p53 Enhances Artemisia annua L. Polyphenols-Induced Cell Death Through Upregulation of p53-Dependent Targets and Cleavage of PARP1 and Lamin A/C in HCT116 Colorectal Cancer Cells


 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
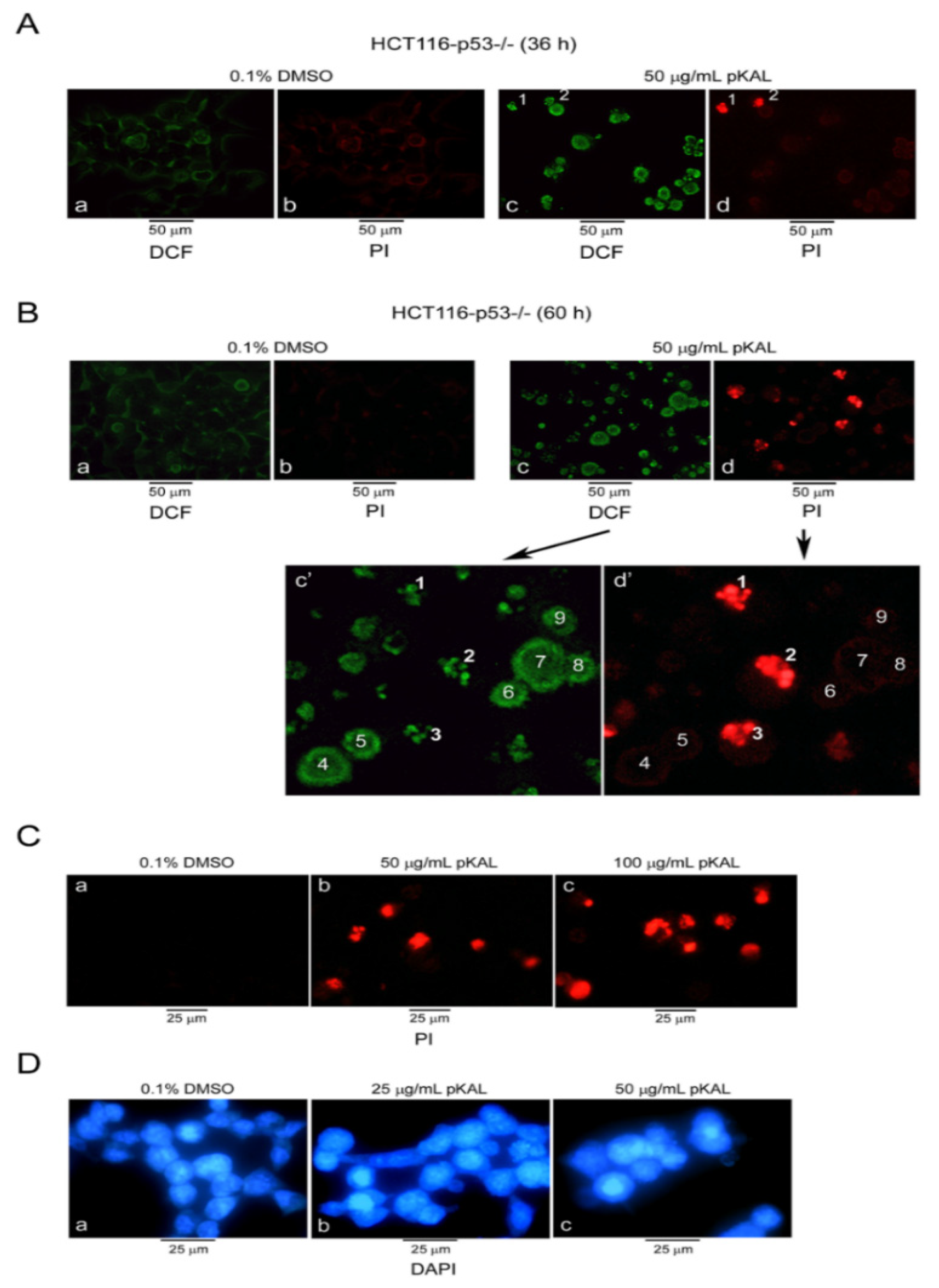
2.1. pKAL Induced ROS Production, PI Uptake, and Nuclear Structure Change in p53-Null HCT116 Colorectal Cancer Cells
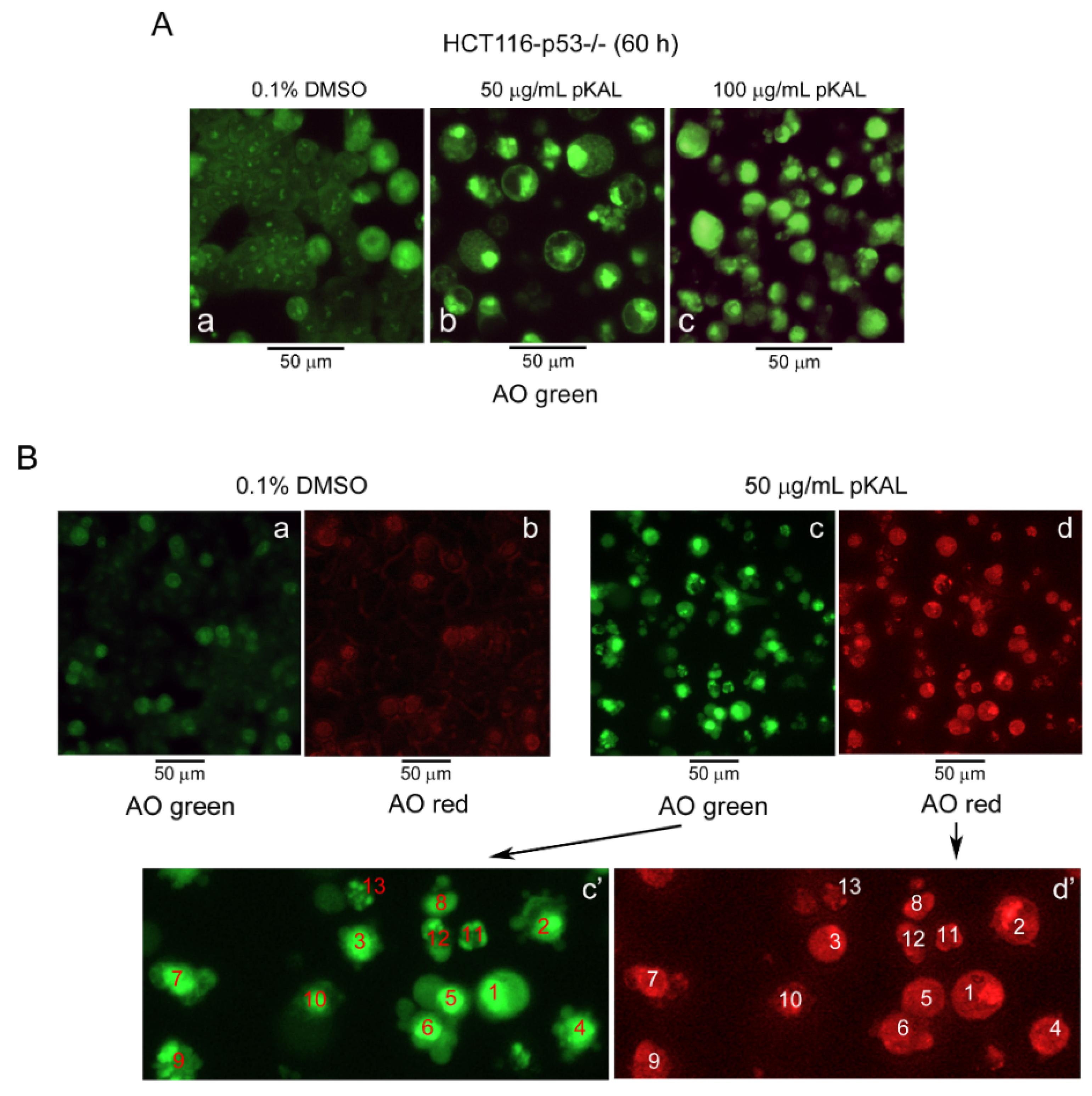
2.2. pKAL Altered DNA Conformation and Acidic Vesicle Formation in p53-Null HCT116 Cells
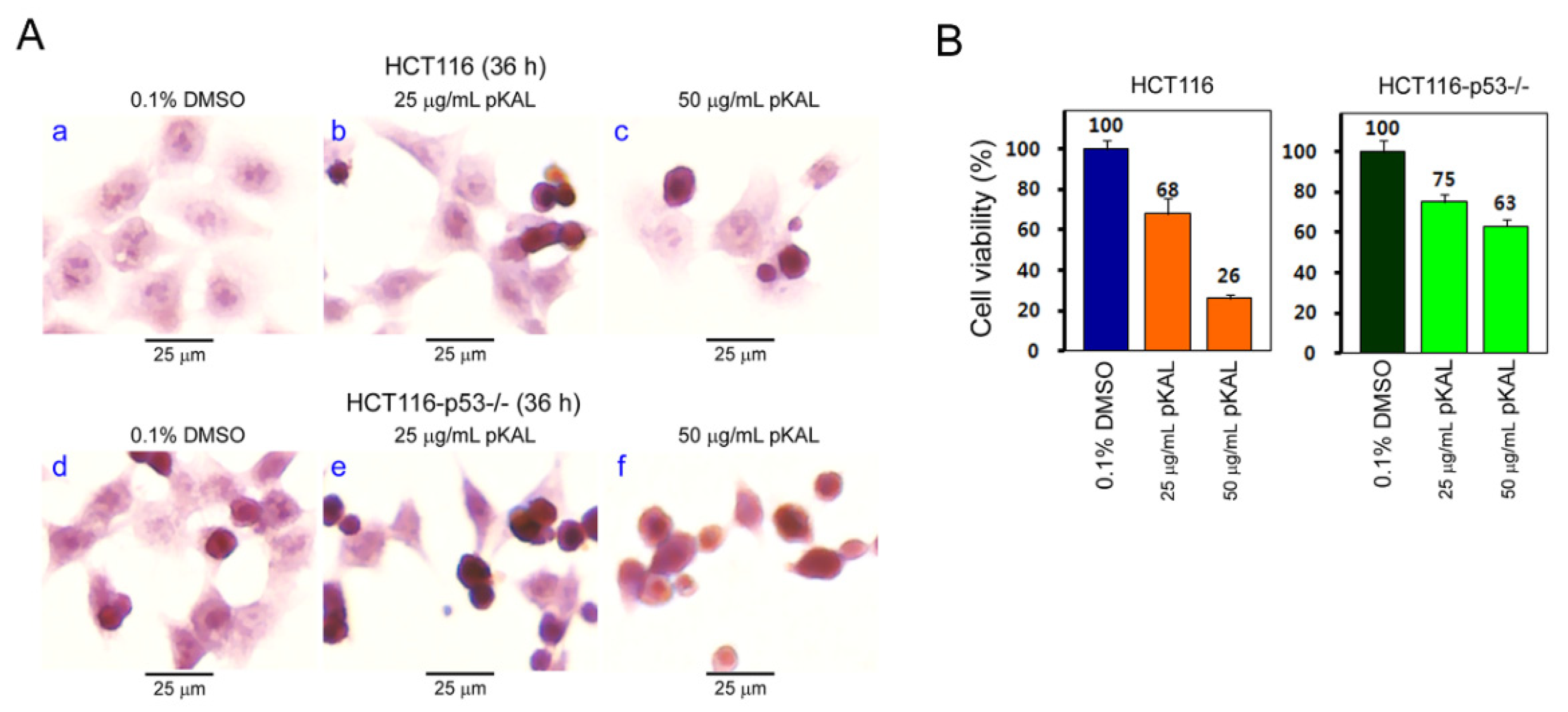
2.3. The Ability of pKAL to Suppress Cell Viability was Higher in p53-Wild HCT116 Cells than in p53-Null
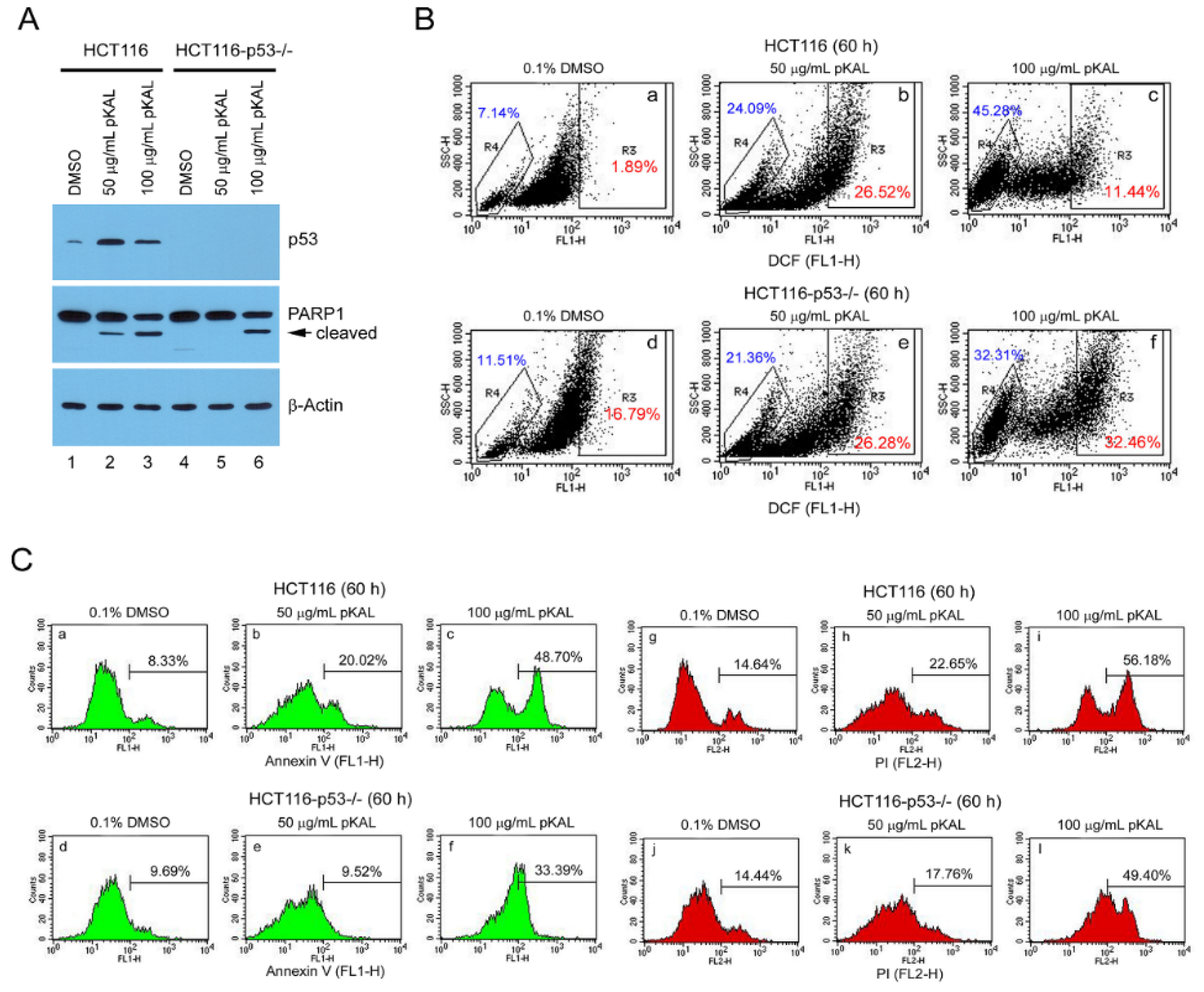
2.4. The Ability of pKAL to Induce Apoptosis was Higher in p53-Wild HCT116 Cells than in p53-Null
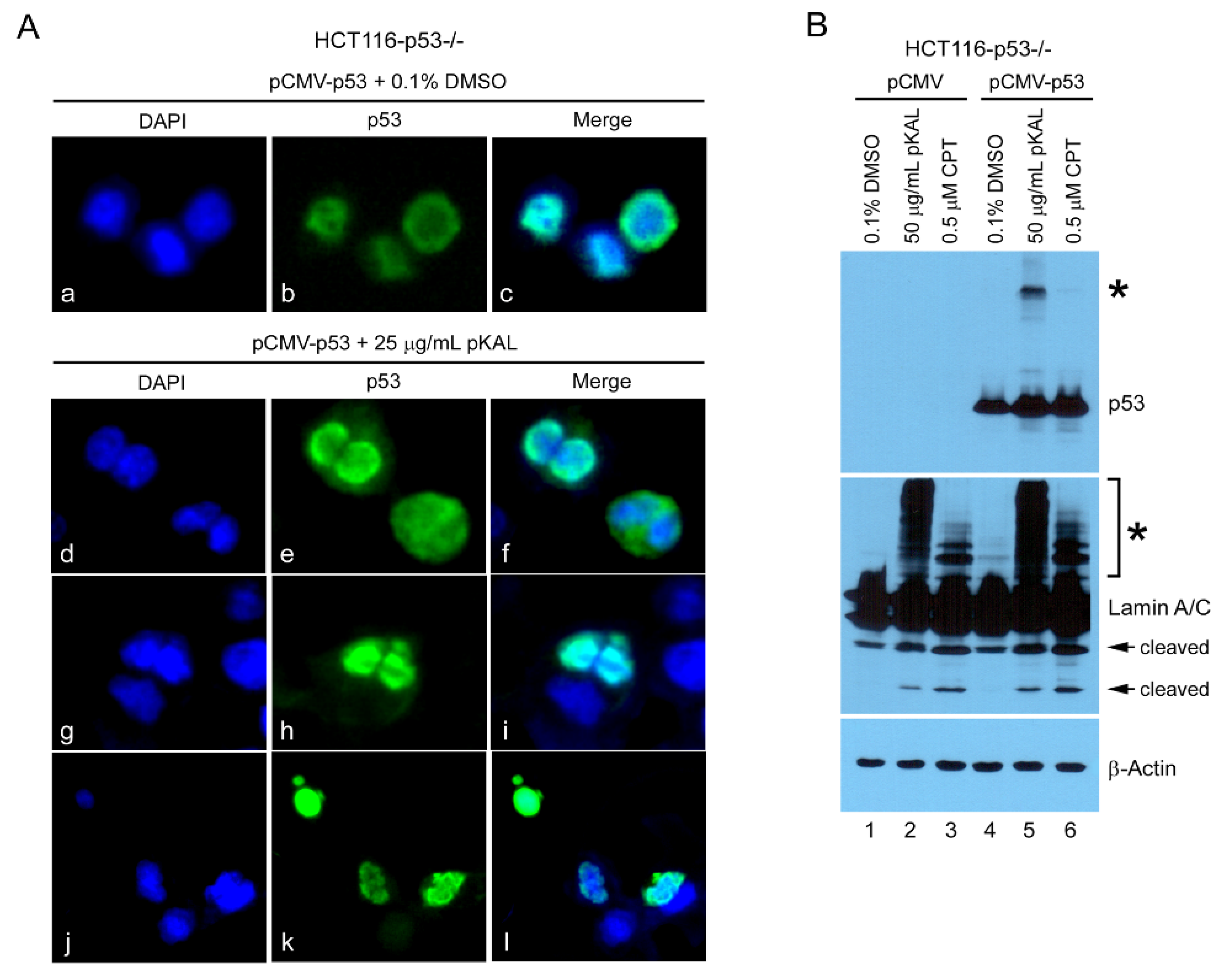
2.5. Upregulation of Ectopic p53 by pKAL Resulted in the Increase of pKAL-Induced Nuclear Structure Change and Post-Translational Modification of Lamin A/C
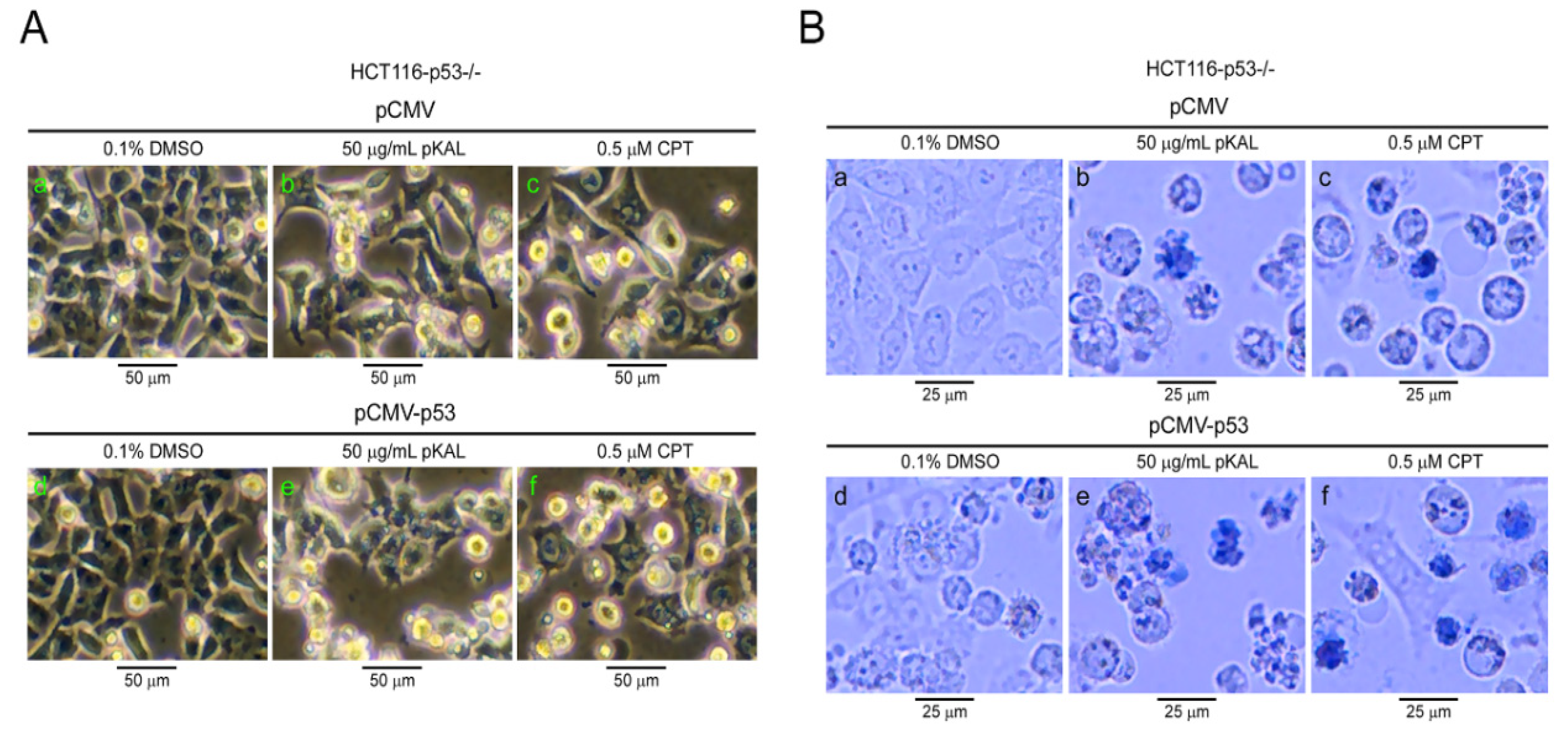
2.6. pKAL-Induced Cell Death was Enhanced by Ectopic p53 in p53-Null HCT116 Cells
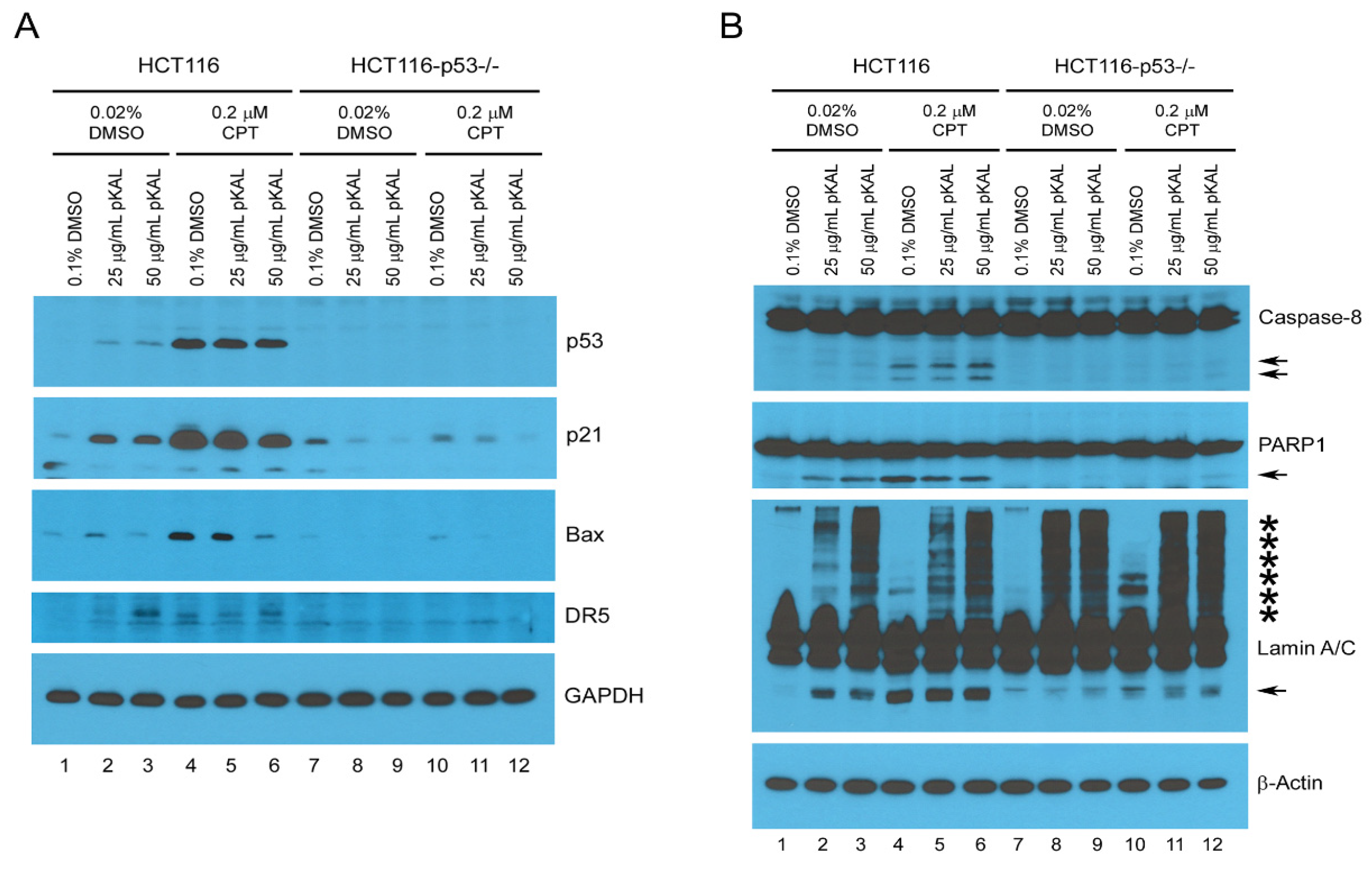
2.7. Upregulation of p53-Dependent Targets and Cleavage of PARP1 and Lamin A/C Were Associated with pKAL-Induced Cell Death Enhanced by p53
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. pKAL Compounds
4.3. Cell Culture
4.4. Fluorescence Microscopy of DCF-Stained Cells
4.5. Fluorescence Microscopy of PI-Stained Cells
4.6. Fluorescence Microscopy of DAPI-Stained Cells
4.7. Fluorescence Microscopy of AO-Stained Cells
4.8. Light Microscopy of Hematoxylin-Stained Cells
4.9. Cell Viability Assay
4.10. Western Blot Analysis
4.11. Flow Cytometric Analysis of DCF- and Annexin V/PI-Stained Cells
4.12. DNA Transfection
4.13. Immunofluorescence Microscopy
4.14. Phase-Contrast Light Microscopy
4.15. Light Microscopy of Trypan Blue Stained-Cells
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PARP1 | Poly (ADP-ribose) polymerase 1 |
| pKAL | Polyphenols isolated from Korean Artemisia annua L. |
