, J.F.; Long, G.V.; et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: Implications for anti-PD-1/PD-L1 clinical trials. Pigment. Cell Melanoma Res. 2014, 28, 245–253. [Google Scholar] [CrossRef]
- Hutarew, G. PD-L1 testing, fit for routine evaluation? From a pathologist’s point of view. Memo 2016, 9, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Kitano, S.; Nakayama, T.; Yamashita, M. Biomarkers for Immune Checkpoint Inhibitors in Melanoma. Front. Oncol. 2018, 8, 270. [Google Scholar] [CrossRef]
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B’s double life: Intracellular regulator and extracellular signal. Biochim. et Biophys. Acta (BBA) Bioenerg. 2009, 1793, 1008–1022. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Yang, Q.; Wilder, P.T.; Carrier, F.; Weber, D.J. The Calcium-binding Protein S100B Down-regulates p53 and Apoptosis in Malignant Melanoma. J. Biol. Chem. 2010, 285, 27487–27498. [Google Scholar] [CrossRef] [Green Version]
- Harpio, R.; Einarsson, R. S100 proteins as cancer biomarkers with focus on S100B in malignant melanoma. Clin. Biochem. 2004, 37, 512–518. [Google Scholar] [CrossRef]
- Hauschild, A.; Engel, G.; Brenner, W.; Gläser, R.; Mönig, H.; Henze, E.; Christophers, E. S100B protein detection in serum is a significant prognostic factor in metastatic melanoma. Oncology 1999, 56, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Bouwhuis, M.; Suciu, S.; Kruit, W.; Sales, F.; Stoitchkov, K.; Patel, P.; Cocquyt, V.; Thomas, J.; Liénard, D.; Eggermont, A.M.; et al. Prognostic value of serial blood S100B determinations in stage IIB–III melanoma patients: A corollary study to EORTC trial 18952. Eur. J. Cancer 2011, 47, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.B.; Forschner, A.; Leiter, U.; Garbe, C.; Eigentler, T. S100B and LDH as early prognostic markers for response and overall survival in melanoma patients treated with anti-PD-1 or combined anti-PD-1 plus anti-CTLA-4 antibodies. Br. J. Cancer 2018, 119, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Shannan, B.; Perego, M.; Somasundaram, R.; Herlyn, M. Heterogeneity in Melanoma. Infect. Complicat. Cancer Patients 2015, 167, 1–15. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, R.I.; Lin, J.Y. Cutaneous Melanoma—A Review in Detection, Staging, and Management. Hematol. Clin. N. Am. 2019, 33, 25–38. [Google Scholar] [CrossRef]
- Garbe, C.; Eigentler, T.K.; Keilholz, U.; Hauschild, A.; Kirkwood, J.M. Systematic Review of Medical Treatment in Melanoma: Current Status and Future Prospects. Oncologist 2011, 16, 5–24. [Google Scholar] [CrossRef] [Green Version]
- Domingues, B.; Lopes, J.M.; Soares, P.; Pópulo, H. Melanoma treatment in review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Davies, N.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.W.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Wu, P.K.; Park, J.I. MEK1/2 inhibitors: Molecular activity and resistance mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Brugnara, S.; Sicher, M.; Bonandini, E.M.; Donner, D.; Chierichetti, F.; Barbareschi, M.; Girardelli, C.R.; Caffo, O. Treatment with combined dabrafenib and trametinib in BRAF(V600E)-mutated metastatic malignant melanoma: A case of long-term complete response after treatment cessation. Drugs Context 2018, 7, 212515. [Google Scholar] [PubMed] [Green Version]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chen, F.; Zhou, B. Therapeutic efficacy and safety of combined BRAF and MEK inhibition in patients with malignant melanoma: A meta-analysis. OncoTargets Ther. 2017, 10, 5391–5403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrscher, H.; Robert, C. Immune checkpoint inhibitors in melanoma in the metastatic, neoadjuvant, and adjuvant setting. Curr. Opin. Oncol. 2020, 32, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Menzies, A.M.; Long, G.V. Dabrafenib and Trametinib, Alone and in Combination for BRAF-Mutant Metastatic Melanoma. Clin. Cancer Res. 2014, 20, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.; Arance, A.; Mandalà, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Sugaya, K.; Fukagawa, T.; Matsumoto, K.-I.; Mita, K.; Takahashi, E.-I.; Ando, A.; Inoko, H.; Ikemura, T. Three Genes in the Human MHC Class III Region near the Junction with the Class II: Gene for Receptor of Advanced Glycosylation End Products, PBX2 Homeobox Gene and a Notch Homolog, Human Counterpart of Mouse Mammary Tumor Gene int-3. Genomics 1994, 23, 408–419. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Stern, D.M. Receptor for age (RAGE) is a gene within the major histocompatibility class III region: Implications for host response mechanisms in homeostasis and chronic disease. Front. Biosci. 2001, 6, D1151–D1160. [Google Scholar] [PubMed]
- Ostendorp, T.; Weibel, M.; Leclerc, E.; Kleinert, P.; Kroneck, P.M.; Heizmann, C.W.; Fritz, G. Expression and purification of the soluble isoform of human receptor for advanced glycation end products (sRAGE) from Pichia pastoris. Biochem. Biophys. Res. Commun. 2006, 347, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Sawma, P.; Duneau, J.P.; Khao, J.; Hénin, J.; Bagnard, D.; Sturgis, J. Single-spanning transmembrane domains in cell growth and cell-cell interactions: More than meets the eye? Cell Adhes. Migr. 2010, 4, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Verweij, C.L. How RAGE turns in rage. Genes Immun. 2002, 3, 117–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Lampe, L.; Park, S.; Vangara, B.S.; Waldo, G.S.; Cabantous, S.; Subaran, S.S.; Yang, N.; Lakatta, E.G.; Lin, L. Disulfide Bonds within the C2 Domain of RAGE Play Key Roles in Its Dimerization and Biogenesis. PLoS ONE 2012, 7, e50736. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Manigrasso, M.; Scalabrin, M.; Rai, V.; Reverdatto, S.; Burz, D.S.; Fabris, D.; Schmidt, A.M.; Shekhtman, A. Change in the Molecular Dimension of a RAGE-Ligand Complex Triggers RAGE Signaling. Structure 2016, 24, 1509–1522. [Google Scholar] [CrossRef] [Green Version]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.S.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [PubMed]
