Triangular Relationship between p53, Autophagy, and Chemotherapy Resistance



Abstract
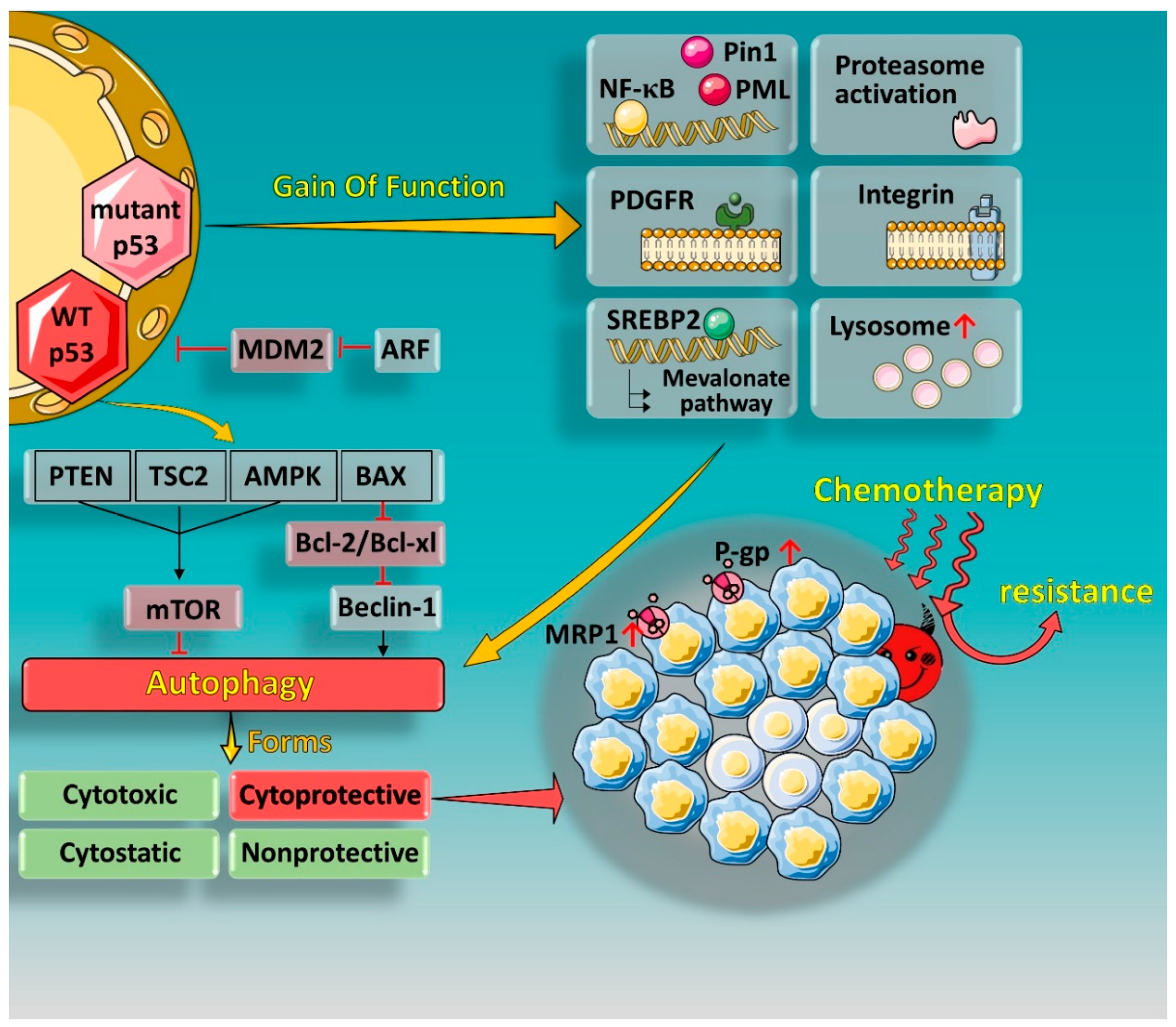
:1. Introduction
1.1. p53 and Drug Resistance
1.2. Autophagy and p53 in Cancer Treatment
1.3. Autophagy and Multidrug Resistance (MDR)
2. Effect of p53 Status on Autophagy and MDR
2.1. Leukemia
2.2. Gastric Cancer
2.3. Pancreatic Cancer
2.4. Colorectal Cancer
2.5. Liver Cancer
2.6. Lung Cancer
2.7. Breast Cancer
3. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GOF | Gain of function |
| MDR | Multidrug resistance |
| TIS | Therapy-induced senescence |
| MDR1 | Multidrug resistance gene 1 |
| wt | Wildtype |
| NSCLC | Non-small cell lung cancer |
| CQ | Chloroquine |
| HCQ | Hydroxychloroquine |
| ATG | Autophagy-related genes |
| MOMP | Mitochondrial outer membrane permeabilization |
| HMGB1 | High mobility group box 1 |
| PTX | Paclitaxel |
| EPI | Epirubicin |
| ABC | ATP-binding cassette |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| CLL | Chronic lymphocytic leukemia |
| MDS | Myelodysplastic syndromes |
| BCP | B-cell precursor acute lymphoblastic leukemia |
| SAHA | Suberoylanilide hydroxamic acid |
| HP | Helicobacter pylori |
| GC | Gastric cancer |
| KLK6 | Kallikrein-related peptidase 6 |
| GA | Gemcitabine hydrochloride and nab-paclitaxel |
| 5-FU | 5-fluorouracil |
| BA | Betulinic acid |
| Act D | Actinomycin D |
| TET2 | Ten-eleven-translocation 2 |
| HCC | Hepatocellular carcinoma |
| HBV | Hepatitis B |
| HCV | Hepatitis C |
| AFB1 | Aflatoxin |
| HBx | X gene of HBV |
| 3-MA | 3-methyladenine |
| SCLC | Small cell lung cancer |
| ALLN | Peptide aldehyde N-acetyl-leu-leu-norleucinal |
| ICI | Fulvestrant or falsodex |
| TAM | Tamoxifen |
| CTL | Cytotoxic T lymphocytes |
| TNBC | Triple negative breast cancer |
References
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [Green Version]
- Bargonetti, J.; Manfredi, J.J. Multiple roles of the tumor suppressor p53. Curr. Opin. Oncol. 2002, 14, 86–91. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikhanskaya, F.; Siddique, M.M.; Kei Lee, M.; Broggini, M.; Sabapathy, K. Evaluation of the combined effect of p53 codon 72 polymorphism and hotspot mutations in response to anticancer drugs. Clin. Cancer Res. 2005, 11, 4348–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P.; et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nau, M.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. p53: A frequent target for genetic abnormalities in lung cancer. Science 1989, 246, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Teoh, W.W.; Phang, B.H.; Tong, W.M.; Wang, Z.Q.; Sabapathy, K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell 2012, 22, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [Green Version]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell Biol. 2019, 11, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Scian, M.J.; Stagliano, K.E.; Anderson, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol. Cell. Biol. 2005, 25, 10097–10110. [Google Scholar] [CrossRef] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.t.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; di Agostino, S.; Mizrahi, I.; Alsheich-Bartok, O.; Voorhoeve, M.; Damalas, A.; Blandino, G.; Haupt, Y. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 2009, 69, 4818–4826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Kojima, K.; Duvvuri, S.; Ruvolo, V.; Samaniego, F.; Younes, A.; Andreeff, M. Decreased sensitivity of 17p-deleted chronic lymphocytic leukemia cells to a small molecule BCL-2 antagonist ABT-737. Cancer 2012, 118, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.T.; Lung, M.L. Mutant p53 expression enhances drug resistance in a hepatocellular carcinoma cell line. Cancer Chemother. Pharm. 2004, 53, 519–526. [Google Scholar] [CrossRef]