| DR5 | Death receptor 5 |
| ROS | Reactive oxygen species |
| PI | Propidium iodide |
| DCF-DA | 2′,7′-dichlorofluorescein diacetate |
| DAPI | 4′6-diamidino-2-phenylindole |
| AO | Acridine orange |
| CCK-8 | Cell counting kit-8 |
| CPT | Camptothecin |
| FBS | Fetal bovine serum |
| RT | Room temperature |
| PBS | Phosphate-buffered saline |
| BSA | Bovine serum albumin |
| PTEN | Phosphatase and tensin homolog |
| PDK1 | Phosphoinositide-dependent protein kinase-1 |
| EMT | Epithelial-mesenchymal transition |
| CMV | Cytomegalovirus |
| DMSO | Dimethyl sulfoxide |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| HRP | Horseradish-peroxidase |
References
- Lee, J.E.; Mannisto, S.; Spiegelman, D.; Hunter, D.J.; Bernstein, L.; Van den Brandt, P.A.; Buring, J.E.; Cho, E.; English, D.R.; Flood, A.; et al. Intakes of fruit, vegetables, and carotenoids and renal cell cancer risk: A pooled analysis of 13 prospective studies. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1730–1739. [Google Scholar] [CrossRef] [Green Version]
- Gandini, S.; Merzenich, H.; Robertson, C.; Boyle, P. Meta-analysis of studies on breast cancer risk and diet: The role of fruit and vegetable consumption and the intake of associated micronutrients. Eur. J. Cancer 2000, 36, 636–646. [Google Scholar] [CrossRef]
- Alam, M.N.; Almoyad, M.; Huq, F. Polyphenols in Colorectal Cancer: Current State of Knowledge including Clinical Trials and Molecular Mechanism of Action. BioMed Res. Int. 2018, 2018, 4154185. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Rankin, G.O.; Li, Z.; Depriest, L.; Chen, Y.C. Kaempferol induces apoptosis in ovarian cancer cells through activating p53 in the intrinsic pathway. Food Chem. 2011, 128, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Sliva, D. Suppression of cancer invasiveness by dietary compounds. Mini Rev. Med. Chem. 2008, 8, 677–688. [Google Scholar] [CrossRef]