- Park, H.; Boyington, J.C. The 1.5 Å Crystal Structure of Human Receptor for Advanced Glycation Endproducts (RAGE) Ectodomains Reveals Unique Features Determining Ligand Binding. J. Biol. Chem. 2010, 285, 40762–40770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Galichet, A.; Weibel, M.; Heizmann, C.W. Calcium-regulated intramembrane proteolysis of the RAGE receptor. Biochem. Biophys. Res. Commun. 2008, 370, 1–5. [Google Scholar] [CrossRef]
- Kalea, A.Z.; Reiniger, N.; Yang, H.; Arriero, M.; Schmidt, A.M.; Hudson, B.I. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009, 23, 1766–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malherbe, P.; Richards, J.; Gaillard, H.; Thompson, A.; Diener, C.; Schuler, A.; Huber, G. cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and characterization of cells co-expressing cell-surface scavenger receptors and Swedish mutant amyloid precursor protein. Mol. Brain Res. 1999, 71, 159–170. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Schlueter, C.; Hauke, S.; Flohr, A.M.; Rogalla, P.; Bullerdiek, J. Tissue-specific expression patterns of the RAGE receptor and its soluble forms—A result of regulated alternative splicing? Biochim. Biophys. Acta 2003, 1630, 1–6. [Google Scholar] [CrossRef]
- Sterenczak, K.A.; Willenbrock, S.; Barann, M.; Klemke, M.; Soller, J.T.; Eberle, N.; Nolte, I.; Bullerdiek, J.; Escobar, H.M. Cloning, characterisation, and comparative quantitative expression analyses of receptor for advanced glycation end products (RAGE) transcript forms. Gene 2009, 434, 35–42. [Google Scholar] [CrossRef]
- Jules, J.; Maiguel, D.; Hudson, B.I. Alternative Splicing of the RAGE Cytoplasmic Domain Regulates Cell Signaling and Function. PLoS ONE 2013, 8, e78267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Madden, A.; Ward, M.; Mooney, M.H.; Elliott, C.T.; Stitt, A.W. Homodimerization Is Essential for the Receptor for Advanced Glycation End Products (RAGE)-mediated Signal Transduction. J. Biol. Chem. 2010, 285, 23137–23146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostendorp, T.; Leclerc, E.; Galichet, A.; Koch, M.; Demling, N.; Weigle, B.; Heizmann, C.W.; Kroneck, P.M.H.; Fritz, G. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 2007, 26, 3868–3878. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Reverdatto, S.; Frolov, A.; Hoffmann, R.; Burz, D.S.; Shekhtman, A. Structural Basis for Pattern Recognition by the Receptor for Advanced Glycation End Products (RAGE). J. Biol. Chem. 2008, 283, 27255–27269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Rai, V.; Singer, D.; Chabierski, S.; Xie, J.; Reverdatto, S.; Burz, D.S.; Schmidt, A.M.; Hoffmann, R.; Shekhtman, A. Advanced Glycation End Product Recognition by the Receptor for AGEs. Structure 2011, 19, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Young, J.H.; Krahn, J.M.; Song, D.; Corbett, K.D.; Chazin, W.J.; Pedersen, L.C.; Esko, J.D. Stable RAGE-Heparan Sulfate Complexes Are Essential for Signal Transduction. ACS Chem. Biol. 2013, 8, 1611–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitkiewicz, E.; Tarnowski, K.; Poznański, J.; Kulma, M.; Dadlez, M. Oligomerization Interface of RAGE Receptor Revealed by MS-Monitored Hydrogen Deuterium Exchange. PLoS ONE 2013, 8, e76353. [Google Scholar] [CrossRef] [Green Version]
- Moysa, A.; Hammerschmid, D.; Szczepanowski, R.H.; Sobott, F.; Dadlez, M. Enhanced oligomerization of full-length RAGE by synergy of the interaction of its domains. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Burz, D.S.; He, W.; Bronstein, I.B.; Lednev, I.; Shekhtman, A. Hexameric Calgranulin C (S100A12) Binds to the Receptor for Advanced Glycated End Products (RAGE) Using Symmetric Hydrophobic Target-binding Patches. J. Biol. Chem. 2006, 282, 4218–4231. [Google Scholar] [CrossRef] [Green Version]
- Yatime, L.; Andersen, G.R. Structural insights into the oligomerization mode of the human receptor for advanced glycation end-products. FEBS J. 2013, 280, 6556–6568. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.; Chitayat, S.; Dattilo, B.M.; Schiefner, A.; Diez, J.; Chazin, W.J.; Fritz, G. Structural Basis for Ligand Recognition and Activation of RAGE. Structure 2010, 18, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessa, L.; Gatti, E.; Zeni, F.; Antonelli, A.; Catucci, A.; Koch, M.; Pompilio, G.; Fritz, G.; Raucci, A.; Bianchi, M.E. The Receptor for Advanced Glycation End-Products (RAGE) Is Only Present in Mammals, and Belongs to a Family of Cell Adhesion Molecules (CAMs). PLoS ONE 2014, 9, e86903. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.G.; Sepuru, K.M.; Yu, C. Interaction of S100A13 with C2 domain of receptor for advanced glycation end products (RAGE). Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Sorci, G.; Riuzzi, F.; Giambanco, I.; Donato, R. RAGE in tissue homeostasis, repair and regeneration. Biochim. et Biophys. Acta (BBA) Bioenerg. 2013, 1833, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riuzzi, F.; Sorci, G.; Sagheddu, R.; Chiappalupi, S.; Salvadori, L.; Donato, R. RAGE in the pathophysiology of skeletal muscle. J. Cachexia Sarcopenia Muscle 2018, 9, 1213–1234. [Google Scholar] [CrossRef] [Green Version]
