The Pathophysiological Role of CoA

 , , , , and
, , , , and
Abstract
:1. Introduction
2. CoA Synthesis and Degradation in Mammalian Cells
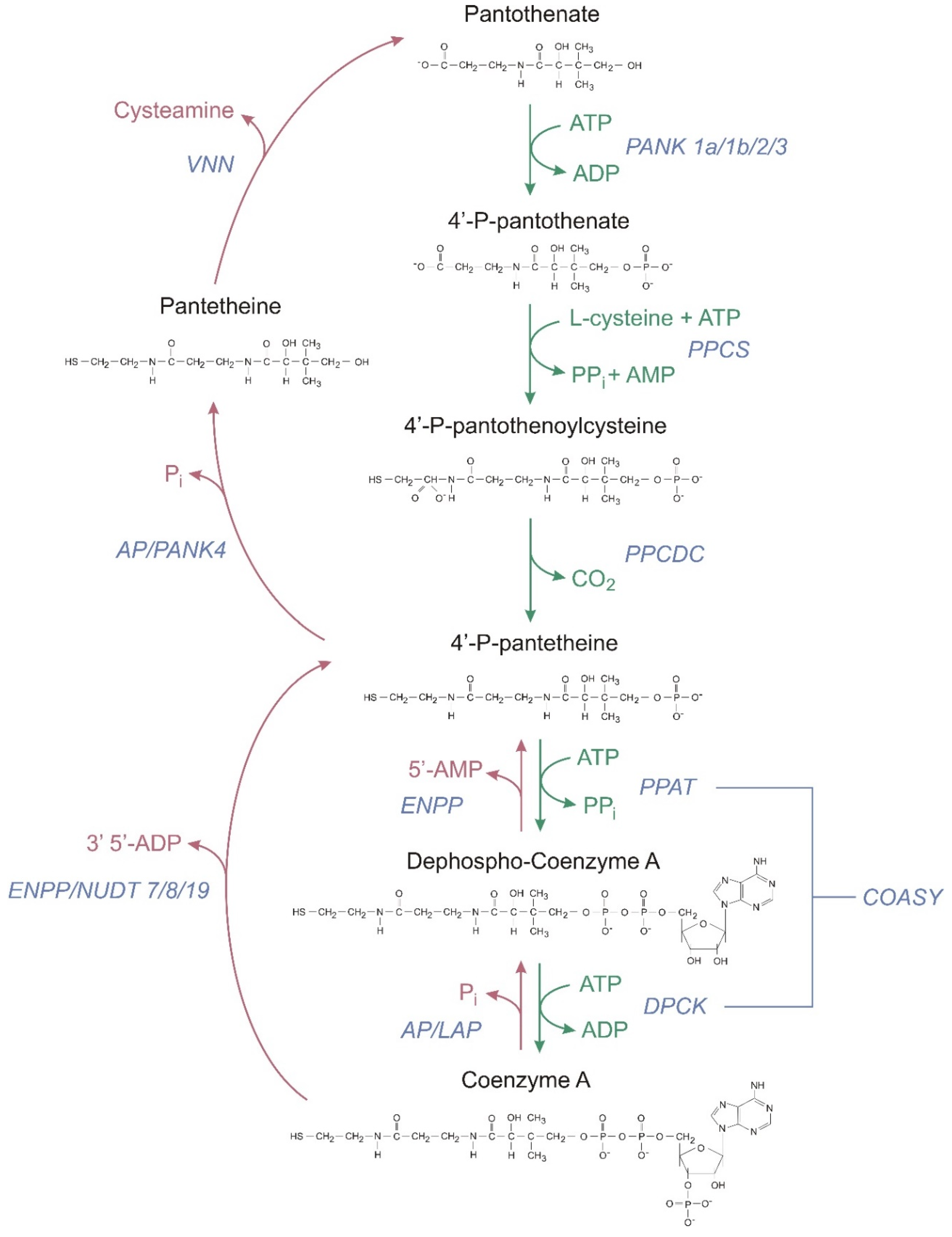
2.1. CoA Synthesis
2.2. CoA Degradation
2.2.1. Extracellular Degradation of CoA (Known as Intestinal or Systemic)
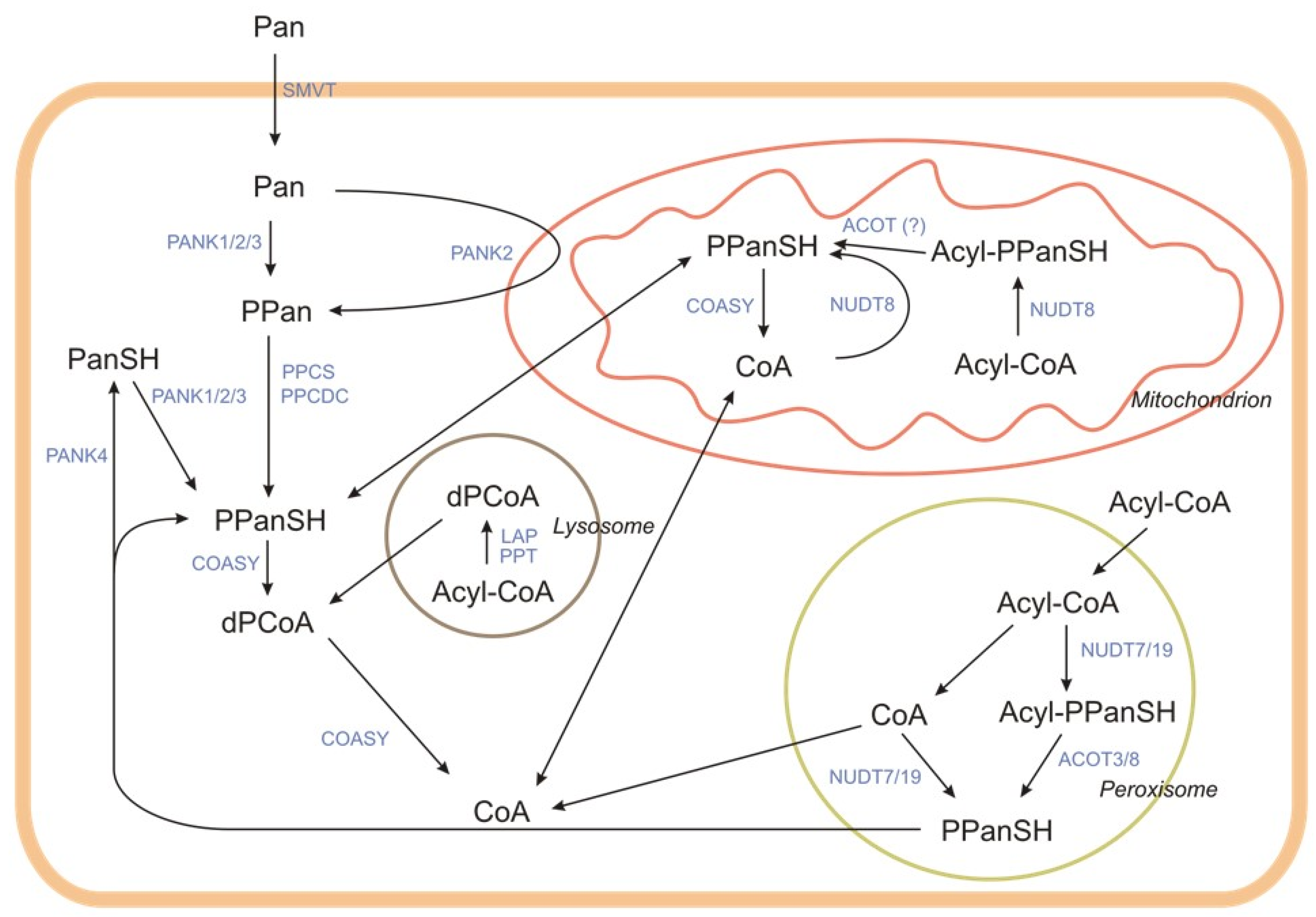
2.2.2. Intracellular Degradation of CoA
3. Tissue Levels and Intracellular Distribution
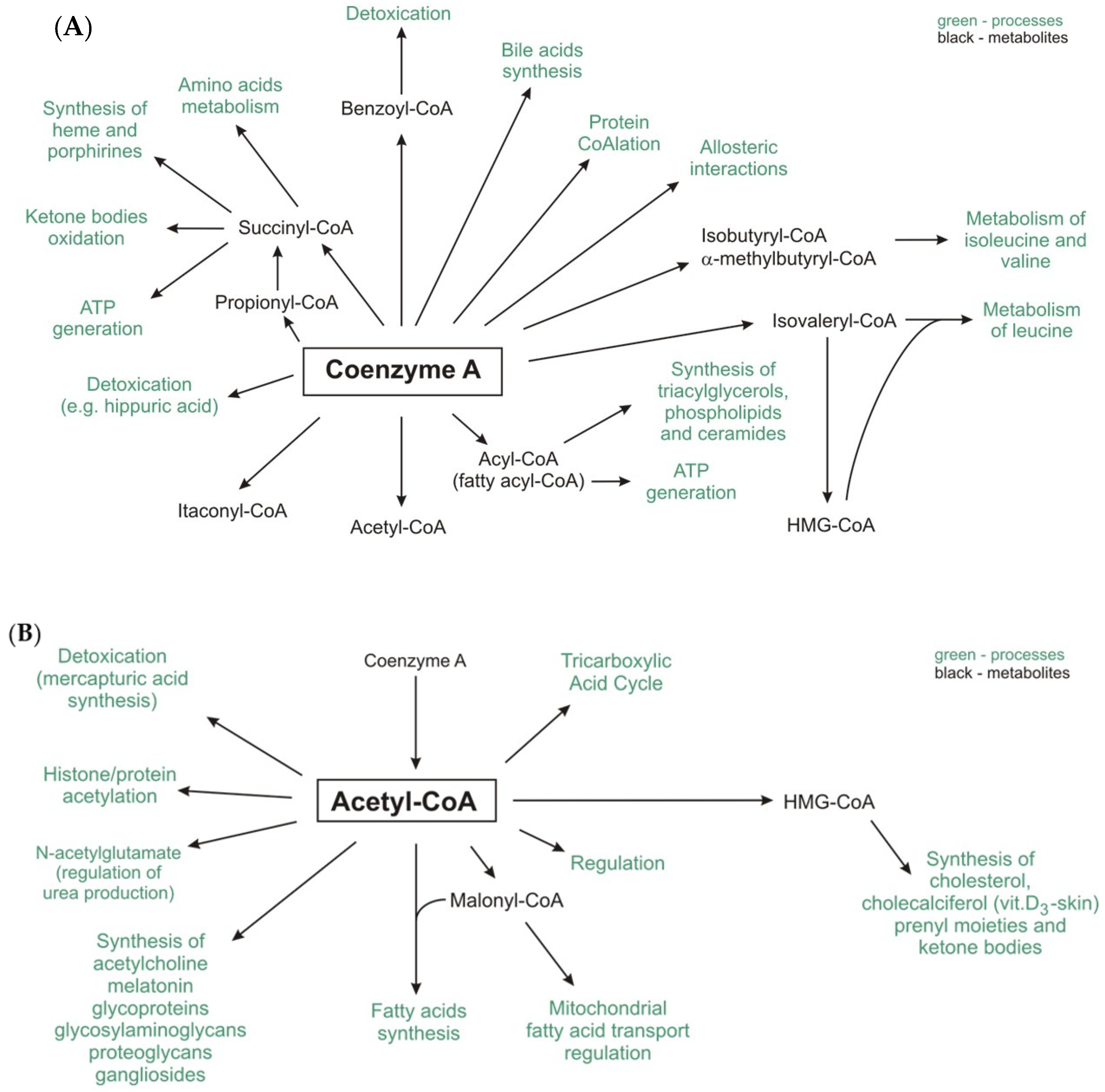
4. Protein CoAlation and Other Protein Modifications Related to CoA
5. CoA and Pathologies
5.1. Neurodegenerative Diseases
5.2. Cancers
5.3. Colitis
5.4. Myopathies
5.5. Infectious Diseases
5.6. Diabetes
5.7. Other Diseases
6. Single-Nucleotide Polymorphisms in Genes Involved in CoA Metabolism
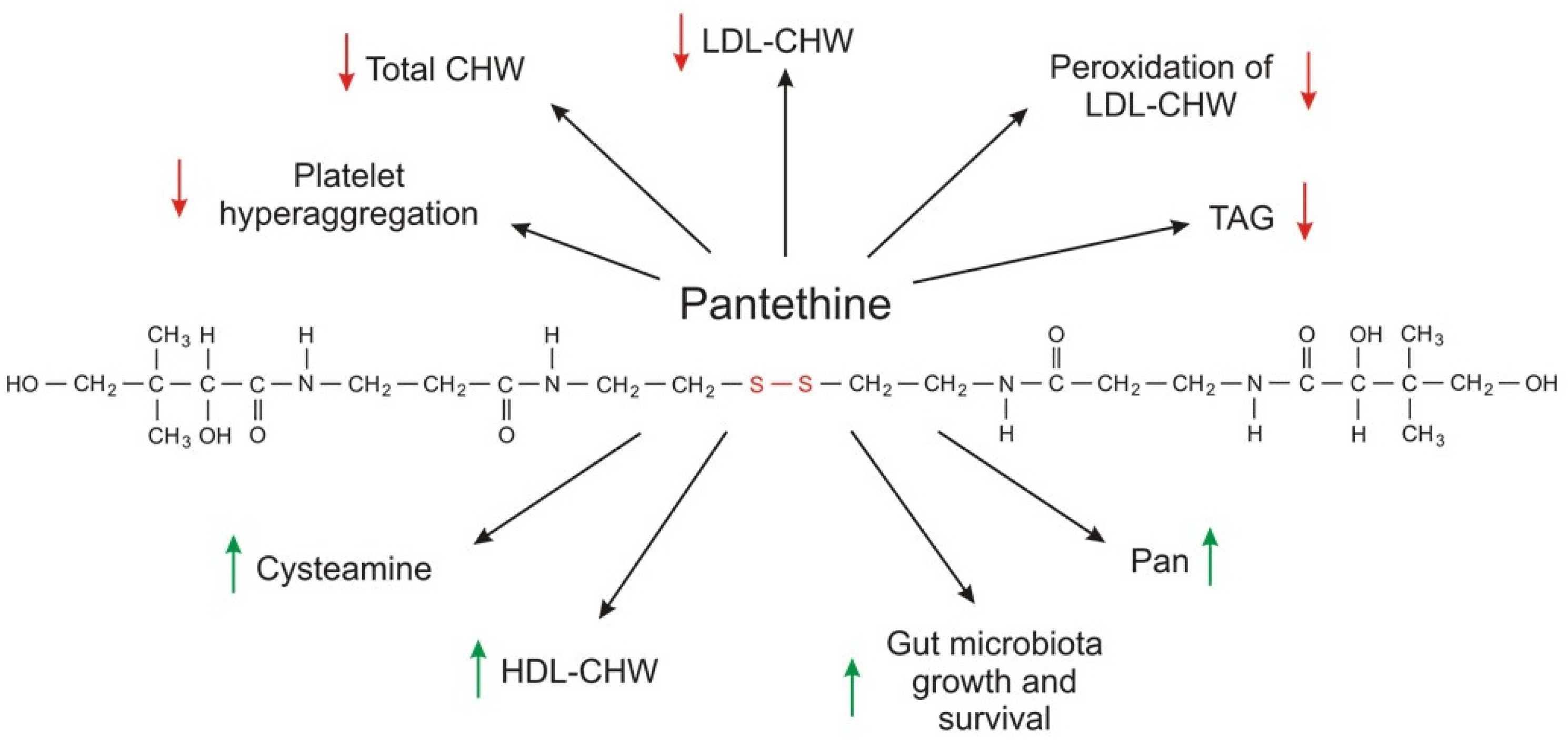
7. CoA and Its Precursor Pantethine as Circulating Lipid-Lowering Supplemental Agents
8. Conclusions and Further Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCD | ATP-binding cassette subfamily D |
| ACAT | Acyl-CoA:cholesterol acyltransferase |
| ACBP | acyl-CoA binding protein |
| ACOT | Acyl-CoA thioesterase |
| ACLY | ATP-citrate lyase |
| ACP | Acyl carrier protein |
| ACS | Acyl-CoA synthetase |
| AD | Alzheimer’s Disease |
| AML | Acute myeloid leukemia |
| AP | Alkaline phosphate |
| apoA | Apolipiprotein A |
| apoB | Apolipoprotein B |
| BMI | Body mass index |
| CDK8 | Cyclin dependent kinase 8 |
| CoA | Coenzyme A |
| COASY | Coenzyme A synthase |
| CoPAN | CoA synthase protein-associated neurodegeneration |
| CPT1 | Carnitine palmitoyltransferase 1 |
| CPT2 | Carnitine palmitoyltransferase 2 |
| DPCK | Dephospho-CoA kinase |
| dPCoA | Dephospho-CoA |
| ELOVL | Fatty acid elongase |
| ENPP | Ectonucleotide pyrophosphatases/phosphodiesterases |
| ER | Estrogen receptor |
| FASN | Fatty acid synthase |
| GSH | Glutathione |
| GWAS | Genome-wide association studies |
| HD | Huntington’s Disease |
| HER2 | Human epidermal growth factor receptor |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-CoA |
| HMGR | 3-hydroxy-3-methylglutaryl-CoA reductase |
| IBD | Inflammatory bowel disease |
| IGFBP3 | Insulin-like growth factor-binding protein |
| IMM | Inner mitochondrial membrane |
| LAP | Lysosomal acid phosphatase |
| NSCLC | Non-small cell lung cancer |
| NUDT | Nucleoside diphosphate linked moiety X-type motif |
| OMM | Outer mitochondrial membrane |
| Pan | Pantothenate, pantothenic acid |
| PANK | Pantothenate kinase |
| PCH | Pontocerebellar hypoplasia |
| PDC | Pyruvate dehydrogenase complex |
| PDK | Pyruvate dehydrogenase kinase |
| PKAN | Pantothenate Kinase Associated Neurodegeneration |
| PPan | 4′phosphopantothenate |
| PPanSH | 4′-phosphopantetheine |
| PPCDC | Phosphopantothenoylcysteine decarboxylase |
| PPCS | Phosphopantothenoylcysteine synthetase |