- Wong, R.P.; Tsang, W.P.; Chau, P.Y.; Co, N.N.; Tsang, T.Y.; Kwok, T.T. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol. Cancer Ther. 2007, 6, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Lozano, G. Restoring p53 in cancer: The promises and the challenges. J. Mol. Cell Biol. 2019, 11, 615–619. [Google Scholar] [CrossRef]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Wang, Y.; Suh, Y.A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintas-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J. Clin. Investig. 2011, 121, 893–904. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zhang, Y.; El-Naggar, A.K.; Xiong, S.; Yang, P.; Jackson, J.G.; Chau, G.; Lozano, G. Therapeutic efficacy of p53 restoration in Mdm2-overexpressing tumors. Mol. Cancer Res. MCR 2014, 12, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Kondo, S. Autophagy and cancer therapy. Autophagy 2006, 2, 85–90. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Briceno, E.; Reyes, S.; Sotelo, J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg. Focus 2003, 14, e3. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Shingu, T.; Fujiwara, K.; Bogler, O.; Akiyama, Y.; Moritake, K.; Shinojima, N.; Tamada, Y.; Yokoyama, T.; Kondo, S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int. J. Cancer 2009, 124, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.T.; Meng, X.J.; Li, L.L.; Liu, Y.; Li, W.F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 17. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer 2010, 46, 1900–1909. [Google Scholar] [CrossRef]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J.L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014, 21, 92–99. [Google Scholar] [CrossRef]
- Michaud, M.; Xie, X.; Bravo-San Pedro, J.M.; Zitvogel, L.; White, E.; Kroemer, G. An autophagy-dependent anticancer immune response determines the efficacy of melanoma chemotherapy. Oncoimmunology 2014, 3, e944047. [Google Scholar] [CrossRef] [Green Version]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Frohlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. p53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchenko, N.D.; Moll, U.M. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle 2007, 6, 1718–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Marchenko, N.; Erster, S.; Nemajerova, A.; Dehner, A.; Klein, C.; Pan, H.; Kessler, H.; Pancoska, P.; Moll, U.M. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J. Biol. Chem. 2006, 281, 8600–8606. [Google Scholar] [CrossRef] [Green Version]
- Ulloa, L.; Messmer, D. High-mobility group box 1 (HMGB1) protein: Friend and foe. Cytokine Growth Factor Rev. 2006, 17, 189–201. [Google Scholar] [CrossRef]
- Muller, S.; Ronfani, L.; Bianchi, M.E. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J. Intern. Med. 2004, 255, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin. Immunol. 2018, 38, 40–48. [Google Scholar] [CrossRef]
- Ellerman, J.E.; Brown, C.K.; de Vera, M.; Zeh, H.J.; Billiar, T.; Rubartelli, A.; Lotze, M.T. Masquerader: High mobility group box-1 and cancer. Clin. Cancer Res. 2007, 13, 2836–2848. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L.; Tang, D.; Lotze, M.T. Life after death: Targeting high mobility group box 1 in emergent cancer therapies. Am. J. Cancer Res. 2013, 3, 1–20. [Google Scholar]
- Kang, R.; Livesey, K.M.; Zeh, H.J.; Loze, M.T.; Tang, D. HMGB1: A novel Beclin 1-binding protein active in autophagy. Autophagy 2010, 6, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D.; Ni, J. Targeting HMGB1-mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy 2012, 8, 275–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J., 3rd; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Lorin, S.; Pierron, G.; Ryan, K.M.; Codogno, P.; Djavaheri-Mergny, M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy 2010, 6, 153–154. [Google Scholar] [CrossRef] [Green Version]
- Eby, K.G.; Rosenbluth, J.M.; Mays, D.J.; Marshall, C.B.; Barton, C.E.; Sinha, S.; Johnson, K.N.; Tang, L.; Pietenpol, J.A. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol. Cancer 2010, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Gillet, J.P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [CrossRef]
- Ahn, J.H.; Jang, G.H.; Lee, M. Defective autophagy in multidrug resistant cells may lead to growth inhibition by BH3-mimetic gossypol. J. Cell. Physiol. 2013, 228, 1496–1505. [Google Scholar] [CrossRef]