- Chellan, B.; Sutton, N.R.; Bowman, M.A.H. S100/RAGE-Mediated Inflammation and Modified Cholesterol Lipoproteins as Mediators of Osteoblastic Differentiation of Vascular Smooth Muscle Cells. Front. Cardiovasc. Med. 2018, 5, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, A.; Zhu, Q.; Smith, E.A. Lateral diffusion and signaling of receptor for advanced glycation end-products (RAGE): A receptor involved in chronic inflammation. Eur. Biophys. J. 2017, 47, 39–48. [Google Scholar] [CrossRef]
- Deng, X.; Sun, L.; Lai, X.; Xiang, L.; Li, Q.; Zhang, W.; Zhang, L.; Sun, S. Tea polypeptide ameliorates diabetic nephropathy through RAGE and NF-κB signaling pathway in type 2 diabetes mice. J. Agric. Food Chem. 2018, 66, 11957–11967. [Google Scholar] [CrossRef]
- Hongwei, Y.; Ruiping, C.; Yingyan, F.; Guanjun, Z.; Jie, H.; Xingyu, L.; Jie, T.; Zhenghong, L.; Qin, G.; Junfeng, H.; et al. Effect of Irbesartan on AGEs-RAGE and MMPs systems in rat type 2 diabetes myocardial-fibrosis model. Exp. Biol. Med. 2019, 244, 612–620. [Google Scholar] [CrossRef]
- Toth, C.; Martinez, J.; Zochodne, D.W. RAGE, Diabetes, and the Nervous System. Curr. Mol. Med. 2007, 7, 766–776. [Google Scholar] [CrossRef]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Gąsiorowski, K.; Brokos, B.; Echeverria, V.; Barreto, G.E.; Leszek, J. RAGE-TLR Crosstalk Sustains Chronic Inflammation in Neurodegeneration. Mol. Neurobiol. 2018, 55, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Bangert, A.; Andrassy, M.; Müller, A.-M.; Bockstahler, M.; Fischer, A.; Volz, C.H.; Leib, C.; Göser, S.; Korkmaz-Icöz, S.; Zittrich, S.; et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc. Natl. Acad. Sci. USA 2016, 113, E155–E164. [Google Scholar] [CrossRef] [Green Version]
- Fukami, K.; Yamagishi, S.-I.; Okuda, S. Role of AGEs-RAGE system in cardiovascular disease. Curr. Pharm. Des. 2014, 20, 2395–2402. [Google Scholar] [CrossRef]
- Lee, T.-W.; Kao, Y.-H.; Chen, Y.-J.; Chao, T.-F.; Lee, T.-W. Therapeutic potential of vitamin D in AGE/RAGE-related cardiovascular diseases. Cell. Mol. Life Sci. 2019, 76, 4103–4115. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, P.-W.; Zhou, Y.; Dai, B.; Zhang, P.-F.; Zhang, Y.-H.; Liu, Y.; Shi, X.-L. Rage induces hepatocellular carcinoma proliferation and sorafenib resistance by modulating autophagy. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Leclerc, E.; Vetter, S.W. The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2706–2711. [Google Scholar] [CrossRef] [Green Version]
- Sousa, M.M.; Du Yan, S.; Stern, D.; Saraiva, M.J. Interaction of the Receptor for Advanced Glycation End Products (RAGE) with Transthyretin Triggers Nuclear Transcription Factor kB (NF-kB) Activation. Lab. Investig. 2000, 80, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Rai, V.; Hudson, B.I.; Song, F.; Schmidt, A.M.; Barile, G.R. RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol. 2012, 274, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Ruan, B.H.; Li, X.; Winkler, A.R.; Cunningham, K.M.; Kuai, J.; Greco, R.M.; Nocka, K.H.; Fitz, L.J.; Wright, J.F.; Pittman, D.D.; et al. Complement C3a, CpG Oligos, and DNA/C3a Complex Stimulate IFN-α Production in a Receptor for Advanced Glycation End Product-Dependent Manner. J. Immunol. 2010, 185, 4213–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.-M.; Hofmann, M.; Taguchi, A.; Du Yan, S.; Stern, D.M. RAGE: A Multiligand Receptor Contributing to the Cellular Response in Diabetic Vasculopathy and Inflammation. Semin. Thromb. Hemost. 2000, 26, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [Green Version]
- Salama, I.; Malone, P.; Mihaimeed, F.; Jones, J. A review of the S100 proteins in cancer. Eur. J. Surg. Oncol. (EJSO) 2008, 34, 357–364. [Google Scholar] [CrossRef]
- Leclerc, E. The Roles of S100 Proteins and RAGE in Melanoma. In Breakthroughs in Melanoma Research; IntechOpen: London, UK, 2011; pp. 331–356. [Google Scholar]
- Leclerc, E.; Fritz, G.; Weibel, M.; Heizmann, C.W.; Galichet, A. S100B and S100A6 Differentially Modulate Cell Survival by Interacting with Distinct RAGE (Receptor for Advanced Glycation End Products) Immunoglobulin Domains. J. Biol. Chem. 2007, 282, 31317–31331. [Google Scholar] [CrossRef] [Green Version]
- Gaynor, R.; Irie, R.; Morton, D.; Herschman, H.R. S100 protein is present in cultured human malignant melanomas. Nat. Cell Biol. 1980, 286, 400–401. [Google Scholar] [CrossRef]
- Xiong, T.-F.; Pan, F.-Q.; Li, D. Expression and clinical significance of S100 family genes in patients with melanoma. Melanoma Res. 2019, 29, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Karonidis, A.; Mantzourani, M.; Gogas, H.; Tsoutsos, D. Serum S100B levels correlate with stage, N status, mitotic rate and disease outcome in melanoma patients independent to LDH. J. B. U. ON. Off. J. Balk. Union Oncol. 2017, 22, 1296–1302. [Google Scholar]
- Meghnani, V.; Wagh, A.; Indurthi, V.S.; Koladia, M.; Vetter, S.W.; Law, B.; Leclerc, E. The receptor for advanced glycation end products influences the expression of its S100 protein ligands in melanoma tumors. Int. J. Biochem. Cell Biol. 2014, 57, 54–62. [Google Scholar] [CrossRef]
- Delphin, C.; Ronjat, M.; Deloulme, J.-C.; Garin, G.; Debussche, L.; Higashimoto, Y.; Sakaguchi, K.; Baudier, J. Calcium-dependent interaction of S100B with the C-terminal domain of the tumor suppressor p53. J. Biol. Chem. 1999, 274, 10539–10544. [Google Scholar] [CrossRef] [Green Version]
