Unraveling the Role of Leptin in Liver Function and Its Relationship with Liver Diseases


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
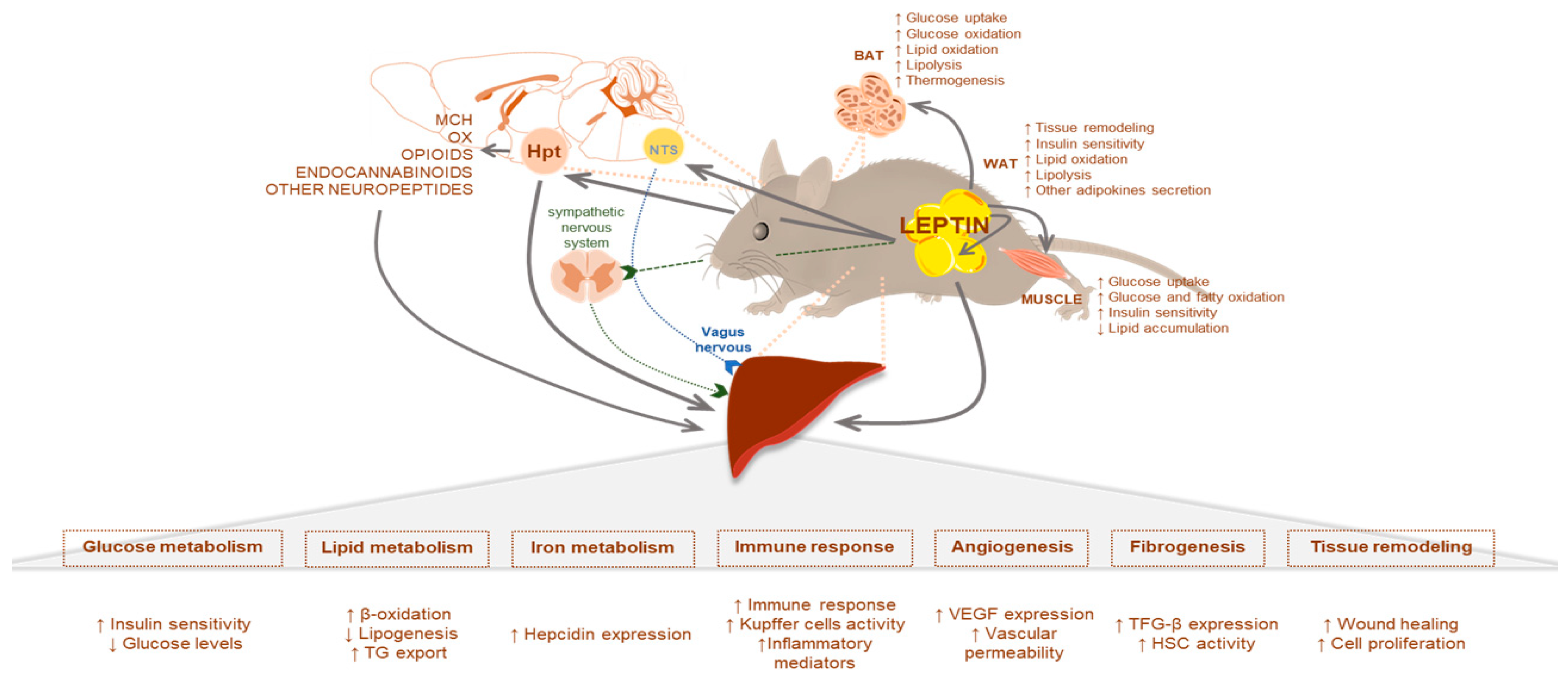
2. Leptin Signaling in Liver Metabolism: The Physiological Role
2.1. Leptin Effects Mediated by the CNS
2.2. Leptin and Glucose Homeostasis
2.3. Leptin and Lipid Homeostasis
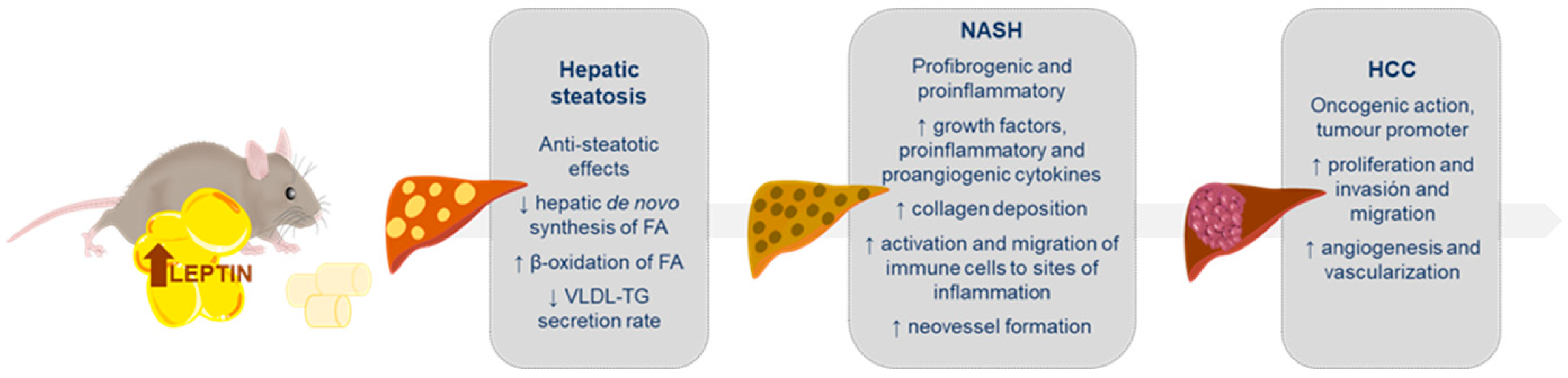
3. Leptin in Non-Alcoholic Fatty Liver Disease
3.1. Leptin in Liver Steatosis
3.2. Leptin in Non-Alcoholic Steatohepatitis (NASH)
3.3. Leptin in Hepatocarcinoma (HCC)
3.4. Leptin and NAFLD in Human Studies
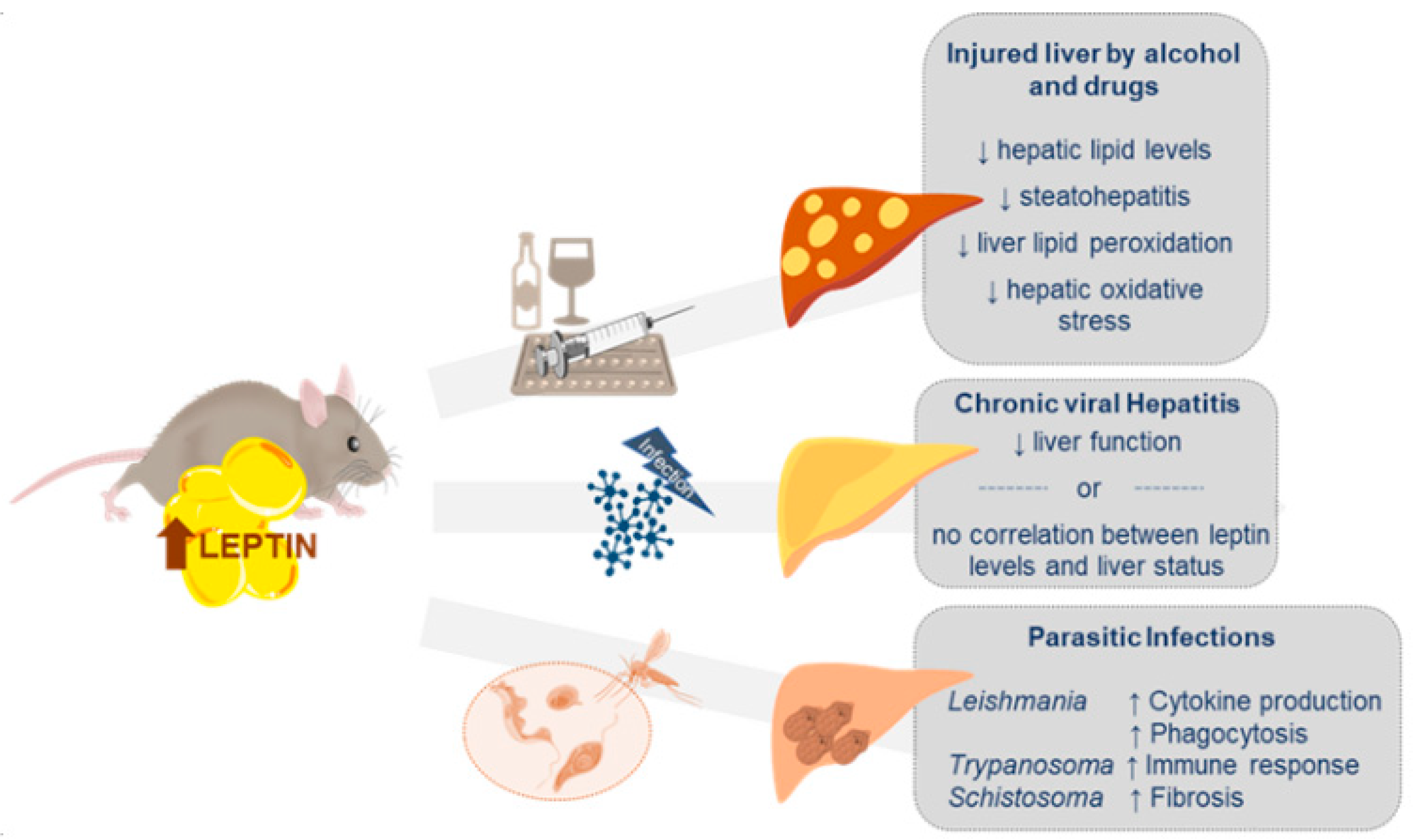
4. Leptin and Other Liver Pathologies
4.1. Liver Injury by Alcohol and Drugs
4.2. Chronic Viral Hepatitis (CVH)
4.3. Other Liver Diseases
5. Leptin Role in the Liver Regeneration Process
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AgRP | Agoutí-related protein |
| AKT | Protein kinase B |
| ALD | Alcoholic liver disease |
| BAT | Brown adipose tissue |
| BMI | body mass index |
| CCK | Cholecystokinin |
| CD | Cluster of differentiation |
| ChREBP | Carbohydrate responsive element binding protein |
| CNS | Central nervous system |
| Con A | Concanavalin A |
| CVH | Chronic viral hepatitis |
| DAG | Diacylglycerol |
| DC | Dendritic cells |
| DEN | Diethylnitrosamine |
| DNL | De novo lipogenesis |
| EGFR | Epidermal growth factor receptor |
| ELOVL5 | Elongation of very-long-chain fatty acids protein 5 |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FA | Fatty acid |
| FoxO1 | Forkhead box protein O1 |
| GA | Guanabenz acetate |
| GABA | γ-aminobutyric acid |
| GLP1 | Glucagon-like-protein 1 |
| GS3Kβ | Glycogen synthase kinase 3 beta |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HepG2 | Hepatoma G2 |
| HF1a | Hypoxia-inducible factor 1a |
| HPA | Hypothalamic–pituitary–adrenal |
| HSC | Hepatic stellate cells |
| ICAM-1 | Intercellular adhesion molecule- 1 |