- Sun, W.L.; Lan, D.; Gan, T.Q.; Cai, Z.W. Autophagy facilitates multidrug resistance development through inhibition of apoptosis in breast cancer cells. Neoplasma 2015, 62, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, S.Y.; Hwang, S.H.; Yeom, H.; Lee, M. An ATG5 knockout promotes paclitaxel resistance in v-Ha-ras-transformed NIH 3T3 cells. Biochem. Biophys. Res. Commun. 2019, 513, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef] [PubMed]
- Sirichanchuen, B.; Pengsuparp, T.; Chanvorachote, P. Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol. Cell. Biochem. 2012, 364, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Meschini, S.; Condello, M.; Marra, M.; Formisano, G.; Federici, E.; Arancia, G. Autophagy-mediated chemosensitizing effect of the plant alkaloid voacamine on multidrug resistant cells. Toxicol. Vitr. 2007, 21, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eum, K.H.; Lee, M. Targeting the autophagy pathway using ectopic expression of Beclin 1 in combination with rapamycin in drug-resistant v-Ha-ras-transformed NIH 3T3 cells. Mol. Cells 2011, 31, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Chang, Z.; Wang, W.; Wang, B. Rapamycin-Mediated mTOR Inhibition Reverses Drug Resistance to Adriamycin in Colon Cancer Cells. Hepatogastroenterology 2015, 62, 880–886. [Google Scholar]
- Leisching, G.R.; Loos, B.; Botha, M.H.; Engelbrecht, A.M. The role of mTOR during cisplatin treatment in an in vitro and ex vivo model of cervical cancer. Toxicology 2015, 335, 72–78. [Google Scholar] [CrossRef]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.K.; Li, T.; Law, B.Y.; Ma, E.D.; Yip, N.C.; Michelangeli, F.; Law, C.K.; Zhang, M.M.; Lam, K.Y.; Chan, P.L.; et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis. 2013, 4, e720. [Google Scholar] [CrossRef] [PubMed]
- Law, B.Y.; Mok, S.W.; Chan, W.K.; Xu, S.W.; Wu, A.G.; Yao, X.J.; Wang, J.R.; Liu, L.; Wong, V.K. Hernandezine, a novel AMPK activator induces autophagic cell death in drug-resistant cancers. Oncotarget 2016, 7, 8090–8104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, A.C.; Cutler, D.J. The potential role of lysosomes in tissue distribution of weak bases. Biopharm. Drug Dispos. 1988, 9, 513–526. [Google Scholar] [CrossRef]
- Herlevsen, M.; Oxford, G.; Owens, C.R.; Conaway, M.; Theodorescu, D. Depletion of major vault protein increases doxorubicin sensitivity and nuclear accumulation and disrupts its sequestration in lysosomes. Mol. Cancer Ther. 2007, 6, 1804–1813. [Google Scholar] [CrossRef] [Green Version]
- Hrabeta, J.; Belhajova, M.; Subrtova, H.; Merlos Rodrigo, M.A.; Heger, Z.; Eckschlager, T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. Int. J. Mol. Sci. 2020, 21, 4392. [Google Scholar] [CrossRef]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, A.; Simon, S.M. Subcellular localization and activity of multidrug resistance proteins. Mol. Biol. Cell 2003, 14, 3389–3399. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.S.; Terada, K.; Furukawa, T.; Yang, X.L.; Gao, H.; Miura, N.; Sugiyama, T.; et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316. [Google Scholar]
- Petruzzelli, R.; Polishchuk, R.S. Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs. Cells 2019, 8, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, C.A.; Vargas, L.; Martinez, V.; Bravo, S.; Brown, N.E. Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp. Cell Res. 2017, 360, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Kern, W.; Haferlach, T.; Meggendorfer, M.; Fasan, A.; Haferlach, C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: An analysis of 3307 cases. Leukemia 2017, 31, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Rucker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [Green Version]
- Skah, S.; Richartz, N.; Duthil, E.; Gilljam, K.M.; Bindesboll, C.; Naderi, E.H.; Eriksen, A.B.; Ruud, E.; Dirdal, M.M.; Simonsen, A.; et al. cAMP-mediated autophagy inhibits DNA damage-induced death of leukemia cells independent of p53. Oncotarget 2018, 9, 30434–30449. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.W.; Kim, Y.; Eom, J.I.; Jeung, H.K.; Min, Y.H. Enhanced autophagy in cytarabine arabinoside-resistant U937 leukemia cells and its potential as a target for overcoming resistance. Mol. Med. Rep. 2016, 13, 3433–3440. [Google Scholar] [CrossRef] [Green Version]