- Baudier, J.; Delphin, C.; Grunwald, D.; Khochbin, S.; Lawrence, J.J. Characterization of the tumor suppressor protein p53 as a protein kinase C substrate and a S100b-binding protein. Proc. Natl. Acad. Sci. USA 1992, 89, 11627–11631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Blake, M.; Tang, C.; Zimmer, D.; Rustandi, R.R.; Weber, D.J.; Carrier, F. Inhibition of p53 Transcriptional Activity by the S100B Calcium-binding Protein. J. Biol. Chem. 2001, 276, 35037–35041. [Google Scholar] [CrossRef] [Green Version]
- Keijser, S.; Missotten, G.S.; Bonfrer, J.M.; De Wolff-Rouendaal, D.; Jager, M.J.; De Keizer, R.J.W. Immunophenotypic markers to differentiate between benign and malignant melanocytic lesions. Br. J. Ophthalmol. 2006, 90, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Sviatoha, V.; Tani, E.; Kleina, R.; Sperga, M.; Skoog, L. Immunohistochemical analysis of the S100A1, S100B, CD44 and Bcl-2 antigens and the rate of cell proliferation assessed by Ki-67 antibody in benign and malignant melanocytic tumours. Melanoma Res. 2010, 20, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, E. Measuring Binding of S100 Proteins to RAGE by Surface Plasmon Resonance. Adv. Struct. Saf. Stud. 2012, 963, 201–213. [Google Scholar] [CrossRef]
- Jirku, M.; Lansky, Z.; Bednarova, L.; Sulc, M.; Monincova, L.; Majer, P.; Vyklický, L.; Vondrášek, J.; Teisinger, J.; Bousova, K.; et al. The characterization of a novel S100A1 binding site in the N-terminus of TRPM1. Int. J. Biochem. Cell Biol. 2016, 78, 186–193. [Google Scholar] [CrossRef]
- Khan, I.; Yuan, T.; Chou, R.-H.; Yu, C. S100A4 inhibits cell proliferation by interfering with the S100A1-RAGE V domain. PLoS ONE 2019, 14, e0212299. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.; Heizmann, C.W.; Vetter, S.W. RAGE and S100 protein transcription levels are highly variable in human melanoma tumors and cells. Gen. Physiol. Biophys. 2009, 28, 28. [Google Scholar]
- Maelandsmo, G.M.; Flørenes, V.A.; Mellingsaeter, T.; Hovig, E.; Kerbel, R.S.; Fodstad, O. Differential expression patterns of S100a2, S100a4 and S100a6 during progression of human malignant melanoma. Int. J. Cancer 1997, 74, 464–469. [Google Scholar] [CrossRef]
- Nonaka, D.; Chiriboga, L.; Rubin, B.P. Differential expression of S100 protein subtypes in malignant melanoma, and benign and malignant peripheral nerve sheath tumors. J. Cutan. Pathol. 2008, 35, 1014–1019. [Google Scholar] [CrossRef]
- Dahlmann, M.; Okhrimenko, A.; Marcinkowski, P.; Osterland, M.; Herrmann, P.; Smith, J.; Heizmann, C.W.; Schlag, P.M.; Stein, U. RAGE mediates S100A4-induced cell motility via MAPK/ERK and hypoxia signaling and is a prognostic biomarker for human colorectal cancer metastasis. Oncotarget 2014, 5, 3220–3233. [Google Scholar] [CrossRef] [Green Version]
- Herwig, N.; Belter, B.; Wolf, S.; Haase-Kohn, C.; Pietzsch, J. Interaction of extracellular S100A4 with RAGE prompts prometastatic activation of A375 melanoma cells. J. Cell. Mol. Med. 2016, 20, 825–835. [Google Scholar] [CrossRef]
- Ribé, A.; McNutt, N.S.; Rib, A. S100A6 Protein Expression is Different in Spitz Nevi and Melanomas. Mod. Pathol. 2003, 16, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Byström, S.; Fredolini, C.; Edqvist, P.-H.; Nyaiesh, E.-N.; Drobin, K.; Uhlen, M.; Bergqvist, M.; Ponten, F.; Schwenk, J.M. Affinity Proteomics Exploration of Melanoma Identifies Proteins in Serum with Associations to T-Stage and Recurrence. Transl. Oncol. 2017, 10, 385–395. [Google Scholar] [CrossRef]
- Saha, A.; Lee, Y.-C.; Zhang, Z.; Chandra, G.; Su, S.-B.; Mukherjee, A.B. Lack of an Endogenous Anti-inflammatory Protein in Mice Enhances Colonization of B16F10 Melanoma Cells in the Lungs. J. Biol. Chem. 2010, 285, 10822–10831. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, R.; Sato, H.; Yamauchi, A.; Takahashi, Y.; Inoue, Y.; Sumardika, I.W.; Chen, Y.; Tomonobu, N.; Araki, K.; Shien, K.; et al. exSSSRs (extracellular S100 soil sensor receptors)-Fc fusion proteins work as prominent decoys to S100A8/A9-induced lung tropic cancer metastasis. Int. J. Cancer 2019, 144, 3138–3145. [Google Scholar] [CrossRef]
- Wagner, N.B.; Weide, B.; Gries, M.; Reith, M.; Tarnanidis, K.; Schuermans, V.; Kemper, C.; Kehrel, C.; Funder, A.; Lichtenberger, R.; et al. Tumor microenvironment-derived S100A8/A9 is a novel prognostic biomarker for advanced melanoma patients and during immunotherapy with anti-PD-1 antibodies. J. Immunother. Cancer 2019, 7, 343. [Google Scholar] [CrossRef] [PubMed]
- Massi, D.; Landriscina, M.; Piscazzi, A.; Cosci, E.; Kirov, A.; Paglierani, M.; Di Serio, C.; Mourmouras, V.; Fumagalli, S.; Biagioli, M.; et al. S100A13 is a new angiogenic marker in human melanoma. Mod. Pathol. 2010, 23, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landriscina, M.; Soldi, R.; Bagalá, C.; Micucci, I.; Bellum, S.; Tarantini, F.; Prudovsky, I.; Maciag, T. S100A13 participates in the release of fibroblast growth factor 1 in response to heat shock in vitro. J. Biol. Chem. 2001, 276, 22544–22552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, A.; Pernemalm, M.; Stolt, M.F.; Hansson, J.; Lehtiö, J.; Brage, S.E.; Johansson, C.H. Proteomics analysis of melanoma metastases: Association between S100A13 expression and chemotherapy resistance. Br. J. Cancer 2014, 110, 2489–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.-L.; Wang, J.-W.; Guan, Y.; Yin, Y.; Wei, G.; Cui, J.; Zhou, D.; Zhu, Y.-R.; Quan, W.; Xi, M.-M.; et al. Safflor yellow A protects neonatal rat cardiomyocytes against anoxia/reoxygenation injury in vitro. Acta Pharmacol. Sin. 2013, 34, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Ito, T.; Nakahara, T.; Nagae, K.; Fuyuno, Y.; Nakao, M.; Akahoshi, M.; Nakagawa, R.; Tu, Y.; Uchi, H.; et al. Upregulation of S100P, receptor for advanced glycation end products and ezrin in malignant melanoma. J. Dermatol. 2013, 40, 973–979. [Google Scholar] [CrossRef]