| ICV | Intracerebroventricular |
| IFN-γ | Interferon gamma |
| IP | Intraperitoneal |
| IR | Insulin resistance |
| IRS1 | Insulin receptor substrate 1 |
| JAK | Janus kinase |
| LEPRs | Leptin receptors |
| LHA | Lateral hypothalamus area |
| LPL | Lipoprotein lipase |
| LPS | Lipopolysaccharide |
| MCH | Melanin-concentrating hormone |
| miR | MicroRNA |
| mTOR | Mammalian target of rapamycin |
| MMP | Matrix metalloproteinase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NPY | Neuropeptide Y |
| OX | Orexin |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PHx | Partial hepatectomy |
| PI3K | Phosphatidylinositol 3-kinase |
| PKCε | Protein kinase C-epsilon |
| POMC | Pro-opiomelanocortin |
| PPARα | Peroxisome proliferator-activated receptors alpha |
| PPARγ | Peroxisome proliferator-activated receptors gamma |
| SS | Simple steatosis |
| STAT-3 | Signal transducer and activator of transcription 3 |
| TG | Triglycerides |
| TGFβ1 | Transforming growth factor β 1 |
| TIMP | Tissue inhibitor of metalloproteinase |
| TLR4 | Toll-like receptor 4 |
| VEGF | Vascular endothelial growth factor |
| VLA-2 | Very late antigen-2 |
| VLDL | Very-low density lipoproteins |
| WAT | White adipose tissue |
References
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B. Is leptin the parabiotic “satiety” factor? Past and present interpretations. Appetite 2013, 61, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervey, G.R. The effects of lesions in the hypothalamus in parabiotic rats. J. Physiol. 1959, 145, 336–352. [Google Scholar] [CrossRef]
- Coleman, D.L. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 1973, 9, 294–298. [Google Scholar] [CrossRef]