- Afshari, K.; Haddadi, N.S.; Haj-Mirzaian, A.; Farzaei, M.H.; Rohani, M.M.; Akramian, F.; Naseri, R.; Sureda, A.; Ghanaatian, N.; Abdolghaffari, A.H. Natural flavonoids for the prevention of colon cancer: A comprehensive review of preclinical and clinical studies. J. Cell. Physiol. 2019, 234, 21519–21546. [Google Scholar] [CrossRef]
- Jung, K.W.; Won, Y.J.; Oh, C.M.; Kong, H.J.; Lee, D.H.; Lee, K.H. Prediction of Cancer Incidence and Mortality in Korea, 2017. Cancer Res. Treat. 2017, 49, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.R.; Patel, H.D. p53: An Attractive Therapeutic Target for Cancer. Curr. Med. Chem. 2020, 27, 3706–3734. [Google Scholar] [CrossRef]
- Sakanashi, F.; Shintani, M.; Tsuneyoshi, M.; Ohsaki, H.; Kamoshida, S. Apoptosis, necroptosis and autophagy in colorectal cancer: Associations with tumor aggressiveness and p53 status. Pathol. Res. Pract. 2019, 215, 152425. [Google Scholar] [CrossRef]
- Yoon, M.H.; Kang, S.M.; Lee, S.J.; Woo, T.G.; Oh, A.Y.; Park, S.; Ha, N.C.; Park, B.J. p53 induces senescence through Lamin A/C stabilization-mediated nuclear deformation. Cell Death Dis. 2019, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; Karnezis, A.N.; Garcia, D.; Madriles, F.; Kortlever, R.M.; Rostker, F.; Swigart, L.B.; Pham, D.M.; Seo, Y.; Evan, G.I.; et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010, 468, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.; Reale, M.; Ullah, H.; Sureda, A.; Tejada, S.; Wang, Y.; Zhang, Z.J.; Xiao, J. Anti-cancer effects of polyphenols via targeting p53 signaling pathway: Updates and future directions. Biotechnol. Adv. 2020, 38, 107385. [Google Scholar] [CrossRef]
- Lin, S.T.; Tu, S.H.; Yang, P.S.; Hsu, S.P.; Lee, W.H.; Ho, C.T.; Wu, C.H.; Lai, Y.H.; Chen, M.Y.; Chen, L.C. Apple Polyphenol Phloretin Inhibits Colorectal Cancer Cell Growth via Inhibition of the Type 2 Glucose Transporter and Activation of p53-Mediated Signaling. J. Agric. Food Chem. 2016, 64, 6826–6837. [Google Scholar] [CrossRef]