- Tímár, J.; Udvarhelyi, N.; Bánfalvi, T.; Gilde, K.; Orosz, Z. Accuracy of the determination of S100B protein expression in malignant melanoma using polyclonal or monoclonal antibodies. Histopathology 2004, 44, 180–184. [Google Scholar] [CrossRef]
- De Lecea, M.; Palomares, T.; Al Kassam, D.; Cavia-Saiz, M.; Geh, J.; De Llano, P.; Muñiz, P.; Armesto, D.; Martinez-Indart, L.; Alonso-Varona, A. Indoleamine 2,3 dioxygenase as a prognostic and follow-up marker in melanoma. A comparative study with LDH and S100B. J. Eur. Acad. Dermatol. Venereol. 2016, 31, 636–642. [Google Scholar] [CrossRef]
- Gebhardt, C.; Lichtenberger, R.; Utikal, J. Biomarker value and pitfalls of serum S100B in the follow-up of high-risk melanoma patients. J. Dtsch. Dermatol. Ges. 2016, 14, 158–164. [Google Scholar] [CrossRef]
- Meghnani, V.; Vetter, S.W.; Leclerc, E. RAGE overexpression confers a metastatic phenotype to the WM115 human primary melanoma cell line. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Carlson, J.A.; Slominski, A.T. Role of TRPM in melanocytes and melanoma. Exp. Dermatol. 2012, 21, 650–654. [Google Scholar] [CrossRef] [Green Version]
- Boye, K.; Maelandsmo, G.M. S100A4 and metastasis: A small actor playing many roles. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Garrett, S.C.; Varney, K.M.; Weber, D.J.; Bresnick, A.R. S100A4, a Mediator of Metastasis. J. Biol. Chem. 2005, 281, 677–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, F.; Qu, J.; Zhang, S.; Li, Y.; Zhang, S. S100A4 in cancer progression and metastasis: A systematic review. Oncotarget 2017, 8, 73219–73239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, N.; Belter, B.; Pietzsch, J. Extracellular S100A4 affects endothelial cell integrity and stimulates transmigration of A375 melanoma cells. Biochem. Biophys. Res. Commun. 2016, 477, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Fullen, D.R.; Reed, J.A.; Finnerty, B.; McNutt, N.S. S100A6 expression in fibrohistiocytic lesions. J. Cutan. Pathol. 2001, 28, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Weterman, M.A.; Stoopen, G.M.; Van Muijen, G.N.; Kuznicki, J.; Ruiter, D.J.; Bloemers, H.P. Expression of calcyclin in human melanoma cell lines correlates with metastatic behavior in nude mice. Cancer Res. 1992, 52, 1291–1296. [Google Scholar]
- Mitamura, Y.; Ito, T.; Nakano-Nakamura, M.; Uchi, H.; Furue, M. S100A6 and c-Kit-Positive Spindle Cell Melanoma of the Dorsal Foot. Case Rep. Dermatol. 2014, 6, 140–144. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Tomonobu, N.; Kinoshita, R.; Sakaguchi, M. S100 Soil Sensor Receptors and Molecular Targeting Therapy Against Them in Cancer Metastasis. Transl. Oncol. 2020, 13, 100753. [Google Scholar] [CrossRef]
- Hibino, T.; Sakaguchi, M.; Miyamoto, S.; Yamamoto, M.; Motoyama, A.; Hosoi, J.; Shimokata, T.; Ito, T.; Tsuboi, R.; Huh, N.-H. S100A9 Is a Novel Ligand of EMMPRIN That Promotes Melanoma Metastasis. Cancer Res. 2013, 73, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruma, I.M.; Putranto, E.W.; Kondo, E.; Murata, H.; Watanabe, M.; Huang, P.; Kinoshita, R.; Futami, J.; Inoue, Y.; Yamauchi, A.; et al. MCAM, as a novel receptor for S100A8/A9, mediates progression of malignant melanoma through prominent activation of NF-κB and ROS formation upon ligand binding. Clin. Exp. Metastasis 2016, 33, 609–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sumardika, I.W.; Tomonobu, N.; Ruma, I.M.W.; Kinoshita, R.; Kondo, E.; Inoue, Y.; Sato, H.; Yamauchi, A.; Murata, H.; et al. Melanoma cell adhesion molecule is the driving force behind the dissemination of melanoma upon S100A8/A9 binding in the original skin lesion. Cancer Lett. 2019, 452, 178–190. [Google Scholar] [CrossRef]
- Jiang, H.; Hu, H.; Tong, X.; Jiang, Q.; Zhu, H.; Zhang, S.-Y. Calcium-binding protein S100P and cancer: Mechanisms and clinical relevance. J. Cancer Res. Clin. Oncol. 2011, 138, 1–9. [Google Scholar] [CrossRef]
- Ilmonen, S.; Vaheri, A.; Asko-Seljavaara, S.; Carpén, O. Ezrin in primary cutaneous melanoma. Mod. Pathol. 2004, 18, 503–510. [Google Scholar] [CrossRef]
- Mäkitie, T.; Carpén, O.; Vaheri, A.; Kivelä, T. Ezrin as a prognostic indicator and its relationship to tumor characteristics in uveal malignant melanoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2442–2449. [Google Scholar]
- Vicentino, A.R.R.; Carneiro, V.C.; Allonso, D.; Guilherme, R.D.F.; Benjamim, C.F.; Dos Santos, H.A.M.; Xavier, F.; Pyrrho, A.D.S.; Gomes, J.D.A.S.; Fonseca, M.D.C.; et al. Emerging Role of HMGB1 in the Pathogenesis of Schistosomiasis Liver Fibrosis. Front. Immunol. 2018, 9, 1979. [Google Scholar] [CrossRef] [Green Version]
- Gerlitz, G.; Hock, R.; Ueda, T.; Bustin, M. The dynamics of HMG protein-chromatin interactions in living cells. Biochem. Cell Biol. 2009, 87, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Travers, A. Priming the nucleosome: A role for HMGB proteins? EMBO Rep. 2003, 4, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A New Group of Chromatin-Associated Proteins with a High Content of Acidic and Basic Amino Acids. JBIC J. Biol. Inorg. Chem. 1973, 38, 14–19. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Iii, H.J.Z.; Lotze, M.T. High-mobility group box 1 and cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1799, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Harris, H.E.; Yang, H.; Tracey, K.J. HMGB1 as a DNA-binding cytokine. J. Leukoc. Biol. 2002, 72, 1084–1091. [Google Scholar] [PubMed]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef]