- Kanno, S.; Higurashi, A.; Watanabe, Y.; Shouji, A.; Asou, K.; Ishikawa, M. Susceptibility to cytosine arabinoside (Ara-C)-induced cytotoxicity in human leukemia cell lines. Toxicol. Lett. 2004, 152, 149–158. [Google Scholar] [CrossRef]
- Zauli, G.; Celeghini, C.; Melloni, E.; Voltan, R.; Ongari, M.; Tiribelli, M.; di Iasio, M.G.; Lanza, F.; Secchiero, P. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica 2012, 97, 1722–1730. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Amrein, L.; Soulieres, D.; Johnston, J.B.; Aloyz, R. p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Leuk. Res. 2011, 35, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M., Jr.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Corre, S.; Michel, V.; Le Luel, K.; Fernandes, J.; Ziveri, J.; Jouvion, G.; Danckaert, A.; Mouchet, N.; Da Silva Barreira, D.; et al. USF1 defect drives p53 degradation during Helicobacter pylori infection and accelerates gastric carcinogenesis. Gut 2020, 69, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Lee, S.J.; Kim, J.T.; Kim, S.J.; Min, J.K.; Bae, K.H.; Jung, H.; Kim, B.Y.; Lim, J.S.; Yang, Y.; et al. Kallikrein-related peptidase 6 induces chemotherapeutic resistance by attenuating auranofin-induced cell death through activation of autophagy in gastric cancer. Oncotarget 2016, 7, 85332–85348. [Google Scholar] [CrossRef] [Green Version]
- Ke, X.; Qin, Q.; Deng, T.; Liao, Y.; Gao, S.J. Heterogeneous Responses of Gastric Cancer Cell Lines to Tenovin-6 and Synergistic Effect with Chloroquine. Cancers 2020, 12, 365. [Google Scholar] [CrossRef] [Green Version]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Smit, V.T.; Boot, A.J.; Smits, A.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782. [Google Scholar] [CrossRef] [Green Version]
- Redston, M.S.; Caldas, C.; Seymour, A.B.; Hruban, R.H.; da Costa, L.; Yeo, C.J.; Kern, S.E. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994, 54, 3025–3033. [Google Scholar]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar]
- Maurice, D.; Pierreux, C.E.; Howell, M.; Wilentz, R.E.; Owen, M.J.; Hill, C.S. Loss of Smad4 function in pancreatic tumors: C-terminal truncation leads to decreased stability. J. Biol. Chem. 2001, 276, 43175–43181. [Google Scholar] [CrossRef] [Green Version]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lim, D.H.; Jang, K.T.; Lim, T.; Lee, J.; Choi, Y.L.; Jang, H.L.; Yi, J.H.; Baek, K.K.; Park, S.H.; et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol. Cancer Ther. 2011, 10, 1993–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, R.F.; Gordon, E.M.; Anderson, W.F.; Parekh, D. Gene therapy for primary and metastatic pancreatic cancer with intraperitoneal retroviral vector bearing the wild-type p53 gene. Surgery 1998, 124, 143–150, discussion 150-141. [Google Scholar] [CrossRef]
- Kern, S.E.; Pietenpol, J.A.; Thiagalingam, S.; Seymour, A.; Kinzler, K.W.; Vogelstein, B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 1992, 256, 827–830. [Google Scholar] [CrossRef]
- Fujii, S.; Mitsunaga, S.; Yamazaki, M.; Hasebe, T.; Ishii, G.; Kojima, M.; Kinoshita, T.; Ueno, T.; Esumi, H.; Ochiai, A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008, 99, 1813–1819. [Google Scholar] [CrossRef]
- Hashimoto, D.; Blauer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Violette, S.; Poulain, L.; Dussaulx, E.; Pepin, D.; Faussat, A.M.; Chambaz, J.; Lacorte, J.M.; Staedel, C.; Lesuffleur, T. Resistance of colon cancer cells to long-term 5-fluorouracil exposure is correlated to the relative level of Bcl-2 and Bcl-X(L) in addition to Bax and p53 status. Int. J. Cancer 2002, 98, 498–504. [Google Scholar] [CrossRef]
- Benhattar, J.; Cerottini, J.P.; Saraga, E.; Metthez, G.; Givel, J.C. p53 mutations as a possible predictor of response to chemotherapy in metastatic colorectal carcinomas. Int. J. Cancer 1996, 69, 190–192. [Google Scholar] [CrossRef]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef]
- Amin, A.; Bajbouj, K.; Koch, A.; Gandesiri, M.; Schneider-Stock, R. Defective autophagosome formation in p53-null colorectal cancer reinforces crocin-induced apoptosis. Int. J. Mol. Sci. 2015, 16, 1544–1561. [Google Scholar] [CrossRef] [Green Version]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulinic acid, a natural compound with potent anticancer effects. Anti-Cancer Drugs 2010, 21, 215–227. [Google Scholar] [CrossRef]