- Da Costa, D.C.F.; Fialho, E.; Silva, J.L. Cancer Chemoprevention by Resveratrol: The p53 Tumor Suppressor Protein as a Promising Molecular Target. Molecules 2017, 22, 1014. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Thakur, V.S.; Bhaskaran, N.; Nawab, A.; Babcook, M.A.; Jackson, M.W.; Gupta, S. Green tea polyphenols induce p53-dependent and p53-independent apoptosis in prostate cancer cells through two distinct mechanisms. PLoS ONE 2012, 7, e52572. [Google Scholar] [CrossRef]
- Lu, J.N.; Lee, W.S.; Kim, M.J.; Yun, J.W.; Jung, J.H.; Yi, S.M.; Jeong, J.H.; Kim, H.J.; Choi, Y.H.; Kim, G.S.; et al. The inhibitory effect of anthocyanins on Akt on invasion and epithelial-mesenchymal transition is not associated with the anti-EGFR effect of the anthocyanins. Int. J. Oncol. 2014, 44, 1756–1766. [Google Scholar] [CrossRef]
- Lu, J.N.; Lee, W.S.; Yun, J.W.; Kim, M.J.; Kim, H.J.; Kim, D.C.; Jeong, J.H.; Choi, Y.H.; Kim, G.S.; Ryu, C.H.; et al. Anthocyanins from Vitis coignetiae Pulliat Inhibit Cancer Invasion and Epithelial-Mesenchymal Transition, but These Effects Can Be Attenuated by Tumor Necrosis Factor in Human Uterine Cervical Cancer HeLa Cells. Evid. Based Complement. Altern. Med. 2013, 2013, 503043. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.W.; Lee, W.S.; Go, S.I.; Nagappan, A.; Baek, J.Y.; Lee, J.D.; Lee, S.J.; Park, C.; Kim, G.Y.; Kim, H.J.; et al. Pachymic Acid Induces Apoptosis of EJ Bladder Cancer Cells by DR5 Up-Regulation, ROS Generation, Modulation of Bcl-2 and IAP Family Members. Phytother. Res. 2015, 29, 1516–1524. [Google Scholar] [CrossRef]
- Bataille, F.; Rummele, P.; Dietmaier, W.; Gaag, D.; Klebl, F.; Reichle, A.; Wild, P.; Hofstadter, F.; Hartmann, A. Alterations in p53 predict response to preoperative high dose chemotherapy in patients with gastric cancer. Mol. Pathol. 2003, 56, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Bertheau, P.; Lehmann-Che, J.; Varna, M.; Dumay, A.; Poirot, B.; Porcher, R.; Turpin, E.; Plassa, L.F.; De Roquancourt, A.; Bourstyn, E.; et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast 2013, 22 (Suppl. 2), S27–S29. [Google Scholar] [CrossRef]
