High-Throughput Flow Cytometry Combined with Genetic Analysis Brings New Insights into the Understanding of Chromatin Regulation of Cellular Quiescence


Abstract
:
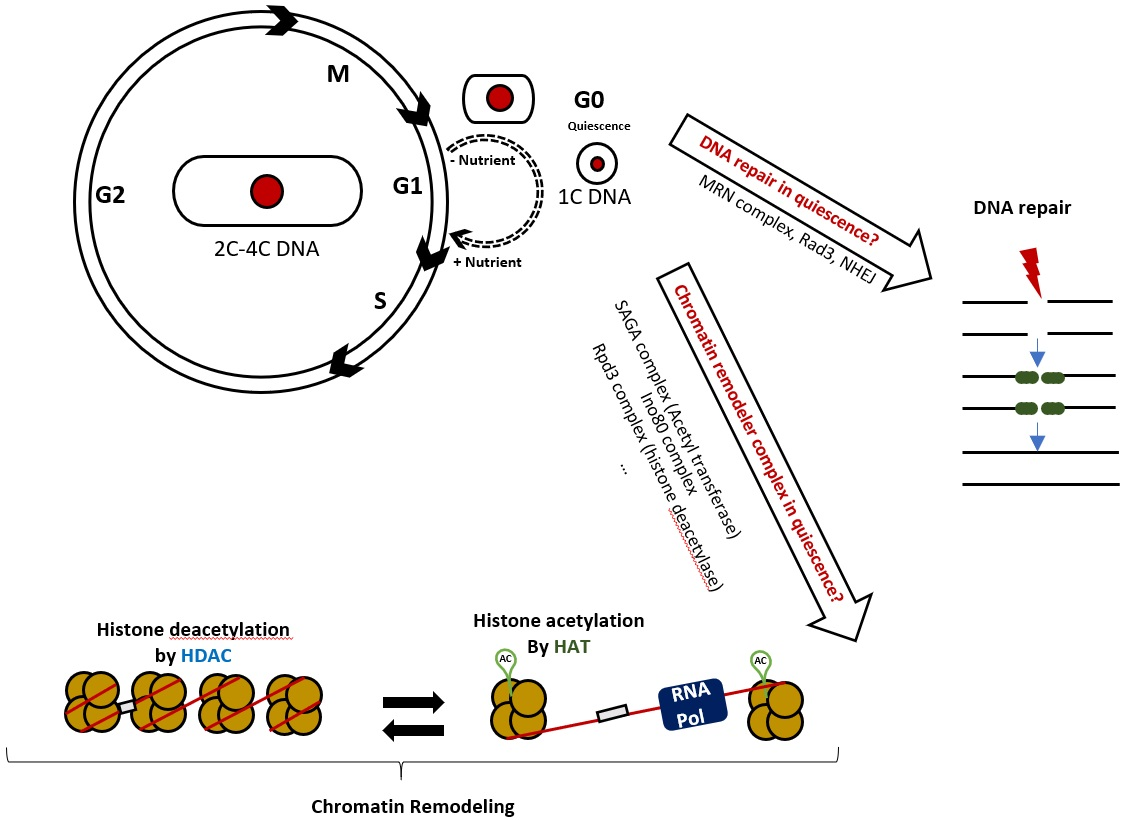{kind=link}
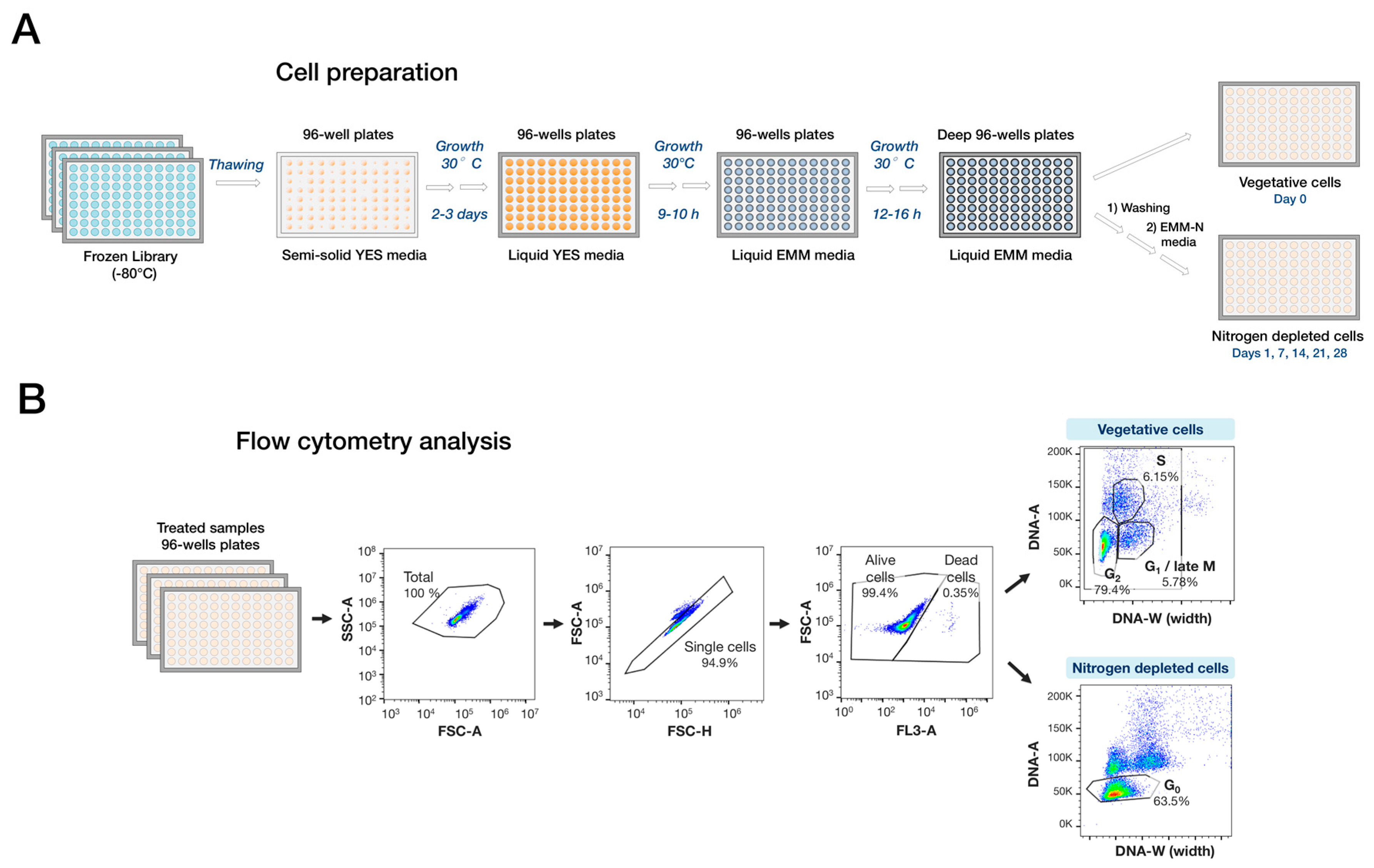{kind=link}
{kind=link}
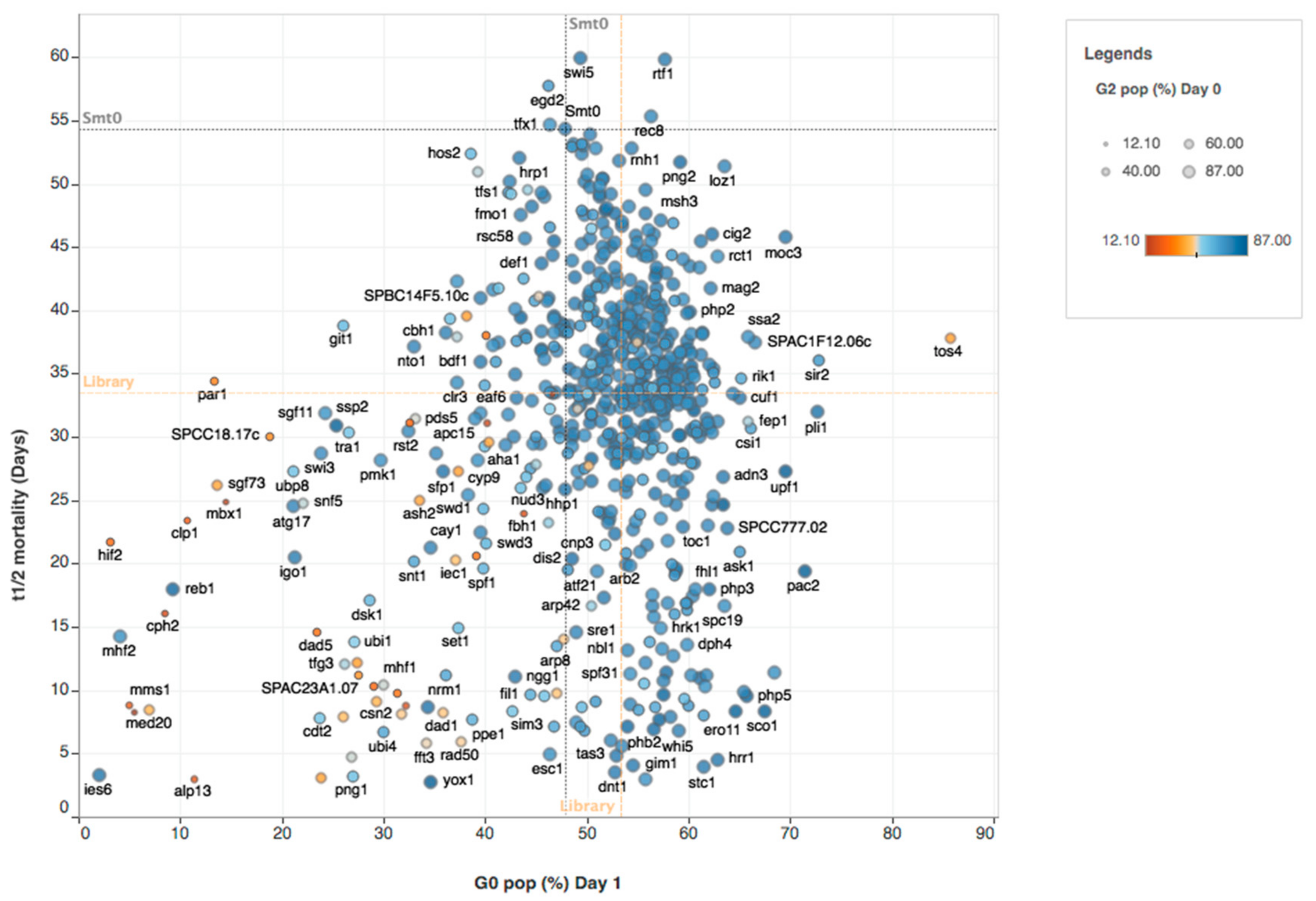{kind=link}
{kind=link}
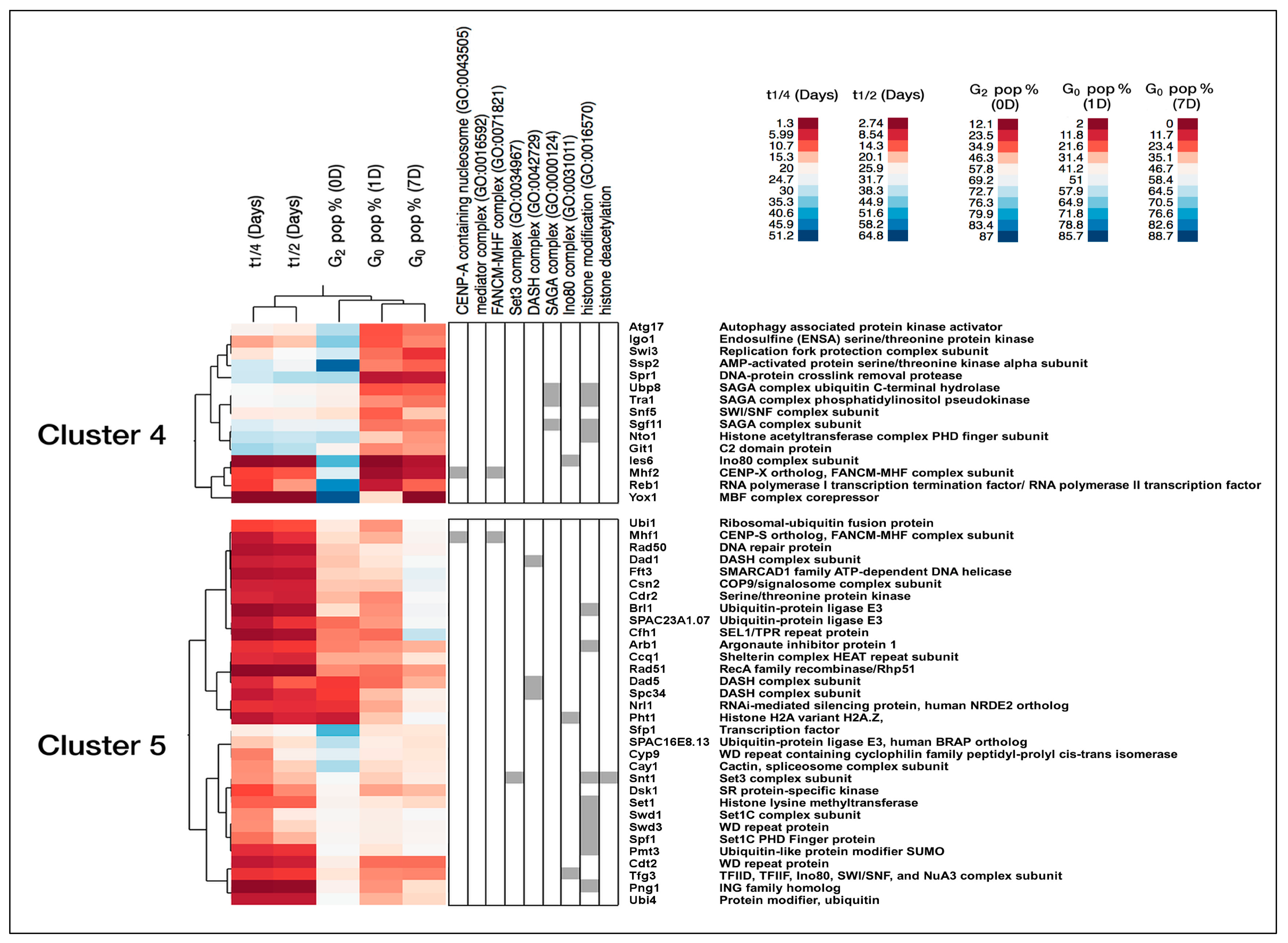{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Rationale of the Experimental Approach
2.2. Hierarchical Clustering of Mutant Phenotype Patterns in Quiescence
2.2.1. Chromatin Factors Required for Entry into G0
2.2.2. Chromatin Factors Required for Viability during Quiescence
2.2.3. A Unique Role for the Histone H3-Encoding Gene hht2
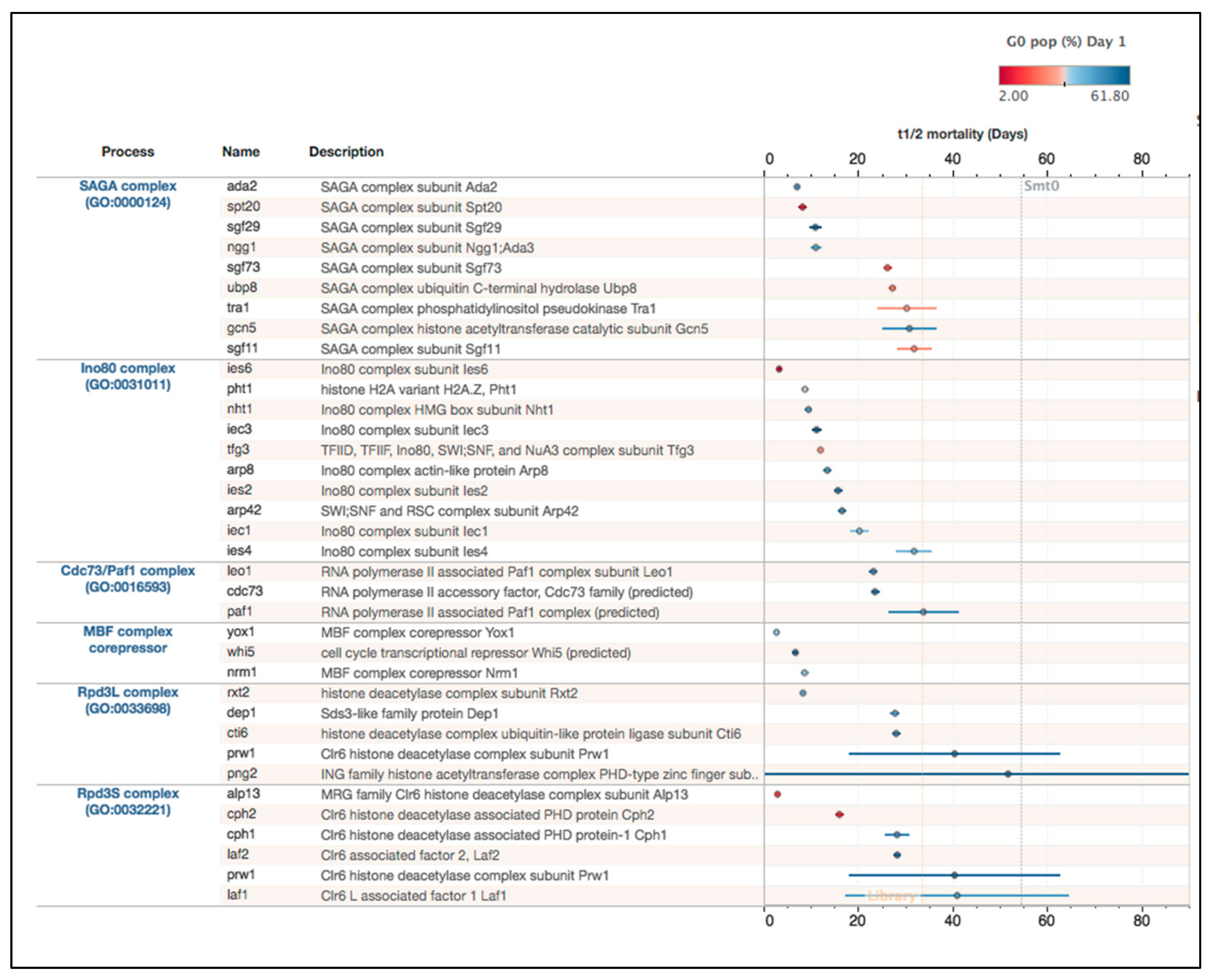
2.3. Chromatin Regulatory Complexes Required for Viability during Quiescence
2.3.1. Validation of Mortality Phenotypes in G0 for Ino80 Mutants
2.3.2. Comparison of Swr1 and Ino80
2.4. DNA Repair Processes Required for Survival in Quiescence
3. Discussion
3.1. Chromatin Regulation during Entry into Quiescence
3.2. Chromatin Factors Essential for Viability during Quiescence
3.3. DNA Repair in G0
4. Materials and Methods
4.1. Preparation of Gene Deletion Library in Prototrophic Strains
4.2. Preparation of Vegetative and Quiescence Cells Samples
4.3. Preparation of Cells for High-Throughput Flow Cytometry
4.4. High-Throughput Flow Cytometry Measurement
4.5. Data Analysis Issued from High-Throughput Flow Cytometry Measurement