| PPT | Palmitoyl-protein thioesterase |
| PR | Progesterone receptor |
| ROS | Reactive oxygen species |
| SLC25A42 | Solute Carrier Family 25 Member 42 |
| SMVT | Sodium dependent multivitamin transporter |
| SNP | Single nucleotide polymorphisms |
| VNN | Pantetheinase |
References
- Lykstad, J.; Sharma, S. Biochemistry, Water Soluble Vitamins; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Miller, J.W.; Rucker, R.B. Pantothenic Acid. Present Knowledge in Nutrition, 10th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2012; pp. 375–390. [Google Scholar]
- Gheita, A.A.; Gheita, T.A.; Kenawy, S.A. The potential role of B5: A stitch in time and switch in cytokine. Phyther. Res. 2020, 34, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Patassini, S.; Begley, P.; Xu, J.; Church, S.J.; Kureishy, N.; Reid, S.J.; Waldvogel, H.J.; Faull, R.L.M.; Snell, R.G.; Unwin, R.D.; et al. Cerebral Vitamin B5 (D-Pantothenic Acid) Deficiency as a Potential Cause of Metabolic Perturbation and Neurodegeneration in Huntington’s Disease. Metabolites 2019, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yale, K.; Juhasz, M.; Mesinkovska, N.A. Medication-Induced Repigmentation of Gray Hair: A Systematic Review. Ski. Appendage Disord. 2020, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Kureishy, N.; Church, S.J.; Scholefield, M.; Unwin, R.D.; Xu, J.; Patassini, S.; Cooper, G.J.S. Vitamin B5 (D-pantothenic acid) localizes in myelinated structures of the rat brain: Potential role for cerebral vitamin B5 stores in local myelin homeostasis. Biochem. Biophys. Res. Commun. 2020, 522, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, N.D.; Mcleod, R.S. (Eds.) Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Anderson, G.; Reiter, R.J. Melatonin: Roles in influenza, Covid-19, and other viral infections. Rev. Med. Virol. 2020, 30, e2109. [Google Scholar] [CrossRef]
- Theodoulou, F.L.; Sibon, O.C.M.; Jackowski, S.; Gout, I. Coenzyme A and its derivatives: Renaissance of a textbook classic. Biochem. Soc. Trans. 2014, 42, 1025–1032. [Google Scholar] [CrossRef]
- Van der Sluis, R.; Erasmus, E. Xenobiotic/medium chain fatty acid: CoA ligase—A critical review on its role in fatty acid metabolism and the detoxification of benzoic acid and aspirin. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1169–1179. [Google Scholar] [CrossRef]
- Gout, I.; Coenzyme, A. A protective thiol in bacterial antioxidant defence. Biochem. Soc. Trans. 2019, 47, 469–476. [Google Scholar] [CrossRef]
- Bhandary, S.; Aguan, K. Pyruvate dehydrogenase complex deficiency and its relationship with epilepsy frequency—An overview. Epilepsy Res. 2015, 116, 40–52. [Google Scholar] [CrossRef]
- UniProt. Available online: https://www.uniprot.org/ (accessed on 25 September 2020).