- Parkkinen, J.; Raulo, E.; Merenmies, J.; Nolo, R.; Kajander, E.O.; Baumann, M.; Rauvala, H. Amphoterin, the 30-kDa protein in a family of HMG1-type polypeptides. Enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J. Biol. Chem. 1993, 268, 19726–19738. [Google Scholar] [PubMed]
- Huttunen, H.J.; Rauvala, H. Amphoterin as an extracellular regulator of cell motility: From discovery to disease. J. Intern. Med. 2004, 255, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Riuzzi, F.; Sorci, G.; Donato, R. The Amphoterin (HMGB1)/Receptor for Advanced Glycation End Products (RAGE) Pair Modulates Myoblast Proliferation, Apoptosis, Adhesiveness, Migration, and Invasiveness. J. Biol. Chem. 2006, 281, 8242–8253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorova, J.; Pasheva, E. High mobility group B1 protein interacts with its receptor RAGE in tumor cells but not in normal tissues. Oncol. Lett. 2011, 3, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Sasahira, T.; Kirita, T.; Oue, N.; Bhawal, U.K.; Yamamoto, K.; Fujii, K.; Ohmori, H.; Luo, Y.; Yasui, W.; Bosserhoff, A.-K.; et al. High mobility group box-1-inducible melanoma inhibitory activity is associated with nodal metastasis and lymphangiogenesis in oral squamous cell carcinoma. Cancer Sci. 2008, 99, 1806–1812. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nat. Cell Biol. 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Huber, R.; Meier, B.; Otsuka, A.; Fenini, G.; Satoh, T.; Gehrke, S.; Widmer, D.; Levesque, M.P.; Mangana, J.; Kerl, K.; et al. Tumour hypoxia promotes melanoma growth and metastasis via High Mobility Group Box-1 and M2-like macrophages. Sci. Rep. 2016, 6, 29914. [Google Scholar] [CrossRef] [Green Version]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The Receptor for Advanced Glycation End Products (RAGE) Is a Cellular Binding Site for Amphoterin: Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xu, Q.; Deng, Y.; Yang, Z.; Xing, S.; Zhao, X.; Zhu, P.; Wang, X.; He, Z.; Gao, Y. High-mobility group box 1 accelerates lipopolysaccharide-induced lung fibroblast proliferation in vitro: Involvement of the NF-κB signaling pathway. Lab. Investig. 2015, 95, 635–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.-C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High Mobility Group 1 Protein (Hmg-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef] [PubMed]
- El Gazzar, M. HMGB1 modulates inflammatory responses in LPS-activated macrophages. Inflamm. Res. 2007, 56, 162–167. [Google Scholar] [CrossRef]
- Fages, C.; Nolo, R.; Huttunen, H.J.; Eskelinen, E.; Rauvala, H. Regulation of cell migration by amphoterin. J. Cell Sci. 2000, 113, 113. [Google Scholar]
- Degryse, B.; Bonaldi, T.; Scaffidi, P.; Müller, S.; Resnati, M.; Sanvito, F.; Arrigoni, G.; Bianchi, M.E. The High Mobility Group (Hmg) Boxes of the Nuclear Protein Hmg1 Induce Chemotaxis and Cytoskeleton Reorganization in Rat Smooth Muscle Cells. J. Cell Biol. 2001, 152, 1197–1206. [Google Scholar] [CrossRef]
- Tang, Q.; Li, J.; Zhu, H.; Li, P.; Zou, Z.; Xiao, Y. Hmgb1-IL-23-IL-17-IL-6-Stat3 Axis Promotes Tumor Growth in Murine Models of Melanoma. Mediat. Inflamm. 2013, 2013, 1–13. [Google Scholar] [CrossRef]
- Zhang, K.; Anumanthan, G.; Scheaffer, S.; Cornelius, L.A. HMGB1/RAGE Mediates UVB-Induced Secretory Inflammatory Response and Resistance to Apoptosis in Human Melanocytes. J. Investig. Dermatol. 2019, 139, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chapman, N.M.; Zhang, B.; Li, M.; Fan, M.; Laribee, R.N.; Zaidi, M.R.; Pfeffer, L.M.; Chi, H.; Wu, Z.-H. Upregulation of PD-L1 via HMGB1-activated IRF3 and NF-kappaB contributes to UV radiation-induced immune suppression. Cancer Res. 2019, 79, 2909–2922. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Detty, S.Q.; Agrawal, D.K. Clinical Implications of High-mobility Group Box-1 (HMGB1) and the Receptor for Advanced Glycation End-products (RAGE) in Cutaneous Malignancy: A Systematic Review. Anticancer Res. 2017, 37, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-H.; Huang, S.-M.; Lin, J.-A.; Yen, G.-C. Inhibition of advanced glycation endproduct formation by foodstuffs. Food Funct. 2011, 2, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautier, M.P.; Guillausseau, P.-J.; Wautier, J.-L. Activation of the receptor for advanced glycation end products and consequences on health. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Zieman, S.J.; Kass, D.A. Advanced glycation endproduct crosslinking in the cardiovascular system: Potential therapeutic target for cardiovascular disease. Drugs 2004, 64, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Vetter, S.W. Glycated Serum Albumin and AGE Receptors. Adv. Clin. Chem. 2015, 72, 205–275. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Shimizu, T.; Sugawara, H.; Watanabe, H.; Nakamura, H.; Choei, H.; Sasaki, N.; Yamagishi, S.-I.; Shimizu, H.; Takeuchi, M. Regulation of Human Melanoma Growth and Metastasis by AGE–AGE Receptor Interactions. J. Investig. Dermatol. 2004, 122, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, S.-I.; Nakamura, K.; Inoue, H.; Kikuchi, S.; Takeuchi, M. Possible participation of advanced glycation end products in the pathogenesis of colorectal cancer in diabetic patients. Med. Hypotheses 2005, 64, 1208–1210. [Google Scholar] [CrossRef]
- Nakamara, N.; Matsui, T.; Ishibashi, Y.; Sotokawauchi, A.; Fukami, K.; Higashimoto, Y.; Yamagishi, S.-I. RAGE-aptamer Attenuates the Growth and Liver Metastasis of Malignant Melanoma in Nude Mice. Mol. Med. 2017, 23, 295–306. [Google Scholar] [CrossRef]