4.6. Gene Onthology (GO) Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Su, S.S.; Tanaka, Y.; Samejima, I.; Tanaka, K.; Yanagida, M. A nitrogen starvation-induced dormant G0 state in fission yeast: The establishment from uncommitted G1 state and its delay for return to proliferation. J. Cell Sci. 1996, 109 Pt 6, 109. [Google Scholar]
- Shimanuki, M.; Chung, S.-Y.; Chikashige, Y.; Kawasaki, Y.; Uehara, L.; Tsutsumi, C.; Hatanaka, M.; Hiraoka, Y.; Nagao, K.; Yanagida, M. Two-step, extensive alterations in the transcriptome from G0 arrest to cell division in Schizosaccharomyces pombe. Genes Cells 2007, 12, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Sajiki, K.; Hatanaka, M.; Nakamura, T.; Takeda, K.; Shimanuki, M.; Yoshida, T.; Hanyu, Y.; Hayashi, T.; Nakaseko, Y.; Yanagida, M. Genetic control of cellular quiescence in S. pombe. J. Cell Sci. 2009, 122, 1418–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Yanagida, M. In quiescence of fission yeast, autophagy and the proteasome collaborate for mitochondrial maintenance and longevity. Autophagy 2010, 6, 564–565. [Google Scholar] [CrossRef] [PubMed]
- Marguerat, S.; Schmidt, A.; Codlin, S.; Chen, W.; Aebersold, R.; Bähler, J. Quantitative Analysis of Fission Yeast Transcriptomes and Proteomes in Proliferating and Quiescent Cells. Cell 2012, 151, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sideri, T.; Rallis, C.; Bitton, D.A.; Lages, B.M.; Suo, F.; Rodríguez-López, M.; Du, L.