- Ruetz, M.; Campanello, G.C.; Purchal, M.; Shen, H.; McDevitt, L.; Gouda, H.; Wakabayashi, S.; Zhu, J.; Rubin, E.J.; Warncke, K.; et al. Itaconyl-CoA forms a stable biradical in methylmalonyl-CoA mutase and derails its activity and repair. Science 2019, 366, 589–593. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Pedro, J.M.B.-S.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [Green Version]
- Zabielska, J.; Sledzinski, T.; Stelmanska, E. Acyl-coenzyme A: Cholesterol acyltransferase inhibition in cancer treatment. Anticancer Res. 2019, 39, 3385–3394. [Google Scholar] [CrossRef]
- Stelmanska, E.; Swierczynski, J. Up-regulation of lipogenic enzyme genes expression in inguinal white adipose tissue of female rats by progesterone. J. Steroid Biochem. Mol. Biol. 2013, 134, 37–44. [Google Scholar] [CrossRef]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Bekeova, C.; Anderson-Pullinger, L.; Boye, K.; Boos, F.; Sharpadskaya, Y.; Herrmann, J.M.; Seifert, E.L. Multiple mitochondrial thioesterases have distinct tissue and substrate specificity and CoA regulation, suggesting unique functional roles. J. Biol. Chem. 2019, 294, 19034–19047. [Google Scholar] [CrossRef]
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of Autophagy by Cytosolic Acetyl-Coenzyme A. Mol. Cell 2014, 53, 710–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naquet, P.; Kerr, E.W.; Vickers, S.D.; Leonardi, R. Regulation of coenzyme A levels by degradation: The ‘Ins and Outs’. Prog. Lipid Res. 2020, 78, 101028. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Baratashvili, M.; Van Der Zwaag, M.; Kanon, B.; Colombelli, C.; Lambrechts, R.A.; Schaap, O.; Nollen, E.A.; Podgoršek, A.; Kosec, G.; et al. Extracellular 4′-phosphopantetheine is a source for intracellular coenzyme A synthesis. Nat. Chem. Biol. 2015, 11, 784–792. [Google Scholar] [CrossRef]
- Shibata, K.; Gross, C.J.; Henderson, L.M. Hydrolysis and absorption of pantothenate and its coenzymes in the rat small intestine. J. Nutr. 1983, 113, 2107–2115. [Google Scholar] [CrossRef]
- Magnúsdóttir, S.; Ravcheev, D.; De Crécy-Lagard, V.; Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests cooperation among gut microbes. Front. Genet. 2015, 6, 148. [Google Scholar] [CrossRef] [Green Version]
- Sabui, S.; Kapadia, R.; Ghosal, A.; Schneider, M.; Lambrecht, N.W.G.; Said, H.M. Biotin and pantothenic acid oversupplementation to conditional SLC5A6 KO mice prevents the development of intestinal mucosal abnormalities and growth defects. Am. J. Physiol. Cell Physiol. 2018, 315, C73–C79. [Google Scholar] [CrossRef]
- Said, H.M. Intestinal absorption of water-soluble vitamins in health and disease. Biochem. J. 2011, 437, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Trumbo, P. Pantothenic Acid. In Modern Nutrition in Health and Disease; Ross, A., Caballero, B., Cousins, R., Eds.; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2014; pp. 351–357. [Google Scholar]
- Lopez Martinez, D.; Tsuchiya, Y.; Gout, I. Coenzyme A biosynthetic machinery in mammalian cells. Biochem. Soc. Trans. 2014, 42, 1112–1117. [Google Scholar] [CrossRef]
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Rock, C.O.; Jackowski, S. Feedback regulation of murine pantothenate kinase 3 by coenzyme A and coenzyme A thioesters. J. Biol. Chem. 2005, 280, 32594–32601. [Google Scholar] [CrossRef] [Green Version]
- Dansie, L.E.; Reeves, S.; Miller, K.; Zano, S.P.; Frank, M.; Pate, C.; Wang, J.; Jackowski, S. Physiological roles of the pantothenate kinases. Biochem. Soc. Trans. 2014, 42, 1033–1036. [Google Scholar] [CrossRef] [Green Version]
- Alfonso-Pecchio, A.; Garcia, M.; Leonardi, R.; Jackowski, S. Compartmentalization of Mammalian Pantothenate Kinases. PLoS ONE 2012, 7, 49509. [Google Scholar] [CrossRef] [Green Version]
- Zhyvoloup, A.; Nemazanyy, I.; Panasyuk, G.; Valovka, T.; Fenton, T.; Rebholz, H.; Wang, M.L.; Foxon, R.; Lyzogubov, V.; Usenko, V.; et al. Subcellular Localization and Regulation of Coenzyme A Synthase. J. Biol. Chem. 2003, 278, 50316–50321. [Google Scholar] [CrossRef] [Green Version]
- Gout, I. Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans. 2018, 46, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Trams, E.G.; Fales, H.A.; Gal, A.E. S-Palmityl pantetheine as an intermediate in the metabolism of palmityl coenzyme A by rat liver plasma membrane preparations. Biochem. Biophys. Res. Commun. 1968, 31, 973–976. [Google Scholar] [CrossRef]
- Franklin, J.E.; Trams, E.G. Metabolism of coenzyme A and related nucleotides by liver plasma membranes. BBA Gen. Subj. 1971, 230, 105–116. [Google Scholar] [CrossRef]
- Skrede, S. The Degradation of CoA: Subcellular Localization and Kinetic Properties of CoA-and Dephospho-CoA Pyrophosphatase. Eur. J. Biochem. 1973, 38, 401–407. [Google Scholar] [CrossRef]
- Lee, S.Y.; Müller, C.E. Nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1) and its inhibitors. Medchemcomm 2017, 8, 823–840. [Google Scholar] [CrossRef]
- Goding, J.W.; Grobben, B.; Slegers, H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim. Biophys. Acta Mol. Basis Dis. 2003, 1638, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wittwer, C.T.; Burkhard, D.; Ririe, K.; Rasmussen, R.; Brown, J.; Wyse, B.W.; Hansen, G.R. Purification and properties of a pantetheine-hydrolyzing enzyme from pig kidney. J. Biol. Chem. 1983, 258, 9733–9738. [Google Scholar]
- Granjeaud, S.; Naquet, P.; Galland, F. An ESTs description of the new Vanin gene family conserved from fly to human. Immunogenetics 1999, 49, 964–972. [Google Scholar] [CrossRef]
- Martin, F.; Malergue, F.; Pitari, G.; Philippe, J.M.; Philips, S.; Chabret, C.; Granjeaud, S.; Mattei, M.G.; Mungall, A.J.; Naquet, P.; et al. Vanin genes are clustered (human 6q22-24 and mouse 10A2B1) and encode isoforms of pantetheinase ectoenzymes. Immunogenetics 2001, 53, 296–306. [Google Scholar] [CrossRef]
- Maras, B.; Barra, D.; Duprè, S.; Pitari, G. Is pantetheinase the actual identity of mouse and human vanin-1 proteins? FEBS Lett. 1999, 461, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Berruyer, C.; Pouyet, L.; Millet, V.; Martin, F.M.; LeGoffic, A.; Canonici, A.; Garcia, S.; Bagnis, C.; Naquet, P.; Galland, F. Vanin-1 licenses inflammatory mediator production by gut epithelial cells and controls colitis by antagonizing peroxisome proliferator-activated receptor γ activity. J. Exp. Med. 2006, 203, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Pitari, G.; Malergue, F.; Martin, F.; Philippe, J.M.; Massucci, M.T.; Chabret, C.; Maras, B.; Duprè, S.; Naquet, P.; Galland, F. Pantetheinase activity of membrane-bound Vanin-1: Lack of free cysteamine in tissues of Vanin-1 deficient mice. FEBS Lett. 2000, 483, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Nitto, T.; Inoue, T.; Node, K. Alternative spliced variants in the pantetheinase family of genes expressed in human neutrophils. Gene 2008, 426, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Rommelaere, S.; Millet, V.; Rihet, P.; Atwell, S.; Helfer, E.; Chasson, L.; Beaumont, C.; Chimini, G.; Sambo, M.D.R.; Viallat, A.; et al. Serum Pantetheinase/Vanin Levels regulate erythrocyte homeostasis and severity of malaria. Am. J. Pathol. 2015, 185, 3039–3052. [Google Scholar] [CrossRef] [PubMed]
- Kerr, E.W.; Shumar, S.A.; Leonardi, R. Nudt8 is a novel CoA diphosphohydrolase that resides in the mitochondria. FEBS Lett. 2019, 593, 1133–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, S.J.; Tillander, V.; Ofman, R.; Alexson, S.E.H.; Hunt, M.C. The nudix hydrolase 7 is an acyl-CoA diphosphatase involved in regulating peroxisomal coenzyme A homeostasis. J. Biochem. 2008, 144, 655–663. [Google Scholar] [CrossRef]
- Shumar, S.A.; Kerr, E.W.; Geldenhuys, W.J.; Montgomery, G.E.; Fagone, P.; Thirawatananond, P.; Saavedra, H.; Gabelli, S.B.; Leonardi, R. Nudt19 is a renal CoA diphosphohydrolase with biochemical and regulatory properties that are distinct from the hepatic Nudt7 isoform. J. Biol. Chem. 2018, 293, 4134–4148. [Google Scholar] [CrossRef] [Green Version]
- Guranowski, A. Fluoride is a strong and specific inhibitor of (asymmetrical) Ap4A hydrolases. FEBS Lett. 1990, 262, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Ofman, R.; Speijer, D.; Leen, R.; Wanders, R.J.A. Proteomic analysis of mouse kidney peroxisomes: Identification of RP2p as a peroxisomal nudix hydrolase with acyl-CoA diphosphatase activity. Biochem. J. 2006, 393, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Hunt, M.C.; Tillander, V.; Alexson, S.E.H. Regulation of peroxisomal lipid metabolism: The role of acyl-CoA and coenzyme A metabolizing enzymes. Biochimie 2014, 98, 45–55. [Google Scholar] [CrossRef]