- Wang, S.; Wang, K.; Zhang, C.; Zhang, W.; Xu, Q.; Wang, Y.; Zhang, Y.; Li, Y.; Zhang, Y.; Zhu, H.; et al. Overaccumulation of p53-mediated autophagy protects against betulinic acid-induced apoptotic cell death in colorectal cancer cells. Cell Death Dis. 2017, 8, e3087. [Google Scholar] [CrossRef] [Green Version]
- Esposito, D.; Crescenzi, E.; Sagar, V.; Loreni, F.; Russo, A.; Russo, G. Human rpL3 plays a crucial role in cell response to nucleolar stress induced by 5-FU and L-OHP. Oncotarget 2014, 5, 11737–11751. [Google Scholar] [CrossRef] [PubMed]
- Pagliara, V.; Saide, A.; Mitidieri, E.; d’Emmanuele di Villa Bianca, R.; Sorrentino, R.; Russo, G.; Russo, A. 5-FU targets rpL3 to induce mitochondrial apoptosis via cystathionine-beta-synthase in colon cancer cells lacking p53. Oncotarget 2016, 7, 50333–50348. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Pagliara, V.; Albano, F.; Esposito, D.; Sagar, V.; Loreni, F.; Irace, C.; Santamaria, R.; Russo, G. Regulatory role of rpL3 in cell response to nucleolar stress induced by Act D in tumor cells lacking functional p53. Cell Cycle 2016, 15, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecoraro, A.; Carotenuto, P.; Franco, B.; De Cegli, R.; Russo, G.; Russo, A. Role of uL3 in the Crosstalk between Nucleolar Stress and Autophagy in Colon Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tan, P.; Guo, L.; Gong, J.; Ma, J.; Li, J.; Lee, M.; Fang, S.; Jing, J.; Johnson, G.; et al. p53-dependent autophagic degradation of TET2 modulates cancer therapeutic resistance. Oncogene 2019, 38, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [Green Version]
- Link, T.; Iwakuma, T. Roles of p53 in extrinsic factor-induced liver carcinogenesis. Hepatoma Res. 2017, 3, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Meng, X.; Franklin, D.A.; Dong, J.; Zhang, Y. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res. 2014, 74, 7161–7167. [Google Scholar] [CrossRef] [Green Version]
- Szymanska, K.; Lesi, O.A.; Kirk, G.D.; Sam, O.; Taniere, P.; Scoazec, J.Y.; Mendy, M.; Friesen, M.D.; Whittle, H.; Montesano, R.; et al. Ser-249TP53 mutation in tumour and plasma DNA of hepatocellular carcinoma patients from a high incidence area in the Gambia, West Africa. Int. J. Cancer 2004, 110, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Kirk, G.D.; Lesi, O.A.; Mendy, M.; Szymanska, K.; Whittle, H.; Goedert, J.J.; Hainaut, P.; Montesano, R. 249(ser) TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinoma. Oncogene 2005, 24, 5858–5867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puisieux, A.; Ji, J.; Guillot, C.; Legros, Y.; Soussi, T.; Isselbacher, K.; Ozturk, M. p53-mediated cellular response to DNA damage in cells with replicative hepatitis B virus. Proc. Natl. Acad. Sci. USA 1995, 92, 1342–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.W.; Gibson, M.K.; Vermeulen, W.; Yeh, H.; Forrester, K.; Sturzbecher, H.W.; Hoeijmakers, J.H.; Harris, C.C. Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res. 1995, 55, 6012–6016. [Google Scholar] [PubMed]
- Elmore, L.W.; Hancock, A.R.; Chang, S.F.; Wang, X.W.; Chang, S.; Callahan, C.P.; Geller, D.A.; Will, H.; Harris, C.C. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 14707–14712. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Abdel-Hafiz, H.; Suhail, M.; Al-Mars, A.; Zakaria, M.K.; Fatima, K.; Ahmad, S.; Azhar, E.; Chaudhary, A.; Qadri, I. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J. Gastroenterol. WJG 2014, 20, 10238–10248. [Google Scholar] [CrossRef]
- Schaeffer, L.; Roy, R.; Humbert, S.; Moncollin, V.; Vermeulen, W.; Hoeijmakers, J.H.; Chambon, P.; Egly, J.M. DNA repair helicase: A component of BTF2 (TFIIH) basic transcription factor. Science 1993, 260, 58–63. [Google Scholar] [CrossRef]
- Kwon, J.A.; Rho, H.M. Transcriptional repression of the human p53 gene by hepatitis B viral core protein (HBc) in human liver cells. Biol. Chem. 2003, 384, 203–212. [Google Scholar] [CrossRef]
- Wang, X.W.; Forrester, K.; Yeh, H.; Feitelson, M.A.; Gu, J.R.; Harris, C.C. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc. Natl. Acad. Sci. USA 1994, 91, 2230–2234. [Google Scholar] [CrossRef] [Green Version]