-L.; Bähler, J. Parallel Profiling of Fission Yeast Deletion Mutants for Proliferation and for Lifespan During Long-Term Quiescence. G3 Genes Genomes Genet. 2014, 5, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, B.; Arcangioli, B.; Martienssen, R.A. RNA interference is essential for cellular quiescence. Science 2016, 354, 354. [Google Scholar] [CrossRef] [Green Version]
- Joh, R.I.; Khanduja, J.S.; Calvo, I.A.; Mistry, M.; Palmieri, C.M.; Savol, A.J.; Ho Sui, S.J.; Sadreyev, R.I.; Aryee, M.J.; Motamedi, M. Survival in quiescence requires the euchromatic deployment of clr4/suv39h by argonaute-associated small rnas. Mol. Cell 2016, 64, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Oya, E.; Durand-Dubief, M.; Cohen, A.; Maksimov, V.; Schurra, C.; Nakayama, J.-I.; Weisman, R.; Arcangioli, B.; Ekwall, K. Leo1 is essential for the dynamic regulation of heterochromatin and gene expression during cellular quiescence. Epigenetics Chromatin 2019, 12, 45. [Google Scholar] [CrossRef]
- Weaver, J.L. Introduction to Flow Cytometry. Methods 2000, 21, 199–201. [Google Scholar] [CrossRef]
- Knutsen, J.H.J.; Rein, I.D.; Rothe, C.; Stokke, T.; Grallert, B.; Boye, E. Cell-Cycle Analysis of Fission Yeast Cells by Flow Cytometry. PLoS ONE 2011, 6, e17175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, J.N.; Boerma, J.W.; Breeden, L.L.; Tsukiyama, T. Global Promoter Targeting of a Conserved Lysine Deacetylase for Transcriptional Shutoff during Quiescence Entry. Mol. Cell 2015, 59, 732–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-U.; Hayles, J.; Kim, D.; Wood, V.; Park, H.