- Antonenkov, V.D.; Hiltunen, J.K. Transfer of metabolites across the peroxisomal membrane. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1374–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieselmann, V.; Hasilik, A.; Figura, K. Von Tartrate-inhibitable acid phosphatase purification from placenta, characterization and subcellular distribution in fibroblasts. Hoppe. Seylers. Z. Physiol. Chem. 1984, 365, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Verkruyse, L.A.; Hofmann, S.L. Lysosomal targeting of palmitoyl-protein thioesterase. J. Biol. Chem. 1996, 271, 15831–15836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camp, L.; Verkruyse, L.A.; Afendis, S.J.; Slaughter, C.; Hofmann, S. Molecular cloning and expression of palmitoyl-protein thioesterase. J. Biol. Chem. 1994, 269, 23212–23219. [Google Scholar] [PubMed]
- Calero, G.; Gupta, P.; Nonato, M.C.; Tandel, S.; Biehl, E.R.; Hofmann, S.L.; Clardy, J. The crystal structure of palmitoyl protein thioesterase-2 (PPT2) reveals the basis for divergent substrate specificities of the two lysosomal thioesterases, PPT1 and PPT2. J. Biol. Chem. 2003, 278, 37957–37964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapel, A.; Kieffer-Jaquinod, S.; Sagné, C.; Verdon, Q.; Ivaldi, C.; Mellal, M.; Thirion, J.; Jadot, M.; Bruley, C.; Garin, J.; et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol. Cell. Proteom. 2013, 12, 1572–1588. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.W.; Subramanian, C.; Rock, C.O.; Jackowski, S. Quantification of Coenzyme A in Cells and Tissues. J. Vis. Exp. 2019, 151. [Google Scholar] [CrossRef]
- Robishaw, J.; Neely, J.R. Coenzyme A metabolism. Am. J. Physiol. Endocrinol. Metab. 1985, 11. [Google Scholar] [CrossRef]
- Shurubor, Y.I.; D’Aurelio, M.; Clark-Matott, J.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Cooper, A.J.L.; Krasnikov, B.F. Determination of Coenzyme A and Acetyl-Coenzyme A in Biological Samples Using HPLC with UV Detection. Molecules 2017, 22, 1388. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, A.G.; Beinlich, C.J. Pantothenic Acid in Health and Disease. Vitam. Horm. 1991, 46, 165–228. [Google Scholar]
- Tubbs, P.K.; Garland, P.B. Variations in tissue contents of coenzyme A thio esters and possible metabolic implications. Biochem. J. 1964, 93, 550–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.M.; Chohnan, S.; Virga, K.G.; Stevens, R.D.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Newgard, C.B.; Lee, R.E.; Rock, C.O.; et al. Chemical Knockout of Pantothenate Kinase Reveals the Metabolic and Genetic Program Responsible for Hepatic Coenzyme A Homeostasis. Chem. Biol. 2007, 14, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapp, G.W. Some systemic effects of malignant tumors. I. Co-enzyme A levels. Cancer 1973, 31, 357–360. [Google Scholar] [CrossRef]
- Peng, Y.; Puglielli, L. Nϵ-lysine acetylation in the lumen of the endoplasmic reticulum: A way to regulate autophagy and maintain protein homeostasis in the secretory pathway. Autophagy 2016, 12, 1051–1052. [Google Scholar] [CrossRef] [Green Version]
- Costantini, C.; Ko, M.H.; Jonas, M.C.; Puglielli, L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem. J. 2007, 407, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Hunt, M.C.; Alexson, S.E.H. The role Acyl-CoA thioesterases play in mediating intracellular lipid metabolism. Prog. Lipid Res. 2002, 41, 99–130. [Google Scholar] [CrossRef]
- Fiermonte, G.; Paradies, E.; Todisco, S.; Marobbio, C.M.T.; Palmieri, F. A novel member of solute carrier family 25 (SLC25A42) is a transporter of coenzyme A and adenosine 3′,5′-diphosphate in human mitochondria. J. Biol. Chem. 2009, 284, 18152–18159. [Google Scholar] [CrossRef] [Green Version]
- Kemp, S.; Theodoulou, F.L.; Wanders, R.J.A. Mammalian peroxisomal ABC transporters: From endogenous substrates to pathology and clinical significance. Br. J. Pharmacol. 2011, 164, 1753–1766. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.; Carrier, D.J.; Schaedler, T.; Waterham, H.R.; Van Roermund, C.W.; Theodoulou, F.L. Peroxisomal ABC transporters: Functions and mechanism. Biochem. Soc. Trans. 2015, 43, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Agrimi, G.; Russo, A.; Scarcia, P.; Palmieri, F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme A, FAD and NAD+. Biochem. J. 2012, 443, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Jonas, M.C.; Pehar, M.; Puglielli, L. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J. Cell Sci. 2010, 123, 3378–3388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beld, J.; Sonnenschein, E.C.; Vickery, C.R.; Noel, J.P.; Burkart, M.D. The phosphopantetheinyl transferases: Catalysis of a post-translational modification crucial for life. Nat. Prod. Rep. 2014, 31, 61–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrechts, R.A.; Schepers, H.; Yu, Y.; van der Zwaag, M.; Autio, K.J.; Vieira-Lara, M.A.; Bakker, B.M.; Tijssen, M.A.; Hayflick, S.J.; Grzeschik, N.A.; et al. CoA-dependent activation of mitochondrial acyl carrier protein links four neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10488. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta Proteins Proteomics 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, Y.; Chen, Q.; Zhang, X.; Wang, H.; Wang, Z.; Wang, J.; Tian, C. The role of acetylation sites in the regulation of p53 activity. Mol. Biol. Rep. 2020, 47, 381–391. [Google Scholar] [CrossRef]
- Popov, D. Protein S-glutathionylation: From current basics to targeted modifications. Arch. Physiol. Biochem. 2014, 120, 123–130. [Google Scholar] [CrossRef]
- Huth, W.; Pauli, C.; Möller, U. Immunochemical detection of CoA-modified mitochondrial matrix proteins. Biochem. J. 1996, 320, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Ley, S.C.; De Carvalho, L.P.S. Protein CoAlation: A redox-linked post-translational modification. Biochem. J. 2017, 474, 2897–2899. [Google Scholar] [CrossRef]
- Van Laer, K.; Hamilton, C.J.; Messens, J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxidants Redox Signal. 2013, 18, 1642–1653. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine modification in protein structure/function and human disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Zhyvoloup, A.; Baković, J.; Thomas, N.; Yu, B.Y.K.; Das, S.; Orengo, C.; Newell, C.; Ward, J.; Saladino, G.; et al. Protein CoAlation and antioxidant function of coenzyme A in prokaryotic cells. Biochem. J. 2018, 475, 1909–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Peak-Chew, S.Y.; Newell, C.; Miller-Aidoo, S.; Mangal, S.; Zhyvoloup, A.; Baković, J.; Malanchuk, O.; Pereira, G.C.; Kotiadis, V.; et al. Protein CoAlation: A redox-regulated protein modification by coenzyme A in mammalian cells. Biochem. J. 2017, 474, 2489–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huth, W.; Arvand, M.; Moller, U. Identification of [1-14C]pantothenic-acid-mediated modified mitochondrial proteins. Eur. J. Biochem. 1988, 172, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Schwerdt, G.; Huth, W. Turnover and transformation of mitochondrial acetyl-CoA acetyltransterase into CoA-modified forms. Biochem. J. 1993, 292, 915–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quandt, L.; Huth, W. Modulation of rat-liver mitochondrial acetyl-CoA acetyltransferase activity by a reversible chemical modification with coenzyme A. Biochim. Biophys. Acta (BBA)/Protein Struct. Mol. 1984, 784, 168–176. [Google Scholar] [CrossRef]
- Baković, J.; Yu, B.Y.K.; Silva, D.; Chew, S.P.; Kim, S.; Ahn, S.H.; Palmer, L.; Aloum, L.; Stanzani, G.; Malanchuk, O.; et al. A key metabolic integrator, coenzyme A, modulates the activity of peroxiredoxin 5 via covalent modification. Mol. Cell. Biochem. 2019, 461, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Byrne, D.P.; Burgess, S.G.; Bormann, J.; Baković, J.; Huang, Y.; Zhyvoloup, A.; Yu, B.Y.K.; Peak-Chew, S.; Tran, T.; et al. Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol. 2020, 28, 101318. [Google Scholar] [CrossRef]
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn errors of coenzyme a metabolism and neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Kim, M.K.; Choi, B.Y. The long-term relationship between dietary pantothenic acid (vitamin B5) intake and C-reactive protein concentration in adults aged 40 years and older. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 806–816. [Google Scholar] [CrossRef]
- Bokhari, M.R.; Zulfiqar, H.; Bokhari, S.R.A. Hallervorden Spatz Disease (Pantothenate Kinase-Associated Neurodegeneration, PKAN)-StatPearls-NCBI Bookshelf; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Subramanian, C.; Yao, J.; Frank, M.W.; Rock, C.O.; Jackowski, S. A pantothenate kinase-deficient mouse model reveals a gene expression program associated with brain coenzyme a reduction. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165663. [Google Scholar] [CrossRef]