- Tchelebi, L.; Ashamalla, H.; Graves, P.R. Mutant p53 and the response to chemotherapy and radiation. Subcell. Biochem. 2014, 85, 133–159. [Google Scholar]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Firestone, G.L.; Sundar, S.N. Anticancer activities of artemisinin and its bioactive derivatives. Expert Rev. Mol. Med. 2009, 11, e32. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Zhang, H.; Yun, J.; Chen, G.G.; Lai, P.B. Dihydroartemisinin exhibits antitumor activity toward hepatocellular carcinoma in vitro and in vivo. Biochem. Pharmacol. 2012, 83, 1278–1289. [Google Scholar] [CrossRef]
- Berdelle, N.; Nikolova, T.; Quiros, S.; Efferth, T.; Kaina, B. Artesunate induces oxidative DNA damage, sustained DNA double-strand breaks, and the ATM/ATR damage response in cancer cells. Mol. Cancer Ther. 2011, 10, 2224–2233. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Zhou, J.Y.; Zhang, D.; Liu, M.H.; Chen, Y.G. Artesunate induces apoptosis and autophagy in HCT116 colon cancer cells, and autophagy inhibition enhances the artesunate-induced apoptosis. Int. J. Mol. Med. 2018, 42, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.J.; Kim, G.T.; Kim, B.M.; Lim, E.G.; Kim, S.Y.; Kim, Y.M. Apoptosis-induced effects of extract from Artemisia annua Linne by modulating PTEN/p53/PDK1/Akt/ signal pathways through PTEN/p53-independent manner in HCT116 colon cancer cells. BMC Complement. Altern. Med. 2017, 17, 236. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Desta, K.T.; Kim, G.S.; Lee, S.J.; Lee, W.S.; Kim, Y.H.; Jin, J.S.; Abd El-Aty, A.M.; Shin, H.C.; Shim, J.H.; et al. Polyphenolic profile and antioxidant effects of various parts of Artemisia annua L. Biomed. Chromatogr. 2016, 30, 588–595. [Google Scholar] [CrossRef]
- Ko, Y.S.; Lee, W.S.; Panchanathan, R.; Joo, Y.N.; Choi, Y.H.; Kim, G.S.; Jung, J.M.; Ryu, C.H.; Shin, S.C.; Kim, H.J. Polyphenols from Artemisia annua L. Inhibit Adhesion and EMT of Highly Metastatic Breast Cancer Cells MDA-MB-231. Phytother. Res. 2016, 30, 1180–1188. [Google Scholar] [CrossRef]