-O.; Won, M.; Yoo, H.-S.; Duhig, T.; Nam, M.; Palmer, G.; et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2010, 28, 617–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestroni, L.; Reyes, C.; Vaurs, M.; Gachet, Y.; Tournier, S.; Geli, V.; Coulon, S. Nuclear envelope attachment of telomeres limits TERRA and telomeric rearrangements in quiescent fission yeast cells. Nucleic Acids Res. 2020, 48, 3029–3041. [Google Scholar] [CrossRef]
- Cheon, Y.; Kim, H.; Park, K.; Kim, M.; Lee, D. Dynamic modules of the coactivator SAGA in eukaryotic transcription. Exp. Mol. Med. 2020, 52, 991–1003. [Google Scholar] [CrossRef]
- Shilatifard, A. The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef] [Green Version]
- Helmlinger, D.; Marguerat, S.; Villén, J.; Gygi, S.P.; Bähler, J.; Winston, F. The S. pombe SAGA complex controls the switch from proliferation to sexual differentiation through the opposing roles of its subunits Gcn5 and Spt8. Genes Dev. 2008, 22, 3184–3195. [Google Scholar] [CrossRef] [Green Version]
- Helmlinger, D.; Marguerat, S.; Villén, J.; Swaney, D.L.; Gygi, S.P.; Bähler, J.; Winston, F. Tra1 has specific regulatory roles, rather than global functions, within the SAGA co-activator complex. EMBO J. 2011, 30, 2843–2852. [Google Scholar] [CrossRef]