- Venco, P.; Dusi, S.; Valletta, L.; Tiranti, V. Alteration of the coenzyme A biosynthetic pathway in neurodegeneration with brain iron accumulation syndromes. Biochem. Soc. Trans. 2014, 42, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A corrects pathological defects in human neurons of PANK 2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Subramanian, C.; Yun, M.K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, T.; Ferdinandusse, S.; Ruiter, J.P.N.; Alders, M.; Mathijssen, I.B.; Parboosingh, J.S.; Innes, A.M.; Meijers-Heijboer, H.; Poll-The, B.T.; Bernier, F.P.; et al. Biallelic loss of function variants in COASY cause prenatal onset pontocerebellar hypoplasia, microcephaly, and arthrogryposis. Eur. J. Hum. Genet. 2018, 26, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Gurney, S.; Pang, D.; Temkin, A.; Park, N.; Janicki, S.C.; Zigman, W.B.; Silverman, W.; Tycko, B.; Schupf, N. Polymorphisms in HSD17B1: Early onset and increased risk of Alzheimer’s disease in women with down syndrome. Curr. Gerontol. Geriatr. Res. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Shinagawa, S.; Nagata, T.; Shimada, K.; Shibata, N.; Ohnuma, T.; Kasanuki, K.; Arai, H.; Yamada, H.; Nakayama, K.; et al. Usefulness of DNA Methylation Levels in COASY and SPINT1 Gene Promoter Regions as Biomarkers in Diagnosis of Alzheimer’s Disease and Amnestic Mild Cognitive Impairment. PLoS ONE 2016, 11, e0168816. [Google Scholar] [CrossRef] [Green Version]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–417. [Google Scholar] [CrossRef]
- Patassini, S.; Begley, P.; Xu, J.; Church, S.J.; Reid, S.J.; Kim, E.H.; Curtis, M.A.; Dragunow, M.; Waldvogel, H.J.; Snell, R.G.; et al. Metabolite mapping reveals severe widespread perturbation of multiple metabolic processes in Huntington’s disease human brain. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 1650–1662. [Google Scholar] [CrossRef]
- Iuso, A.; Alhaddad, B.; Weigel, C.; Kotzaeridou, U.; Mastantuono, E.; Schwarzmayr, T.; Graf, E.; Terrile, C.; Prokisch, H.; Strom, T.M.; et al. A homozygous splice site mutation in SLC25A42, encoding the mitochondrial transporter of coenzyme a, causes metabolic crises and epileptic encephalopathy. JIMD Rep. 2019, 44, 1–7. [Google Scholar]
- Almannai, M.; Alasmari, A.; Alqasmi, A.; Faqeih, E.; Al Mutairi, F.; Alotaibi, M.; Samman, M.M.; Eyaid, W.; Aljadhai, Y.I.; Shamseldin, H.E.; et al. Expanding the phenotype of SLC25A42-associated mitochondrial encephalomyopathy. Clin. Genet. 2018, 93, 1097–1102. [Google Scholar] [CrossRef]
- De Berardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, R. The current state of cancer metabolism. Nat. Rev. Cancer 2016, 16, 613–614. [Google Scholar] [CrossRef]
- Dubuis, S.; Ortmayr, K.; Zampieri, M. A framework for large-scale metabolome drug profiling links coenzyme A metabolism to the toxicity of anti-cancer drug dichloroacetate. Commun. Biol. 2018, 1, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharabsheh, H.A.; Scott, J.E. CoAsy knockdown in TNBC cell lines resulted in no overt effect on cell proliferation in vitro. Biochem. Biophys. Res. Commun. 2020, 530, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-A Kinase: A Potent Oncogene and Target for Cancer Therapy. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, Z.; Li, Q.; Pang, Y.; Cui, L.; Qian, T.; Quan, L.; Dai, Y.; Jiao, Y.; Zhang, Z.; et al. Prognostic significance of the PANK family expression in acute myeloid leukemia. Ann. Transl. Med. 2019, 7, 261. [Google Scholar] [CrossRef]
- Xiang, R.L.; Yang, Y.L.; Zuo, J.; Xiao, X.H.; Chang, Y.S.; De Fang, F. PanK4 inhibits pancreatic β-cell apoptosis by decreasing the transcriptional level of pro-caspase-9. Cell Res. 2007, 17, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Izzi, V.; Lakkala, J.; Devarajan, R.; Savolainen, E.-R.; Koistinen, P.; Heljasvaara, R.; Pihlajaniemi, T. Vanin 1 (VNN1) levels predict poor outcome in acute myeloid leukemia. Am. J. Hematol. 2018, 93, E4–E7. [Google Scholar] [CrossRef] [Green Version]
- Nagai, M.A.; da Rós, N.; Neto, M.M.; de Faria Junior, S.R.; Brentani, M.M.; Hirata, R.; Neves, E.J. Gene expression profiles in breast tumors regarding the presence or absence of estrogen and progesterone receptors. Int. J. Cancer 2004, 111, 892–899. [Google Scholar] [CrossRef]
- Williams, C.; Edvardsson, K.; Lewandowski, S.A.; Ström, A.; Gustafsson, J.Å. A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene 2008, 27, 1019–1032. [Google Scholar] [CrossRef] [Green Version]
- Lapin, V.; Shirdel, E.A.; Wei, X.; Mason, J.M.; Jurisica, I.; Mak, T.W. Kinome-wide screening of HER2+ breast cancer cells for molecules that mediate cell proliferation or sensitize cells to trastuzumab therapy. Oncogenesis 2014, 3, e133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gast, K.C.; Cathcart-Rake, E.J.; Norman, A.D.; Eshraghi, L.; Obidegwu, N.; Nichols, H.B.; Rosenberg, S.; Su, H.I.; Stewart, E.A.; Couch, F.J.; et al. Regimen-specific rates of chemotherapy-related amenorrhea in breast cancer survivors. JNCI Cancer Spectr. 2019, 3. [Google Scholar] [CrossRef]
- Findeklee, S.; Lotz, L.; Heusinger, K.; Hoffmann, I.; Dittrich, R.; Beckmann, M.W. Fertility Protection in Female Oncology Patients: How Should Patients Be Counseled? Geburtshilfe Frauenheilkd. 2015, 75, 1243–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruddy, K.J.; Schaid, D.J.; Partridge, A.H.; Larson, N.B.; Batzler, A.; Häberle, L.; Dittrich, R.; Widschwendter, P.; Fink, V.; Bauer, E.; et al. Genetic predictors of chemotherapy-related amenorrhea in women with breast cancer. Fertil. Steril. 2019, 112, 731–739.e1. [Google Scholar] [CrossRef]
- Czumaj, A.; Zabielska, J.; Pakiet, A.; Mika, A.; Rostkowska, O.; Makarewicz, W.; Kobiela, J.; Sledzinski, T.; Stelmanska, E. In vivo effectiveness of orlistat in the suppression of human colorectal cancer cell proliferation. Anticancer Res. 2019, 39, 3815–3822. [Google Scholar] [CrossRef] [PubMed]
- Ferrandon, S.; Devecchio, J.; Duraes, L.; Chouhan, H.; Karagkounis, G.; Davenport, J.; Orloff, M.; Liska, D.; Kalady, M.F. CoA Synthase (COASY) Mediates Radiation Resistance via PI3K Signaling in Rectal Cancer. Cancer Res. 2020, 80, 334–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferron, S.; Kalady, M.F. Identifying new targets for rectal cancer treatment. Oncoscience 2020, 7, 36–37. [Google Scholar] [CrossRef]
- Chai, C.Y.; Zhang, Y.; Song, J.; Lin, S.C.; Sun, S.; Chang, I.W. VNN1 overexpression is associated with poor response to preoperative chemoradiotherapy and adverse prognosis in patients with rectal cancers. Am. J. Transl. Res. 2016, 8, 4455–4463. [Google Scholar]
- Zhang, L.; Li, L.; Gao, G.; Wei, G.; Zheng, Y.; Wang, C.; Gao, N.; Zhao, Y.; Deng, J.; Chen, H.; et al. Elevation of GPRC5A expression in colorectal cancer promotes tumor progression through VNN-1 induced oxidative stress. Int. J. Cancer 2017, 140, 2734–2747. [Google Scholar] [CrossRef] [Green Version]
- Marshall, K.W.; Mohr, S.; El Khettabi, F.; Nossova, N.; Chao, S.; Bao, W.; Ma, J.; Li, X.J.; Liew, C.C. A blood-based biomarker panel for stratifying current risk for colorectal cancer. Int. J. Cancer 2010, 126, 1177–1186. [Google Scholar] [CrossRef]
- Yip, K.T.; Das, P.K.; Suria, D.; Lim, C.R.; Ng, G.H.; Liew, C.C. A case-controlled validation study of a blood-based seven-gene biomarker panel for colorectal cancer in Malaysia. J. Exp. Clin. Cancer Res. 2010, 29, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Saraya, A.; Gupta, P.; Sharma, R. Decreased levels of circulating and tissue miR-107 in human esophageal cancer. Biomarkers 2013, 18, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Zhong, K.Z.; Chen, W.W.; Hu, X.Y.; Jiang, A.L.; Zhao, J. Clinicopathological and prognostic significance of microRNA-107 in human non small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 4545–4551. [Google Scholar] [PubMed]
- Roldo, C.; Missiaglia, E.; Hagan, J.P.; Falconi, M.; Capelli, P.; Bersani, S.; Calin, G.A.; Volinia, S.; Liu, C.G.; Scarpa, A.; et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol. 2006, 24, 4677–4684. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Luo, Q.F.; Wei, C.K.; Li, D.F.; Li, J.; Fang, L. MiRNA-107 inhibits proliferation and migration by targeting CDK8 in breast cancer. Int. J. Clin. Exp. Med. 2014, 7, 32–40. [Google Scholar] [PubMed]
- Zhang, Z.; Zhang, L.; Yin, Z.Y.; Fan, X.L.; Hu, B.; Wang, L.Q.; Zhang, D. miR-107 regulates cisplatin chemosensitivity of A549 non small cell lung cancer cell line by targeting cyclin dependent kinase 8. Int. J. Clin. Exp. Pathol. 2014, 7, 7236–7241. [Google Scholar] [PubMed]
- Roediger, W.E.W. Causation of human ulcerative colitis: A lead from an animal model that mirrors human disease. JGH Open 2019, 3, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Wintrobe, M.M.; Follis, R.H.J.; Alcayaga, R.; Paulson, M.; Humphreys, S. Pantothenic acid deficiency in swine with particular reference to the effects on growth and on the alimentary tract. Bull. Johns Hopkins Hosp. 1943, 73, 313–341. [Google Scholar]
- Gensollen, T.; Bourges, C.; Rihet, P.; Rostan, A.; Millet, V.; Noguchi, T.; Bourdon, V.; Sobol, H.; Dubuquoy, L.; Bertin, B.; et al. Functional polymorphisms in the regulatory regions of the VNN1 gene are associated with susceptibility to inflammatory bowel diseases. Inflamm. Bowel Dis. 2013, 19, 2315–2325. [Google Scholar] [CrossRef]