- Coleman, D.L. Obese and diabetes: Two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 1978, 14, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; Moore, K.J.; Breitbart, R.E.; et al. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.H.; Proenca, R.; Montez, J.M.; Carroll, K.M.; Darvishzadeh, J.G.; Lee, J.I.; Friedman, J.M. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996, 379, 632–635. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e1220. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia 2016, 59, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Stefanou, N.; Papanikolaou, V.; Furukawa, Y.; Nakamura, Y.; Tsezou, A. Leptin as a critical regulator of hepatocellular carcinoma development through modulation of human telomerase reverse transcriptase. BMC Cancer 2010, 10, 442. [Google Scholar] [CrossRef] [Green Version]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Lollmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Chen, X.; Mozzoli, M.; Ryan, I. Effect of fasting on serum leptin in normal human subjects. J. Clin. Endocrinol. Metab. 1996, 81, 3419–3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.L.; Heist, K.; DePaoli, A.M.; Veldhuis, J.D.; Mantzoros, C.S. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J. Clin. Investig. 2003, 111, 1409–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdan, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef]
- Caron, A.; Dungan Lemko, H.M.; Castorena, C.M.; Fujikawa, T.; Lee, S.; Lord, C.C.; Ahmed, N.; Lee, C.E.; Holland, W.L.; Liu, C.; et al. POMC neurons expressing leptin receptors coordinate metabolic responses to fasting via suppression of leptin levels. Elife 2018, 7. [Google Scholar] [CrossRef]
- Balthasar, N.; Coppari, R.; McMinn, J.; Liu, S.M.; Lee, C.E.; Tang, V.; Kenny, C.D.; McGovern, R.A.; Chua, S.C., Jr.; Elmquist, J.K.; et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 2004, 42, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Flak, J.N.; Myers, M.G., Jr. Minireview: CNS Mechanisms of Leptin Action. Mol. Endocrinol. 2016, 30, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Grill, H.J. Leptin and the systems neuroscience of meal size control. Front. Neuroendocrinol. 