- Franco, A.; Meadows, J.C.; Millar, J.B. The dam1/dash complex is required for the retrieval of unclustered kinetochores in fission yeast. J. Cell Sci. 2007, 120, 3345–3351. [Google Scholar] [CrossRef] [Green Version]
- Steglich, B.; Strålfors, A.; Khorosjutina, O.; Persson, J.; Smialowska, A.; Javerzat, J.-P.; Ekwall, K. The Fun30 Chromatin Remodeler Fft3 Controls Nuclear Organization and Chromatin Structure of Insulators and Subtelomeres in Fission Yeast. PLoS Genet. 2015, 11, e1005101. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Yoshida, T.; Kikuchi, S.; Nagao, K.; Kokubu, A.; Pluskal, T.; Villar-Briones, A.; Nakamura, T.; Yanagida, M. Synergistic roles of the proteasome and autophagy for mitochondrial maintenance and chronological lifespan in fission yeast. Proc. Natl. Acad. Sci. USA 2010, 107, 3540–3545. [Google Scholar] [CrossRef] [Green Version]
- Sajiki, K.; Pluskal, T.; Shimanuki, M.; Yanagida, M. Metabolomic Analysis of Fission Yeast at the Onset of Nitrogen Starvation. Metabolites 2013, 3, 1118–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, S.; Breeden, L.L. A common strategy for initiating the transition from proliferation to quiescence. Curr. Genet. 2016, 63, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aligianni, S.; Lackner, D.H.; Klier, S.; Rustici, G.; Wilhelm, B.T.; Marguerat, S.; Codlin, S.; Brazma, A.; De Bruin, R.A.M.; Bähler, J. The Fission Yeast Homeodomain Protein Yox1p Binds to MBF and Confines MBF-Dependent Cell-Cycle Transcription to G1-S via Negative Feedback. PLoS Genet. 