- Ojima, A.; Matsui, T.; Maeda, S.; Takeuchi, M.; Inoue, H.; Higashimoto, Y.; Yamagishi, S.-I. DNA aptamer raised against advanced glycation end products inhibits melanoma growth in nude mice. Lab. Investig. 2014, 94, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Zhu, L.; Zhang, Z.; Li, H.; Li, P.; Wang, Y.; Leng, M. HMGB1-RAGE signaling pathway in pPROM. Taiwan. J. Obstet. Gynecol. 2018, 57, 211–216. [Google Scholar] [CrossRef]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; El-Ashry, D.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Tempering the wrath of RAGE: An emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann. Med. 2009, 41, 408–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Münch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE ligands in cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Essex, A.L.; Davis, H.M. RAGE Signaling in Skeletal Biology. Curr. Osteoporos. Rep. 2019, 17, 16–25. [Google Scholar] [CrossRef]
- Rojas, A.; Morales, M.; Gonzalez, I.; Araya, P. Inhibition of RAGE Axis Signaling: A Pharmacological Challenge. Curr. Drug Targets 2019, 20, 340–346. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [Green Version]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for Advanced Glycation End Products Mediates Inflammation and Enhanced Expression of Tissue Factor in Vasculature of Diabetic Apolipoprotein E–Null Mice. Arter. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, T.; Yan, S.F.; Belov, D.; Rong, L.L.; Sousa, M.; Andrassy, M.; Marso, S.P.; Duda, S.; Arnold, B.; Liliensiek, B.; et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J. Clin. Investig. 2003, 111, 959–972. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.I.; Kalea, A.Z.; Arriero, M.D.M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE Cytoplasmic Domain with Diaphanous-1 Is Required for Ligand-stimulated Cellular Migration through Activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.-I.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.-H. TIRAP, an Adaptor Protein for TLR2/4, Transduces a Signal from RAGE Phosphorylated upon Ligand Binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinie, N.; Gautreau, A. The Arp2/3 regulatory system and its deregulation in cancer. Physiol. Rev. 2018, 98, 215–238. [Google Scholar] [CrossRef]
- Huang, J.-S.; Guh, J.-Y.; Chen, H.-C.; Hung, W.-C.; Lai, Y.-H.; Chuang, L.-Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh, I.H.J. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 2012, 8, 989–991. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Jing, J.; Yu, S.; Song, M.; Tan, H.; Cui, B.; Huang, L. Advanced glycation endproducts induce apoptosis of endothelial progenitor cells by activating receptor RAGE and NADPH oxidase/JNK signaling axis. Am. J. Transl. Res. 2016, 8, 2169–2178. [Google Scholar] [PubMed]
- Taguchi, A.; Blood, D.C.; Del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE–amphoterin signalling suppresses tumour growth and metastases. Nat. Cell Biol. 2000, 405, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Kislinger, T.; Fu, C.; Huber, B.; Qu, W.; Taguchi, A.; Du Yan, S.; Hofmann, M.; Yan, S.F.; Pischetsrieder, M.; Stern, D.; et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef] [Green Version]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the Receptor for Advanced Glycation End Products Triggers a p21ras-dependent Mitogen-activated Protein Kinase Pathway Regulated by Oxidant Stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, R.; Giambanco, I.; Donato, R. S100B/RAGE-dependent activation of microglia via NF-kappa B and AP-1 Co-regulation of COX-2 expression by S100B, IL-1 beta and TNF-alpha. Neurobiol. Aging 2010, 31, 665–677. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Rauvala, H. Receptor for Advanced Glycation End Products (RAGE) Signaling Induces CREB-dependent Chromogranin Expression during Neuronal Differentiation. J. Biol. Chem. 2002, 277, 38635–38646. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Tang, T.; Wang, S.; Wang, Z.; Ma, Y.; Cai, T.; Cheng, X.; Qi, S.; Zhang, Y.; Qi, Z. The molecular mechanisms of Aloin induce gastric cancer cells apoptosis by targeting High Mobility Group Box 1. Drug Des. Dev. Ther. 2019, 13, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33 Pt 5, 891–895. [Google Scholar] [CrossRef]
- Yamazaki, D.; Kurisu, S.; Takenawa, T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene 2009, 28, 1570–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medapati, M.R.; Dahlmann, M.; Ghavami, S.; Pathak, K.A.; Lucman, L.; Klonisch, T.; Hoang-Vu, C.; Stein, U.; Hombach-Klonisch, S. RAGE Mediates the Pro-Migratory Response of Extracellular S100A4 in Human Thyroid Cancer Cells. Thyroid 2015, 25, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Grosse, R. Cell motility through plasma membrane blebbing. J. Cell Biol. 2008, 181, 879–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluch, E.K.; Sykes, C.; Prost, J.; Bornens, M. Dynamic modes of the cortical actomyosin gel during cell locomotion and division. Trends Cell Biol. 2006, 16, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK- and Myosin-Dependent Matrix Deformation Enables Protease-Independent Tumor-Cell Invasion In Vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar] [CrossRef] [Green Version]
- Rouhiainen, A.; Kuja-Panula, J.; Tumova, S.; Rauvala, H. RAGE-Mediated Cell Signaling. Adv. Struct. Saf. Stud. 2012, 963, 239–263. [Google Scholar] [CrossRef]