- Lewis, G.D.; Farrell, L.; Wood, M.J.; Martinovic, M.; Arany, Z.; Rowe, G.C.; Souza, A.; Cheng, S.; McCabe, E.L.; Yang, E.; et al. Metabolic signatures of exercise in human plasma. Sci. Transl. Med. 2010, 2, 33ra37. [Google Scholar] [CrossRef] [Green Version]
- Corbin, D.R.; Rehg, J.E.; Shepherd, D.L.; Stoilov, P.; Percifield, R.J.; Horner, L.; Frase, S.; Zhang, Y.M.; Rock, C.O.; Hollander, J.M.; et al. Excess coenzyme A reduces skeletal muscle performance and strength in mice overexpressing human PANK2. Mol. Genet. Metab. 2017, 120, 350–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostman-Smith, I.; Brown, G.; Johnson, A.; Land, J.M.; Johnson, L.A. Dilated cardiomyopathy due to type II X-linked 3-methylglutaconic aciduria: Successful treatment with. pantothenic acid. Heart 1994, 72, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Rugolotto, S.; Prioli, M.D.; Toniolo, D.; Pellegrino, P.A.; Catuogno, S.; Burlina, A.B. Long-term treatment of Barth syndrome with pantothenic acid: A retrospective study. Mol. Genet. Metab. 2003, 80, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Brohn, F.H.; Trager, W. Coenzyme A Requirement of Malaria Parasites: Enzymes of Coenzyme A Biosynthesis in Normal Duck Erythrocytes and Erythrocytes Infected with Plasmodium lophurae* (intracellular parasitism/host parasite relations). Proc. Natl. Acad. Sci. USA 1975, 72, 2456–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliba, K.J.; Horner, H.A.; Kirk, K. Transport and metabolism of the essential vitamin pantothenic acid in human erythrocytes infected with the malaria parasite Plasmodium falciparum. J. Biol. Chem. 1998, 273, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Saliba, K.J.; Ferru, I.; Kirk, K. Provitamin B5 (Pantothenol) inhibits growth of the intraerythrocytic malaria parasite. Antimicrob. Agents Chemother. 2005, 49, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Saliba, K.J.; Kirk, K. H+-coupled Pantothenate Transport in the Intracellular Malaria Parasite. J. Biol. Chem. 2001, 276, 18115–18121. [Google Scholar] [CrossRef] [Green Version]
- Spry, C.; Macuamule, C.; Lin, Z.; Virga, K.G.; Lee, R.E.; Strauss, E.; Saliba, K.J. Pantothenamides Are Potent, On-Target Inhibitors of Plasmodium falciparum Growth When Serum Pantetheinase Is Inactivated. PLoS ONE 2013, 8, e54974. [Google Scholar] [CrossRef] [Green Version]
- Tjhin, E.T.; Spry, C.; Sewell, A.L.; Hoegl, A.; Barnard, L.; Sexton, A.E.; Siddiqui, G.; Howieson, V.M.; Maier, A.G.; Creek, D.J.; et al. Mutations in the pantothenate kinase of Plasmodium falciparum confer diverse sensitivity profiles to antiplasmodial pantothenate analogues. PLoS Pathog. 2018, 14, e1006918. [Google Scholar] [CrossRef] [Green Version]
- Spry, C.; van Schalkwyk, D.A.; Strauss, E.; Saliba, K.J. Pantothenate Utilization by Plasmodium as a Target for Antimalarial Chemotherapy. Infect. Disord. Drug Targets 2012, 10, 200–216. [Google Scholar] [CrossRef]
- Min-Oo, G.; Fortin, A.; Pitari, G.; Tam, M.; Stevenson, M.M.; Gros, P. Complex genetic control of susceptibility to malaria: Positional cloning of the Char9 locus. J. Exp. Med. 2007, 204, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min-Oo, G.; Ayi, K.; Bongfen, S.E.; Tam, M.; Radovanovic, I.; Gauthier, S.; Santiago, H.; Rothfuchs, A.G.; Roffê, E.; Sher, A.; et al. Cysteamine, the natural metabolite of pantetheinase, shows specific activity against Plasmodium. Exp. Parasitol. 2010, 125, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naquet, P.; Pitari, G.; Duprè, S.; Galland, F. Role of the Vnn1 pantetheinase in tissue tolerance to stress. Biochem. Soc. Trans. 2014, 42, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Penet, M.-F.; Malergue, F.; Lepidi, H.; Dessein, A.; Galland, F.; de Reggi, M.; Naquet, P.; Gharib, B. Vanin-1–/– mice show decreased NSAID- and Schistosoma-induced intestinal inflammation associated with higher glutathione stores. J. Clin. Investig. 2004, 113, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Meghari, S.; Berruyer, C.; Lepidi, H.; Galland, F.; Naquet, P.; Mege, J.L. Vanin-1 controls granuloma formation and macrophage polarization in Coxiella burnetii infection. Eur. J. Immunol. 2007, 37, 24–32. [Google Scholar] [CrossRef]
- Clifton, G.; Bryant, S.R.; Skinner, C.G. N1-(substituted) pantothenamides, antimetabolites of pantothenic acid. Arch. Biochem. Biophys. 1970, 137, 523–528. [Google Scholar] [CrossRef]
- de Villiers, M.; Barnard, L.; Koekemoer, L.; Snoep, J.L.; Strauss, E. Variation in pantothenate kinase type determines the pantothenamide mode of action and impacts on coenzyme A salvage biosynthesis. FEBS J. 2014, 281, 4731–4753. [Google Scholar] [CrossRef]
- Jansen, P.A.M.; van der Krieken, D.A.; Botman, P.N.M.; Blaauw, R.H.; Cavina, L.; Raaijmakers, E.M.; de Heuvel, E.; Sandrock, J.; Pennings, L.J.; Hermkens, P.H.H.; et al. Stable pantothenamide bioisosteres: Novel antibiotics for Gram-positive bacteria. J. Antibiot. 2019, 72, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Hoegl, A.; Darabi, H.; Tran, E.; Awuah, E.; Kerdo, E.S.C.; Habib, E.; Saliba, K.J.; Auclair, K. Stereochemical modification of geminal dialkyl substituents on pantothenamides alters antimicrobial activity. Bioorganic Med. Chem. Lett. 2014, 24, 3274–3277. [Google Scholar] [CrossRef] [Green Version]
- Maršavelski, A. A novel antimicrobial target—Expanded and revisited mode of action of pantothenamides. RSC Adv. 2016, 6, 44888–44895. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.J.; Barnard, L.; Mottaghi, K.; Tempel, W.; Antoshchenko, T.; Hong, B.S.; Allali-Hassani, A.; Smil, D.; Vedadi, M.; Strauss, E.; et al. Discovery of potent pantothenamide inhibitors of staphylococcus Aureus Pantothenate Kinase through a minimal SAR Study: Inhibition Is Due to Trapping of the Product. ACS Infect. Dis. 2016, 2, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Barnard, L.; Mostert, K.J.; Van Otterlo, W.A.L.; Strauss, E. Developing Pantetheinase-Resistant Pantothenamide Antibacterials: Structural Modification Impacts on PanK Interaction and Mode of Action. ACS Infect. Dis. 2018, 4, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.A.M.; Hermkens, P.H.H.; Zeeuwen, P.L.J.M.; Botman, P.N.M.; Blaauw, R.H.; Burghout, P.; Van Galen, P.M.; Mouton, J.W.; Rutjes, F.P.J.T.; Schalkwijk, J. Combination of Pantothenamides with Vanin Inhibitors as a Novel Antibiotic Strategy against Gram-Positive Bacteria Downloaded from. Antimicrob. Agents Chemother. 2013, 57, 4794–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.M.; Cano, M.L.; Potyraj, J. The relationship between metabolic state and total CoA content of rat liver and heart. J. Nutr. 1978, 108, 854–862. [Google Scholar] [CrossRef]
- Reibel, D.K.; Wyse, B.W.; Berkich, D.A.; Palko, W.M.; Neely, J.R.; Berkich, D.; Palko, W. Effects of diabetes and fasting on pantothenic acid metabolism in rats. Am. J. Physiol. 1981, 240, E597–E601. [Google Scholar] [CrossRef]
- Leonardi, R.; Rock, C.O.; Jackowski, S. Pank1 deletion in leptin-deficient mice reduces hyperglycaemia and hyperinsulinaemia and modifies global metabolism without affecting insulin resistance. Diabetologia 2014, 57, 1466–1475. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, J.; Gupta, G.; De Jesus Andreoli Pinto, T.; Sharma, R.; Pabreja, K.; Matta, Y.; Arora, N.; Mishra, A.; Sharma, R.; Dua, K. Role of microRNAs (miRNAs) in the pathophysiology of Diabetes mellitus. Panminerva Med. 2018, 60, 25–28. [Google Scholar]
- Vaishya, S.; Sarwade, R.D.; Seshadri, V. MicroRNA, proteins, and metabolites as novel biomarkers for prediabetes, diabetes, and related complications. Front. Endocrinol. 2018, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Ashoori, M.R.; Rahmati-Yamchi, M.; Ostadrahimi, A.; Fekri Aval, S.; Zarghami, N. MicroRNAs and adipocytokines: Promising biomarkers for pharmacological targets in diabetes mellitus and its complications. Biomed. Pharmacother. 2017, 93, 1326–1336. [Google Scholar] [CrossRef]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Chen, Z.; Feng, H.; Chen, Y.; Zhang, C.; Yu, J.; Luo, Y.; Zhao, L.; Jiang, X.; Shi, F. Sphingomyelin synthase 2 promotes an aggressive breast cancer phenotype by disrupting the homoeostasis of ceramide and sphingomyelin. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Smith, M.; Flodman, P.L.; Gargus, J.J.; Simon, M.T.; Verrell, K.; Haas, R.; Reiner, G.E.; Naviaux, R.; Osann, K.; Spence, M.A.; et al. Mitochondrial and ion channel gene alterations in autism. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 1796–1802. [Google Scholar] [CrossRef] [Green Version]
- Hollis, F.; Kanellopoulos, A.K.; Bagni, C. Mitochondrial dysfunction in Autism Spectrum Disorder: Clinical features and perspectives. Curr. Opin. Neurobiol. 2017, 45, 178–187. [Google Scholar] [CrossRef]
- Jackowski, S.; Leonardi, R. Deregulated coenzyme A, loss of metabolic flexibility and diabetes. Biochem. Soc. Trans. 2014, 42, 1118–1122. [Google Scholar] [CrossRef] [Green Version]
- GWAS Catalog. Available online: https://www.ebi.ac.uk/gwas/search?query=CoAlevel (accessed on 3 August 2020).