2009, 5, e1000626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eustermann, S.; Schall, K.; Kostrewa, D.; Lakomek, K.; Strauss, M.; Moldt, M.; Hopfner, K.-P. Structural basis for ATP-dependent chromatin remodelling by the INO80 complex. Nat. Cell Biol. 2018, 556, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Brahma, S.; Udugama, M.I.; Kim, J.; Hada, A.; Bhardwaj, S.K.; Hailu, S.G.; Lee, T.-H.; Bartholomew, B. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nat. Commun. 2017, 8, 15616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, C.J.; Aligianni, S.; Durand-Dubief, M.; Persson, J.; Will, W.R.; Webster, J.; Wheeler, L.; Mathews, C.K.; Elderkin, S.; Oxley, D.; et al. Fission Yeast Iec1-Ino80-Mediated Nucleosome Eviction Regulates Nucleotide and Phosphate Metabolism. Mol. Cell. Biol. 2009, 30, 657–674. [Google Scholar] [CrossRef] [Green Version]
- Knezevic, I.; Medina, A.G.; Gaspa, L.; Hidalgo, E.; Ayté, J. The INO80 complex activates the transcription of S-phase genes in a cell cycle-regulated manner. FEBS J. 2018, 285, 3870–3881. [Google Scholar] [CrossRef]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160290. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.P.; Shukla, M.; White, S.A.; Lafos, M.; Tong, P.; Auchynnikava, T.; Spanos, C.; Rappsilber, J.; Pidoux, A.L.; Allshire, R.C. Hap2–Ino80-facilitated transcription promotes de novo establishment of