- Oh, H.-N.; Seo, J.-H.; Lee, M.-H.; Yoon, G.; Cho, S.-S.; Liu, K.; Choi, H.; Oh, K.B.; Cho, Y.-S.; Kim, H.; et al. Oridonin induces apoptosis in oral squamous cell carcinoma probably through the generation of reactive oxygen species and the p38/JNK MAPK pathway. Int. J. Oncol. 2018, 52, 1749–1759. [Google Scholar] [CrossRef] [Green Version]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Dukic-Stefanovic, S.; Gasic-Milenkovic, J.; Deuther-Conrad, W.; Münch, G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs). J. Neurochem. 2003, 87, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Devi, T.S.; Hosoya, K.-I.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Sbai, O.; Devi, T.S.; Melone, M.A.B.; Féron, F.; Khrestchatisky, M.; Singh, L.P.; Perrone, L. RAGE-TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J. Cell Sci. 2010, 123, 4332–4339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Ott, C.; Hörlacher, M.; Weber, D.; Höhn, A.; Grune, T. Advanced-glycation-end-product-induced formation of immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.H.; Keng, Y.T.; Liu, J.F. HMGB-1 induces cell motility and alpha 5 beta 1 integrin expression in human chondrosarcoma cells. Cancer Lett. 2012, 322, 98–106. [Google Scholar] [CrossRef]
- Popa, I.; Ganea, E.; Petrescu, S.M. Expression and subcellular localization of RAGE in melanoma cells. Biochem. Cell Biol. 2014, 92, 127–136. [Google Scholar] [CrossRef]
- Syed, D.N.; Aljohani, A.; Waseem, D.; Mukhtar, H. Ousting RAGE in melanoma: A viable therapeutic target? Semin. Cancer Biol. 2018, 49, 20–28. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.-H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef]
- Ito, N.; Demarco, R.A.; Mailliard, R.B.; Han, J.; Rabinowich, H.; Kalinski, P.; Stolz, N.B.; Zeh, H.J.; Lotze, M.T. Cytolytic cells induce HMGB1 release from melanoma cell lines. J. Leukoc. Biol. 2006, 81, 75–83. [Google Scholar] [CrossRef]
- Treutiger, C.J.; Mullins, G.E.; Johansson, A.-S.M.; Rouhiainen, A.; Rauvala, H.M.E.; Erlandsson-Harris, H.; Andersson, U.; Yang, H.; Tracey, K.J.; Palmblad, J.E.W. High mobility group 1 B-box mediates activation of human endothelium. J. Intern. Med. 2003, 254, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Caveda, L.; Romay, C.; López, E.; Váldes, S.; Padrón, J.; Glaría, L.; Martínez, O.; Delgado, R. Effect of Advanced Glycosylation End Products on the Induction of Nitric Oxide Synthase in Murine Macrophages. Biochem. Biophys. Res. Commun. 1996, 225, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Huang, C.M.; Lin, C.H.; Ho, Y.S.; Chen, C.M.; Lee, H.M. Advanced glycosylation end products induce NF-kappa B dependent NOS expression in RAW 264.7 cells. Mol. Cell. Endocrinol. 2002, 194, 9–17. [Google Scholar] [CrossRef]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Harris, R.A.; Harris, H.E. RAGE is the Major Receptor for the Proinflammatory Activity of HMGB1 in Rodent Macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef]
- Siveen, K.; Kuttan, G. Role of macrophages in tumour progression. Immunol. Lett. 2009, 123, 97–102. [Google Scholar] [CrossRef]
- Wild, C.A.; Bergmann, C.; Fritz, G.; Schuler, P.; Hoffmann, T.K.; Lotfi, R.; Westendorf, A.; Brandau, S.; Lang, S. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int. Immunol. 2012, 24, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Dubois, R.N. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 2015, 36, 1085–1093. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Melanoma | Types and Groups | ||
|---|---|---|---|
| Cutaneous |
| ||
| Extra-cutaneous | Mucosal |
| |
| Ocular | Uveal tract |
| |
| Conjunctiva | |||
| Leptomeningeal |
| ||
| Drug | MOA | OS | Approval Year |
|---|---|---|---|
| Dacarbazine | Alkylating agent | 9.1 months [78] | 1975 |
| Vemurafenib | BRAF V600E inhibitor | 15.9 months [79] | 2011 |
| Vemurafenib + Cobimetinib 1 | BRAF V600E inhibitor MEK inhibitor | 22.5 months [80] | 2020 |
| Ipilimumab | CTL-4 blocking antibody | 19.9 months [81] | 2011 |
| Trametinib | MEK inhibitor | 14.2 months [82] | 2013 |
| Dabrafenib | BRAF V600E inhibitor | 13.1 months [82] | 2017 |
| Dabrafenib + Trametinib 1 | BRAF V600E inhibitor MEK inhibitor | 25.9 months [83] | 2019 |
| Nivolumab | PD-1 antibody | 36.9 months [81] | 2015 |
| Ipilimumab + Nivolumab 1 | CTL-4 blocking antibody PD-1 antibody | 60 months [81] | 2015 |
| Encorafenib + Binimetinib 1 | BRAF V600E or V600K inhibitor MEK inhibitor | 33.6 months [84] | 2018 |
| Pembrolizumab | PD-1 antibody | 32.7 months [85] | 2019 |
| S 100 Protein | Roles and Main Target Proteins in Melanoma | References |
|---|---|---|
| S 100B |
| [145] |
| [62,65] | |
| [60,105,113,146] | |
| [147,148,149] | |
| S 100A1 |
| [150,151] |
| [152,153,154] | |
| S 100A2 |
| [144,155,156,157] |
| S 100A4 |
| [158,159] |
| S 100A6 |
| [142,146,156,160,161] |
| S 100A8/A9 |
| [162,163,164] |
| S 100A13 |
| [144,165,166,167] |
| S 100P |
| [168,169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olaoba, O.T.; Kadasah, S.; Vetter, S.W.; Leclerc, E. RAGE Signaling in Melanoma Tumors. Int. J. Mol. Sci. 2020, 21, 8989. https://doi.org/10.3390/ijms21238989
Olaoba OT, Kadasah S, Vetter SW, Leclerc E. RAGE Signaling in Melanoma Tumors. International Journal of Molecular Sciences. 2020; 21(23):8989. https://doi.org/10.3390/ijms21238989
Chicago/Turabian StyleOlaoba, Olamide T., Sultan Kadasah, Stefan W. Vetter, and Estelle Leclerc. 2020. "RAGE Signaling in Melanoma Tumors" International Journal of Molecular Sciences 21, no. 23: 8989. https://doi.org/10.3390/ijms21238989
APA StyleOlaoba, O. T., Kadasah, S., Vetter, S. W., & Leclerc, E. (2020). RAGE Signaling in Melanoma Tumors. International Journal of Molecular Sciences, 21(23), 8989. https://doi.org/10.3390/ijms21238989