2010, 31, 61–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J. The long road to leptin. J. Clin. Investig. 2016, 126, 4727–4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalamaga, M.; Chou, S.H.; Shields, K.; Papageorgiou, P.; Polyzos, S.A.; Mantzoros, C.S. Leptin at the intersection of neuroendocrinology and metabolism: Current evidence and therapeutic perspectives. Cell Metab. 2013, 18, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantzoros, C.S.; Magkos, F.; Brinkoetter, M.; Sienkiewicz, E.; Dardeno, T.A.; Kim, S.Y.; Hamnvik, O.P.; Koniaris, A. Leptin in human physiology and pathophysiology. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E567–E584. [Google Scholar] [CrossRef]
- Varela, L.; Horvath, T.L. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012, 13, 1079–1086. [Google Scholar] [CrossRef]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef]
- D’Souza, A.M.; Neumann, U.H.; Glavas, M.M.; Kieffer, T.J. The glucoregulatory actions of leptin. Mol. Metab. 2017, 6, 1052–1065. [Google Scholar] [CrossRef]
- Martinez-Sanchez, N. There and Back Again: Leptin Actions in White Adipose Tissue. Int. J. Mol. Sci. 2020, 21, 6039. [Google Scholar] [CrossRef]
- Allen, A.M.; Therneau, T.M.; Larson, J.J.; Coward, A.; Somers, V.K.; Kamath, P.S. Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: A 20 year-community study. Hepatology 2018, 67, 1726–1736. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.A.; Ahmed, A.; Kim, D. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Gut Liver 2020, 14, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Danford, C.J.; Lai, M. NAFLD: A multisystem disease that requires a multidisciplinary approach. Frontline Gastroenterol. 2019, 10, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Mikolasevic, I.; Milic, S.; Turk Wensveen, T.; Grgic, I.; Jakopcic, I.; Stimac, D.; Wensveen, F.; Orlic, L. Nonalcoholic fatty liver disease - A multisystem disease? World J. Gastroenterol. 2016, 22, 9488–9505. [Google Scholar] [CrossRef] [PubMed]