CENP-A chromatin. Genes Dev. 2020, 34, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Mellone, B.G.; Ball, L.; Suka, N.; Grunstein, M.R.; Partridge, J.F.; Allshire, R.C. Centromere Silencing and Function in Fission Yeast Is Governed by the Amino Terminus of Histone H3. Curr. Biol. 2003, 13, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, V.R. The specificity of H2A.Z occupancy in the yeast genome and its relationship to transcription. Curr. Genet. 2020, 66, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, L.; Durand-Dubief, M.; Roguev, A.; Sakalar, C.; Wilhelm, B.; Strålfors, A.; Shevchenko, A.; Aasland, R.; Shevchenko, A.; Ekwall, K.; et al. The Schizosaccharomyces pombe JmjC-Protein, Msc1, Prevents H2A.Z Localization in Centromeric and Subtelomeric Chromatin Domains. PLoS Genet. 2009, 5, e1000726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, S.; Yanagida, M. Distinct modes of DNA damage response in S. pombe G0 and vegetative cells. Genes Cells 2006, 11, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, S.; Achaz, G.; Francesconi, S.; Villain, A.; Miled, S.; Denis, C.; Arcangioli, B. Quiescence unveils a novel mutational force in fission yeast. eLife 2017, 6, e27469. [Google Scholar] [CrossRef]
- Ben Hassine, S.; Arcangioli, B. Tdp1 protects against oxidative DNA damage in non-dividing fission yeast. EMBO J. 2009, 28, 632–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Paull, T.T. Direct activation of the atm protein kinase by the mre11/rad50/nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- Ekwall, K.; Thon, G. Mating-Type Determination in Schizosaccharomyces pombe. Cold Spring Harb. Protoc. 