- Amado, A.M.; Pazin, W.M.; Ito, A.S.; Kuzmin, V.A.; Borissevitch, I.E. Acridine orange interaction with DNA: Effect of ionic strength. Biochim Biophys Acta BBA-Gen. Subj. 2017, 1861, 900–909. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Krigerts, J.; Salmina, K.; Selga, T.; Sorokins, H.; Freivalds, T. Differential staining of peripheral nuclear chromatin with Acridine orange implies an A-form epichromatin conformation of the DNA. Nucleus 2018, 9, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Kusuzaki, K.; Murata, H.; Takeshita, H.; Hashiguchi, S.; Nozaki, T.; Emoto, K.; Ashihara, T.; Hirasawa, Y. Intracellular binding sites of acridine orange in living osteosarcoma cells. Anticancer Res. 2000, 20, 971–975. [Google Scholar]
- Thome, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, E.; Rudolf, K.; Cervinka, M. Camptothecin induces p53-dependent and -independent apoptogenic signaling in melanoma cells. Apoptosis 2011, 16, 1165–1176. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, M.S.; Fornace, A.J., Jr. Death and decoy receptors and p53-mediated apoptosis. Leukemia 2000, 14, 1509–1513. [Google Scholar] [CrossRef] [Green Version]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000547. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; Perez, D.; White, E. Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. 1996, 135, 1441–1455. [Google Scholar] [CrossRef]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—the p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Yin, H.; Zhang, H.; Wu, F.; Liu, Y.; Fu, Y.; Yu, D.; Zong, L. p53 mediates hydroxyurea resistance in aneuploid cells of colon cancer. Exp. Cell Res. 2019, 376, 39–48. [Google Scholar] [CrossRef]
- Yang, R.; Huang, B.; Zhu, Y.; Li, Y.; Liu, F.; Shi, J. Cell type-dependent bimodal p53 activation engenders a dynamic mechanism of chemoresistance. Sci. Adv. 2018, 4, eaat5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertozzi, D.; Marinello, J.; Manzo, S.G.; Fornari, F.; Gramantieri, L.; Capranico, G. The natural inhibitor of DNA topoisomerase I, camptothecin, modulates HIF-1alpha