- Miura, N.; Horikawa, I.; Nishimoto, A.; Ohmura, H.; Ito, H.; Hirohashi, S.; Shay, J.W.; Oshimura, M. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet. Cytogenet. 1997, 93, 56–62. [Google Scholar] [CrossRef]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizui, T.; Yamashina, S.; Tanida, I.; Takei, Y.; Ueno, T.; Sakamoto, N.; Ikejima, K.; Kitamura, T.; Enomoto, N.; Sakai, T.; et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J. Gastroenterol. 2010, 45, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Kairat, A.; Melino, G.; Krammer, P.H.; Stremmel, W.; Oren, M.; Muller, M. Interference with the p53 family network contributes to the gain of oncogenic function of mutant p53 in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2010, 394, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, L.; Li, X.; Li, B.; Li, Y.; Zhang, X.; Ma, Y.; Peng, X.; Jin, H.; Li, H. New insights into autophagy in hepatocellular carcinoma: Mechanisms and therapeutic strategies. Am. J. Cancer Res. 2019, 9, 1329–1353. [Google Scholar]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Qu, Z.; Guo, X.; Zhao, Q.; Zhao, X.; Gao, L.; Sun, K.; Shen, F.; Wu, M.; Wei, L. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 2009, 5, 1131–1144. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Yang, W.; Chen, L.; Shi, M.; Seewoo, V.; Wang, J.; Lin, A.; Liu, Z.; Qiu, W. Role of autophagy in resistance to oxaliplatin in hepatocellular carcinoma cells. Oncol. Rep. 2012, 27, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Yin, X.; Sui, S. Resveratrol inhibited the progression of human hepatocellular carcinoma by inducing autophagy via regulating p53 and the phosphoinositide 3kinase/protein kinase B pathway. Oncol. Rep. 2018, 40, 2758–2765. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xia, B.; Lan, J.; Hu, S.; Huang, L.; Chen, C.; Zeng, X.; Lou, H.; Lin, C.; Pan, W. Design, Synthesis and Anticancer Evaluation of Fangchinoline Derivatives. Molecules 2017, 22, 1923. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Pan, W.; Zhu, M.; Zhang, M.; Hao, X.; Liang, G.; Feng, Y. Fangchinoline induces autophagic cell death via p53/sestrin2/AMPK signalling in human hepatocellular carcinoma cells. Br. J. Pharmacol. 2011, 164, 731–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaper, L.A.; Ofele, K.; Kadyrov, R.; Bechlars, B.; Drees, M.; Cokoja, M.; Herrmann, W.A.; Kuhn, F.E. N-Heterocyclic carbenes via abstraction of ammonia: ‘normal’ carbenes with ‘abnormal’ character. Chem. Commun. 2012, 48, 3857–3859. [Google Scholar] [CrossRef]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Dy, G.K.; Adjei, A.A. Small cell lung cancer. Mayo Clin. Proc. 2008, 83, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo. Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 mutation, and lung cancer. Mol. Cancer Res. MCR 2014, 12, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Ahrendt, S.A.; Hu, Y.; Buta, M.; McDermott, M.P.; Benoit, N.; Yang, S.C.; Wu, L.; Sidransky, D. p53 mutations and survival in stage I non-small-cell lung cancer: Results of a prospective study. J. Natl. Cancer Inst. 2003, 95, 961–970. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Zhou, Y.; Huang, L.; Ou, W.; Wu, J.; Li, S.; Xu, J.; Feng, J.; Liu, B. TP53 mutation is associated with a poor clinical outcome for non-small cell lung cancer: Evidence from a meta-analysis. Mol. Clin. Oncol. 2016, 5, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Li, Y.Y.; Lam, S.K.; Mak, J.C.; Zheng, C.Y.; Ho, J.C. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2013, 81, 354–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusch, V.; Klimstra, D.; Venkatraman, E.; Oliver, J.; Martini, N.; Gralla, R.; Kris, M.; Dmitrovsky, E. Aberrant p53 expression predicts clinical resistance to cisplatin-based chemotherapy in locally advanced non-small cell lung cancer. Cancer Res. 1995, 55, 5038–5042. [Google Scholar] [PubMed]