2017, 2017, 091728. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahedi, Y.; Durand-Dubief, M.; Ekwall, K. High-Throughput Flow Cytometry Combined with Genetic Analysis Brings New Insights into the Understanding of Chromatin Regulation of Cellular Quiescence. Int. J. Mol. Sci. 2020, 21, 9022. https://doi.org/10.3390/ijms21239022
Zahedi Y, Durand-Dubief M, Ekwall K. High-Throughput Flow Cytometry Combined with Genetic Analysis Brings New Insights into the Understanding of Chromatin Regulation of Cellular Quiescence. International Journal of Molecular Sciences. 2020; 21(23):9022. https://doi.org/10.3390/ijms21239022
Chicago/Turabian StyleZahedi, Yasaman, Mickael Durand-Dubief, and Karl Ekwall. 2020. "High-Throughput Flow Cytometry Combined with Genetic Analysis Brings New Insights into the Understanding of Chromatin Regulation of Cellular Quiescence" International Journal of Molecular Sciences 21, no. 23: 9022. https://doi.org/10.3390/ijms21239022
APA StyleZahedi, Y., Durand-Dubief, M., & Ekwall, K. (2020). High-Throughput Flow Cytometry Combined with Genetic Analysis Brings New Insights into the Understanding of Chromatin Regulation of Cellular Quiescence. International Journal of Molecular Sciences, 21(23), 9022. https://doi.org/10.3390/ijms21239022