activity by changing miR expression patterns in human cancer cells. Mol. Cancer Ther. 2014, 13, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.H.; Hsu, S.H.; Hsu, H.W.; Huang, K.C.; Liu, W.; Wu, C.Y.; Huang, W.P.; Chen, J.Y.; Chen, B.H.; Chiu, C.C. Human nonsmall cell lung cancer cells can be sensitized to camptothecin by modulating autophagy. Int. J. Oncol. 2018, 53, 1967–1979. [Google Scholar] [PubMed] [Green Version]
- Tai, C.J.; Liu, C.H.; Pan, Y.C.; Wong, S.H.; Tai, C.J.; Richardson, C.D.; Lin, L.T. Chemovirotherapeutic Treatment Using Camptothecin Enhances Oncolytic Measles Virus-Mediated Killing of Breast Cancer Cells. Sci. Rep. 2019, 9, 6767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Li, Y.; Zamyatnin, A.A., Jr.; Werner, J.; Bazhin, A.V. Reactive oxygen species and colorectal cancer. J. Cell. Physiol. 2018, 233, 5119–5132. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.N.; Wilson, K.L. Partners and post-translational modifications of nuclear lamins. Chromosoma 2013, 122, 13–31. [Google Scholar] [CrossRef] [Green Version]
- Chao, C.C. Mechanisms of p53 degradation. Clin. Chim. Acta 2015, 438, 139–147. [Google Scholar] [CrossRef]
- Solomon, H.; Brauning, B.; Fainer, I.; Ben-Nissan, G.; Rabani, S.; Goldfinger, N.; Moscovitz, O.; Shakked, Z.; Rotter, V.; Sharon, M. Post-translational regulation of p53 function through 20S proteasome-mediated cleavage. Cell Death Differ. 2017, 24, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Roper, M.G. Measurement of DCF fluorescence as a measure of reactive oxygen species in murine islets of Langerhans. Anal. Methods 2014, 6, 3019–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, L.C.; Scott, A.P.; Marfell, B.J.; Boughaba, J.A.; Chojnowski, G.; Waterhouse, N.J. Measuring Cell Death by Propidium Iodide Uptake and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Chazotte, B. Labeling nuclear DNA using DAPI. Cold Spring Harb. Protoc. 2011, 2011, 5556. [Google Scholar] [CrossRef] [Green Version]