- Saini, H.; Hakeem, I.; Mukherjee, S.; Chowdhury, S.; Chowdhury, R. Autophagy Regulated by Gain of Function Mutant p53 Enhances Proteasomal Inhibitor-Mediated Cell Death through Induction of ROS and ERK in Lung Cancer Cells. J. Oncol. 2019, 2019, 6164807. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, M.C.; Jing, L.; Liu, Z.Y.; Guo, H.; Liu, Y.; Bai, Y.Y.; Cheng, Y.Z.; Nan, K.J.; Liang, X. Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway. Drug Des. Dev. Ther. 2015, 9, 6421–6431. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.H.; Xu, J.; Saleh, T.; Wu, Y.; Lima, S.; Gewirtz, D.A. Influence of nonprotective autophagy and the autophagic switch on sensitivity to cisplatin in non-small cell lung cancer cells. Biochem. Pharm. 2020, 175, 113896. [Google Scholar] [CrossRef]
- Bertheau, P.; Lehmann-Che, J.; Varna, M.; Dumay, A.; Poirot, B.; Porcher, R.; Turpin, E.; Plassa, L.F.; de Roquancourt, A.; Bourstyn, E.; et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast 2013, 22 (Suppl. S2), S27–S29. [Google Scholar] [CrossRef]
- Schimmelpenning, H.; Eriksson, E.T.; Zetterberg, A.; Auer, G.U. Association of immunohistochemical p53 tumor suppressor gene protein overexpression with prognosis in highly proliferative human mammary adenocarcinomas. World J. Surg. 1994, 18, 827–832. [Google Scholar] [CrossRef]
- Cui, L.; Song, Z.; Liang, B.; Jia, L.; Ma, S.; Liu, X. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol. Rep. 2016, 35, 3639–3647. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Yu, X.; Luo, A.; Liu, Y.; Wang, S.; Li, Y.; Shi, W.; Liu, Z.; Qu, X. MiR-214 increases the sensitivity of breast cancer cells to tamoxifen and fulvestrant through inhibition of autophagy. Mol. Cancer 2015, 14, 208. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Warri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chollat-Namy, M.; Ben Safta-Saadoun, T.; Haferssas, D.; Meurice, G.; Chouaib, S.; Thiery, J. The pharmalogical reactivation of p53 function improves breast tumor cell lysis by granzyme B and NK cells through induction of autophagy. Cell Death Dis. 2019, 10, 695. [Google Scholar] [CrossRef] [PubMed]
- Brenton, J.D.; Carey, L.A.; Ahmed, A.A.; Caldas, C. Molecular classification and molecular forecasting of breast cancer: Ready for clinical application? J. Clin. Oncol. 2005, 23, 7350–7360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Chittaranjan, S.; Bortnik, S.; Dragowska, W.H.; Xu, J.; Abeysundara, N.; Leung, A.; Go, N.E.; DeVorkin, L.; Weppler, S.A.; Gelmon, K.; et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 2014, 20, 3159–3173. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Wang, L.; Zhang, L.; Lu, L.; Shen, J.; Chan, R.L.; Li, M.; Wu, W.K.; To, K.K.; Cho, C.H. Sensitivity of apoptosis-resistant colon cancer cells to tanshinones is mediated by autophagic cell death and p53-independent cytotoxicity. Phytomedicine 2015, 22, 536–544. [Google Scholar] [CrossRef]
- Ding, Z.B.; Hui, B.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Gu, C.Y.; Yang, H.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin. Cancer Res. 2011, 17, 6229–6238. [Google Scholar] [CrossRef] [Green Version]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Tumor | Models/Cells | Drug/Agent | Sensitivity | Autophagy Level | The Sensitivity after Use Autophagy Inhibitor | Autophagy & p53 | Reference |
|---|---|---|---|---|---|---|---|
| AMLs | Patients; HL60, K562, THP1, OCIM3, MOLM13, and NB4 | HCQ | mutp53 < wtp53 | mutp53 > wtp53 (but the status of p53 have no effect on autophagy flux) | N/A | TP53mut AML cells show decreased sensitivity for short-term treatment with HCQ and an impaired upregulation of the apoptotic genes PUMA and BAX, indicating that the initial apoptotic response in these cells is strongly impaired | [105] |
| Primary acute myeloid leukemia blasts and OCI-AML3, MOLM, MV4-11, HL60, or NB4 | Sorafenib | p53-independent | N/A | Cells lack of p53 function, increased sensitivity (online Supplementary Figure S5) | N/A | [109] | |
| BCP-ALL | cytarabine-resistant U937 leukemia cells | Cytarabine | N/A | N/A | Cells lack of p53 function, increased sensitivity | N/A | [106] |
| CML | Ba/F3 p210 and Ba/F3 T315I cells | suberoylanilide hydroxamic acid (SAHA) | p53-independent | N/A | Increased sensitivity, independent of p53 | N/A | [110] |
| CLL | Patients | Dasatinib | mutp53 > wtp53 | mutp53 < wtp53 | mutp53 (non-protect); wtp53 (CQ increase sensitivity; 3-MA or pifithrin same trend increase or no change) | mutp53 CLL lymphocytes are hypersensitive to dasatinib because of the lack of dasatinib-induced p53 dependent autophagy where mutated p53 exerts an inhibitory effect on dasatinib-induced p53-independent autophagy (wtp53 induces autophagy, mutp53 low autophagy) | [111] |