- Sabatti, C.; Service, S.K.; Hartikainen, A.L.; Pouta, A.; Ripatti, S.; Brodsky, J.; Jones, C.G.; Zaitlen, N.A.; Varilo, T.; Kaakinen, M.; et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 2009, 41, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Karlsson Linnér, R.; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.R.I.; Peyrot, W.J.; Purves, K.L.; Davis, K.A.S.; Rayner, C.; Choi, S.W.; Hübel, C.; Gaspar, H.A.; Kan, C.; Van der Auwera, S.; et al. Genome-wide gene-environment analyses of major depressive disorder and reported lifetime traumatic experiences in UK Biobank. Mol. Psychiatry 2020, 25, 1430–1446. [Google Scholar] [CrossRef] [PubMed]
- Comuzzie, A.G.; Cole, S.A.; Laston, S.L.; Voruganti, V.S.; Haack, K.; Gibbs, R.A.; Butte, N.F. Novel Genetic Loci Identified for the Pathophysiology of Childhood Obesity in the Hispanic Population. PLoS ONE 2012, 7, e51954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, T.J.; Choquet, H.; Yin, J.; Banda, Y.; Kvale, M.N.; Glymour, M.; Schaefer, C.; Risch, N.; Jorgenson, E. A large multiethnic genome-wide association study of adult body mass index identifies novel loci. Genetics 2018, 210, 499–515. [Google Scholar] [CrossRef] [Green Version]
- Winkler, T.W.; Justice, A.E.; Graff, M.; Barata, L.; Feitosa, M.F.; Chu, S.; Czajkowski, J.; Esko, T.; Fall, T.; Kilpeläinen, T.O.; et al. The Influence of Age and Sex on Genetic Associations with Adult Body Size and Shape: A Large-Scale Genome-Wide Interaction Study. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef] [Green Version]
- Nagel, M.; Watanabe, K.; Stringer, S.; Posthuma, D.; Van Der Sluis, S. Item-level analyses reveal genetic heterogeneity in neuroticism. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.M.; Zhu, G.; Dy, V.; Heath, A.C.; Madden, P.A.F.; Kemp, J.P.; McMahon, G.; St. Pourcain, B.; Timpson, N.J.; Golding, J.; et al. Genome-wide association study identifies loci affecting blood copper, selenium and zinc. Hum. Mol. Genet. 2013, 26, 3998–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelis, M.C.; Monda, K.L.; Yu, K.; Paynter, N.; Azzato, E.M.; Bennett, S.N.; Berndt, S.I.; Boerwinkle, E.; Chanock, S.; Chatterjee, N.; et al. Genome-wide meta-analysis identifies regions on 7p21 (AHR) and 15q24 (CYP1A2) as determinants of habitual caffeine consumption. PLoS Genet. 2011, 7, 1002033. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, O.J.; Pirastu, N.; Poole, R.; Fallowfield, J.A.; Hayes, P.C.; Grzeszkowiak, E.J.; Taal, M.W.; Wilson, J.F.; Parkes, J.; Roderick, P.J. Coffee Consumption and Kidney Function: A Mendelian Randomization Study. Am. J. Kidney Dis. 2020, 75, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kraja, A.T.; Smith, J.A.; Brody, J.A.; Franceschini, N.; Bis, J.C.; Rice, K.; Morrison, A.C.; Lu, Y.; Weiss, S.; et al. Meta-analysis identifies common and rare variants influencing blood pressure and overlapping with metabolic trait loci. Nat. Genet. 2016, 48, 1162–1170. [Google Scholar] [CrossRef]
- Wu, Y.; Byrne, E.M.; Zheng, Z.; Kemper, K.E.; Yengo, L.; Mallett, A.J.; Yang, J.; Visscher, P.M.; Wray, N.R. Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Wu, B.; Xuan, T.; Liu, Z.; Chen, J. Efficacy and tolerability of adding coenzyme A 400 U/d capsule to stable statin therapy for the treatment of patients with mixed dyslipidemia: An 8-week, multicenter, double-Blind, randomized, placebo-controlled study. Lipids Health Dis. 2014, 13. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Wu, B.; Xuan, T.; Xia, S.; Liu, Z.; Chen, J. Efficacy of Statin Monotherapy or in Combination With Coenzyme A Capsule in Patients With Metabolic Syndrome and Mixed Dyslipidemia. J. Clin. Med. Res. 2015, 7, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Donati, C.; Barbi, G.; Cairo, G.; Prati, G.F.; Degli Esposti, E. Pantethine improves the lipid abnormalities of chronic hemodialysis patients: Results of a multicenter clinical trial. Clin. Nephrol. 1986, 25, 70–74. [Google Scholar]
- Rumberger, J.A.; Napolitano, J.; Azumano, I.; Kamiya, T.; Evans, M. Pantethine, a derivative of vitamin B5 used as a nutritional supplement, favorably alters low-density lipoprotein cholesterol metabolism in low– to moderate–cardiovascular risk North American subjects: A triple-blinded placebo and diet-controlled investig. Nutr. Res. 2011, 31, 608–615. [Google Scholar] [CrossRef]
- Evans, M.; Rumberger, J.; Azumano, I.; Napolitano, J.; Citrolo, D.; Kamiya, T. Pantethine, a derivative of vitamin B5, favorably alters total, LDL and non-HDL cholesterol in low to moderate cardiovascular risk subjects eligible for statin therapy: A triple-blinded placebo and diet-controlled investigation. Vasc. Health Risk Manag. 2014, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Gaddi, A.; Descovich, G.C.; Noseda, G.; Fragiacomo, C.; Colombo, L.; Craveri, A.; Montanari, G.; Sirtori, C.R. Controlled evaluation of pantethine, a natural hypolipidemic compound, in patients with different forms of hyperlipoproteinemia. Atherosclerosis 1984, 50, 73–83. [Google Scholar] [CrossRef]
- Bertolini, S.; Donati, C.; Elicio, N.; Daga, A.; Cuzzolaro, S.; Marcenaro, A.; Saturnino, M.; Balestreri, R. Lipoprotein changes induced by pantethine in hyperlipoproteinemic patients: Adults and children. Int. J. Clin. Pharmacol. Ther. Toxicol. 1986, 24, 630–637. [Google Scholar] [PubMed]
- Murai, A.; Miyahara, T.; Tanaka, T.; Sako, Y.; Nishimura, N.; Kameyama, M. The effects of pantethine on lipid and lipoprotein abnormalities in survivors of cerebral infarction. Artery 1985, 12, 234–243. [Google Scholar] [PubMed]
- Chen, Y.Q.; Zhao, S.P.; Zhao, Y.H. Efficacy and tolerability of coenzyme A vs. pantethine for the treatment of patients with hyperlipidemia: A randomized, double-blind, multicenter study. J. Clin. Lipidol. 2015, 9, 692–697. [Google Scholar] [CrossRef]
- Cighetti, G.; Del Puppo, M.; Paroni, R.; Galli, G.; Kienle, M.G. Effects of pantethine on cholesterol synthesis from mevalonate in isolated rat hepatocytes. Atherosclerosis 1986, 60, 67–77. [Google Scholar] [CrossRef]
- Cighetti, G.; Del Puppo, M.; Paroni, R.; Kienle, M.G. Modulation of HMG-CoA reductase activity by pantetheine/pantethine. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1988, 963, 389–393. [Google Scholar] [CrossRef]
- McCarty, M.F. Inhibition of acetyl-CoA carboxylase by cystamine may mediate the hypotriglyceridemic activity of pantethine. Med. Hypotheses 2001, 56, 314–317. [Google Scholar] [CrossRef]
- Jones, M.L.; Tomaro-Duchesneau, C.; Martoni, C.J.; Prakash, S. Cholesterol lowering with bile salt hydrolase-active probiotic bacteria, mechanism of action, clinical evidence, and future direction for heart health applications. Expert Opin. Biol. Ther. 2013, 13, 631–642. [Google Scholar] [CrossRef]
- Craig, J.A.; Snelll, E.E. The comparative activities of pantethine, pantothenic acid, and coenzyme A for various microorganisms. J. Bacteriol. 1951, 61, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Gyorgy, P.; Rose, C.S. Further observations on the metabolic requirements of Lactobacillus bifidus var. pennsylvanicus. J. Bacteriol. 1955, 69, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultanbawa, Y. Effects of bifidogenic factors on growth of bifidobacterium bifidum in cultured milk yoghurt. J. Natl. Sci. Found. Sri Lanka 2006, 34, 205. [Google Scholar] [CrossRef] [Green Version]