- Atale, N.; Gupta, S.; Yadav, U.C.; Rani, V. Cell-death assessment by fluorescent and nonfluorescent cytosolic and nuclear staining techniques. J. Microsc. 2014, 255, 7–19. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Christensen, M.E.; Waterhouse, N.J. Measuring Cell Death by Trypan Blue Uptake and Light Microscopy. Cold Spring Harb. Protoc. 2016, 7. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, E.J.; Lee, W.S.; Paramanantham, A.; Kim, H.J.; Shin, S.C.; Kim, G.S.; Jung, J.-M.; Ryu, C.H.; Hong, S.C.; Chung, K.H.; et al. p53 Enhances Artemisia annua L. Polyphenols-Induced Cell Death Through Upregulation of p53-Dependent Targets and Cleavage of PARP1 and Lamin A/C in HCT116 Colorectal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9315. https://doi.org/10.3390/ijms21239315
Jung EJ, Lee WS, Paramanantham A, Kim HJ, Shin SC, Kim GS, Jung J-M, Ryu CH, Hong SC, Chung KH, et al. p53 Enhances Artemisia annua L. Polyphenols-Induced Cell Death Through Upregulation of p53-Dependent Targets and Cleavage of PARP1 and Lamin A/C in HCT116 Colorectal Cancer Cells. International Journal of Molecular Sciences. 2020; 21(23):9315. https://doi.org/10.3390/ijms21239315
Chicago/Turabian StyleJung, Eun Joo, Won Sup Lee, Anjugam Paramanantham, Hye Jung Kim, Sung Chul Shin, Gon Sup Kim, Jin-Myung Jung, Chung Ho Ryu, Soon Chan Hong, Ky Hyun Chung, and et al. 2020. "p53 Enhances Artemisia annua L. Polyphenols-Induced Cell Death Through Upregulation of p53-Dependent Targets and Cleavage of PARP1 and Lamin A/C in HCT116 Colorectal Cancer Cells" International Journal of Molecular Sciences 21, no. 23: 9315. https://doi.org/10.3390/ijms21239315
APA StyleJung, E. J., Lee, W. S., Paramanantham, A., Kim, H. J., Shin, S. C., Kim, G. S., Jung, J. -M., Ryu, C. H., Hong, S. C., Chung, K. H., & Kim, C. W. (2020). p53 Enhances Artemisia annua L. Polyphenols-Induced Cell Death Through Upregulation of p53-Dependent Targets and Cleavage of PARP1 and Lamin A/C in HCT116 Colorectal Cancer Cells. International Journal of Molecular Sciences, 21(23), 9315. https://doi.org/10.3390/ijms21239315