| Gastric cancer | AGS | Palbociclib | p53-independent | p53-independent | p53-independent | N/A | [115] |
| Pancreatic cancer | Patients | Gemcitabine and nab-Paclitaxel | p53-independent | NA | With HCQ, p53-independent | N/A | [129] |
| Colon cancer | SW620 Ad300 cells and SW620 cells; p53+/+ and p53−/− HCT116 cells | Cryptotanshinone (CTS) and dihydrotanshinone (DTS) | Null p53 = wtp53 | Resistance cell > SW620 cells (SW620 cells apoptosis > resistance cell) | Null p53 = wtp53, no change | CTS and DTS induced p53-independent apoptosis and autophagy in colon cancer cells | [198] |
| HCT-116 cell | Crocin (the bioactive molecule of saffron) | Null 53 > wtp53 | N/A | Baf A1 increased the sensitivity of p53 wt HCT-116 cells, no change in p53 null HCT-116 cells | N/A | [138] | |
| HCT-116 cell | Betulinic acid (BA), a naturally occurring pentacyclic triterpene | mutp53 expression enhanced HCT-116 resistance to BA | N/A | CQ and ATG5 siRNA increased the BA-induced sensitivity in a p53-independent manner | N/A | [140] | |
| Liver cancer | Huh-7 (mutp53) and SMMC-7721 (wtp53) | Oxaliplatin | wtp53 < mutp53 | wtp53 = mutp53 | Exposure to CQ or 3-MA significantly increased oxaliplatin-induced cell death in both wtp53 and mutp53 cells; genetic autophagy inhibition also concurred with this increase in cell death in both cell lines when autophagy is knocked down | Oxaliplatin induced p53-independent autophagy in HCC | [199] |
| HepG2 (wtp53) and Huh-7 (mu p53) | Sorafenib | p53-independent | N/A | Autophagy inhibition (BafA1 and 3-MA) reduced sensitivity in sorafenib-resistant HepG2 and Huh-7 cells when compared to parental cells; | N/A | [200] | |
| Lung cancer | H1299 (p53 null cells) transfected with wtp53 or R273H GOF p53 | 5-FU and cisplatin; most studies performed with proteasomal inhibitor, peptide aldehyde N-acetyl-leu-leu-norleucinal (ALLN) | R273H GOF p53 > wtp53 | inhibition of R273H GOF p53 increased autophagy induction | treatment with rapamycin (mTOR inhibitor) or serum starvation (autophagy-inducers) enhanced ALLN-induced cytotoxicity in R273H GOF p53 H1299 cells; furthermore, inhibition of autophagy with CQ did not significantly alter ALLN-induced cell death in R273H GOF p53 H1299 cells | Enhancing autophagy can R273H GOF p53 cells sensitize to ALLN treatment by promoting ROS and ERK signaling | [183] |
| Breast cancer | MDA-MB-231(mutp53-R280K) and DLD1 (mutp53-S241F) | Histone DeACetylases inhibitor, suberoylanilide hydroxamic acid (SAHA) | N/A | MDA-MB-231(mutp53-R280K) > DLD1 (mutp53-S241F) | Autophagy inhibition (BafA1) enhanced cytotoxicity of SAHA in MDA-MB-231 cells | SAHA induced autophagy induction, which promoted degradation of mutp53 in MDA-MB-231 cells but not in DLD1 cells; autophagy inhibition stabilized mutp53 in MDA-MB-231 cells | [196] |
| MDA-MB-231 (mutp53-R280K) and SUM159PT | Epirubicin | N/A | anthracycline-resistant > anthracycline-sensitive cell lines | Pharmacological (CQ or BafA1) and genetic inhibition (siATG5 or siATG7) significantly sensitized both anthracycline-sensitive and anthracycline-resistant cell lines; cytoprotective autophagy induced | N/A | [197] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Patel, N.H.; Gewirtz, D.A. Triangular Relationship between p53, Autophagy, and Chemotherapy Resistance. Int. J. Mol. Sci. 2020, 21, 8991. https://doi.org/10.3390/ijms21238991
Xu J, Patel NH, Gewirtz DA. Triangular Relationship between p53, Autophagy, and Chemotherapy Resistance. International Journal of Molecular Sciences. 2020; 21(23):8991. https://doi.org/10.3390/ijms21238991
Chicago/Turabian StyleXu, Jingwen, Nipa H. Patel, and David A. Gewirtz. 2020. "Triangular Relationship between p53, Autophagy, and Chemotherapy Resistance" International Journal of Molecular Sciences 21, no. 23: 8991. https://doi.org/10.3390/ijms21238991
APA StyleXu, J., Patel, N. H., & Gewirtz, D. A. (2020). Triangular Relationship between p53, Autophagy, and Chemotherapy Resistance. International Journal of Molecular Sciences, 21(23), 8991. https://doi.org/10.3390/ijms21238991