- Hiramatsu, K.; Nozaki, H.; Arimori, S. Influence of pantethine on platelet volume, microviscosity, lipid composition and functions in diabetes mellitus with hyperlipidemia. Tokai J. Exp. Clin. Med. 1981, 6, 49–57. [Google Scholar] [PubMed]
- Bittolo Bon, G.; Cazzolato, G.; Zago, S.; Avogaro, P. Effects of pantethine on in-vitro peroxidation of low density lipoproteins. Atherosclerosis 1985, 57, 99–106. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CoA-SH as a Substrate | |||
|---|---|---|---|
| Enzyme | Reaction | Process | |
| Lipid metabolism | acyl-CoA synthetases (ACS) | fatty acid + CoA-SH + ATP → fatty acyl-CoA + AMP + PPi | fatty acids activation |
| carnitine palmitoyltransferase 2 (CPT2) | acylcarnitine + CoA-SH → carnitine + fatty acyl-CoA | carnitine shuttle | |
| thiolases e.g., β-ketoacyl-CoA thiolase | acyl-CoA + CoA-SH → acyl(n carbon-2)-CoA + acetyl-CoA acetoacetyl-CoA + CoA-SH → 2 acetyl-CoA | fatty acids oxidation ketone bodies oxidation | |
| ATP-citrate lyase (ACLY) | citrate + ATP + CoA-SH → oxaloacetate + acetyl-CoA + ADP + Pi | lipogenesis, synthesis of cholesterol and others | |
| Carbohydrate metabolism | pyruvate dehydrogenase complex (PDC) | pyruvate + CoA-SH + NAD+ → acetyl-CoA + NADH + H+ + CO2 | oxidative decarboxylation of pyruvate |
| Amino acids metabolism | branched-chain α-keto acid dehydrogenase complex | α-ketoisovaleric acid + CoA-SH + NAD+ → isobutyryl-CoA + NADH + H+ + CO2 α-ketoisocapronic acid + CoA-SH + NAD+ → iso-valeryl-CoA + NADH + H+ + CO2 α-keto-β-methylvaleric acid + CoA-SH + NAD+ → α-methylbutyryl-CoA + NADH + H+ + CO2 | oxidative decarboxylation of branched-chain α-keto acids |
| Lipid, carbohydrate, amino acids and ethanol metabolism | α-oxoglutarate dehydrogenase complex | α-oxoglutarate + CoA-SH + NAD+ → succinyl-CoA + NADH + H+ + CO2 | tricarboxylic acid cycle |
| acetyl-CoA synthetase | acetate + CoA-SH +ATP → acetyl-CoA +AMP + PPi | ethanol metabolism, acetate formed by gut microbiota metabolism | |
| CoA-SH as a Product | |||
|---|---|---|---|
| Enzyme | Reaction | Process | |
| Lipid metabolism | fatty acid synthase (FASN) | 7 malonyl-CoA + acetyl-CoA + 14 NADPH + 14 H+ → palmitate + 14 NADP+ + 7 CO2 + 6 H2O + 8 CoA-SH | lipogenesis |
| fatty acid elongases (ELOVLs) | fatty acyl-CoA + malonyl-CoA → β-keto-acyl-CoA + CO2 + CoA-SH or fatty acyl-CoA + acetyl-CoA → β-keto-acyl-CoA + CoA-SH | microsomal elongation of fatty acid chains mitochondrial elongation of fatty acid chains | |
| acyltransferases e.g., diacylglycerol O-acyltransferase (DGAT) e.g., acyl-CoA:cholesterol acyltransferase (ACAT) | 1,2-diacylglycerol + fatty acyl-CoA → triacylglycerol + CoA-SH cholesterol + acyl-CoA → cholesteryl ester + CoA-SH | triacylglycerol synthesis cholesterol metabolism | |
| carnitine palmitoyltransferase 1 (CPT1) | carnitine + acyl-CoA → acylcarnitine + CoA-SH | carnitine shuttle | |
| 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR) | HMG-CoA + 2 NADPH + 2 H+ → mevalonate +2 NADP+ + CoA-SH | synthesis of cholesterol, cholecalciferol (skin), prenyl moieties | |
| acyl-CoA thioesterases | fatty acyl-CoA + H2O → free fatty acid + CoA-SH | regulation of intracellular levels of acyl-CoA, free fatty acids and CoASH | |
| Lipid, carbohydrate, amino acids and ethanol metabolism | citrate synthase | acetyl-CoA + oxaloacetate + H2O → citrate + CoA-SH | tricarboxylic acid cycle |
| succinate thiokinase (also called succinyl-CoA synthetase) | succinyl-CoA + ADP (GDP) + Pi → succinate + ATP (GTP) + CoA-SH | tricarboxylic acid cycle | |
| Others | acetyltransferases e.g., choline O-acetyltransferase e.g., histone acetyltransferase (HAT) | choline + acetyl-CoA → acetylcholine + CoA-SH histone-Lys + acetyl-CoA→ histone-Lys-acetyl + CoA-SH | neurotransmitters synthesis protein acetylation |
| Tissue | Total CoA Concentration/Level | ||
|---|---|---|---|
| Liver | 87–434 nmol/g tissue | ||
| Subcellular compartment | cytosol | 0.1–0.14 mM | |
| mitochondria | 5.29 mM | ||
| peroxisomes | 0.7 mM | ||
| Heart | ~100 nmol/g tissue | ||
| Subcellular compartment | cytosol | 0.014 mM | |
| mitochondria | 2.26 mM | ||
| Enzyme | Gene | SNP Variant | Associated Trait | Nature of Change | Tested Population | Reference |
|---|---|---|---|---|---|---|
| Pantothenate kinase | PANK1 | rs11185790-A | Insulin level | Decreased insulin level | European | [172] |
| rs7073802-A | Educational attainments | Increased self-reported math ability | European | [173] | ||
| PANK3 | rs35693458-A | Unipolar depression | Increased probability of major depressive disorder in individuals not exposed to trauma | European | [174] | |
| PANK4 | rs12073504-G | Obesity-related trait | Increased IGFBP3 | Latin American | [175] | |
| rs7535528-G | BMI | Increased BMI | East Asian, African American, European, South Asian, Latin American | [176] | ||
| rs7535528-A | BMI | Decreased BMI | European | [177] | ||
| rs7535528-A | Neuroticism | Increased irritability | European | [178] | ||
| Phosphopantothenoylcysteine decarboxylase | PPCDC | rs2120019-C | Blood trace element | Decreased serum Zn levels | European | [179] |
| rs12148488-T rs8042558-T | Coffee consumption | Decreased consumption | European | [180,181] | ||
| rs147451859-G | Response to chemotherapy | Adverse response to antineoplastic agent in breast cancer | European | [120] | ||
| rs12148488-G | Blood pressure | Decreased mean arterial pressure | African American, Latin American, European | [182] | ||
| Coenzyme A synthase | COASY | rs668799-T | Medication use | Increased drugs used in diabetes | European | [183] |
| rs598126-T | AD | Increased risk of AD | American | [101] | ||
| Pantetheinase | VNN1 | rs3756975-C rs13204527-T rs909977-T | IBD | Increased risk of IBD | European | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czumaj, A.; Szrok-Jurga, S.; Hebanowska, A.; Turyn, J.; Swierczynski, J.; Sledzinski, T.; Stelmanska, E. The Pathophysiological Role of CoA. Int. J. Mol. Sci. 2020, 21, 9057. https://doi.org/10.3390/ijms21239057
Czumaj A, Szrok-Jurga S, Hebanowska A, Turyn J, Swierczynski J, Sledzinski T, Stelmanska E. The Pathophysiological Role of CoA. International Journal of Molecular Sciences. 2020; 21(23):9057. https://doi.org/10.3390/ijms21239057
Chicago/Turabian StyleCzumaj, Aleksandra, Sylwia Szrok-Jurga, Areta Hebanowska, Jacek Turyn, Julian Swierczynski, Tomasz Sledzinski, and Ewa Stelmanska. 2020. "The Pathophysiological Role of CoA" International Journal of Molecular Sciences 21, no. 23: 9057. https://doi.org/10.3390/ijms21239057
APA StyleCzumaj, A., Szrok-Jurga, S., Hebanowska, A., Turyn, J., Swierczynski, J., Sledzinski, T., & Stelmanska, E. (2020). The Pathophysiological Role of CoA. International Journal of Molecular Sciences, 21(23), 9057. https://doi.org/10.3390/ijms21239057