- Targher, G. What’s Past Is Prologue: History of Nonalcoholic Fatty Liver Disease. Metabolites 2020, 10, 397. [Google Scholar] [CrossRef] [PubMed]
- Safar Zadeh, E.; Lungu, A.O.; Cochran, E.K.; Brown, R.J.; Ghany, M.G.; Heller, T.; Kleiner, D.E.; Gorden, P. The liver diseases of lipodystrophy: The long-term effect of leptin treatment. J. Hepatol. 2013, 59, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Bray, G.A. Obesity, a disorder of nutrient partitioning: The MONA LISA hypothesis. J. Nutr. 1991, 121, 1146–1162. [Google Scholar] [CrossRef]
- Cohen, S.M.; Werrmann, J.G.; Tota, M.R. 13C NMR study of the effects of leptin treatment on kinetics of hepatic intermediary metabolism. Proc. Natl. Acad. Sci. USA 1998. [Google Scholar] [CrossRef] [Green Version]
- Huynh, F.K.; Neumann, U.H.; Wang, Y.; Rodrigues, B.; Kieffer, T.J.; Covey, S.D. A role for hepatic leptin signaling in lipid metabolism via altered very low density lipoprotein composition and liver lipase activity in mice. Hepatology 2013, 57, 543–554. [Google Scholar] [CrossRef]
- Gallardo, N.; Bonzón-Kulichenko, E.; Fernández-Agulló, T.; Moltó, E.; Gómez-Alonso, S.; Blanco, P.; Carrascosa, J.M.; Ros, M.; Andrés, A. Tissue-specific effects of central leptin on the expression of genes involved in lipid metabolism in liver and white adipose tissue. Endocrinology 2007, 148, 5604–5610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, F.K.; Levi, J.; Denroche, H.C.; Gray, S.L.; Voshol, P.J.; Neumann, U.H.; Speck, M.; Chua, S.C.; Covey, S.D.; Kieffer, T.J. Disruption of hepatic leptin signaling protects mice from age- and diet-related glucose intolerance. Diabetes 2010, 59, 3032–3040. [Google Scholar] [CrossRef] [Green Version]
- Fujikawa, T.; Coppari, R. Living without insulin: The role of leptin signaling in the hypothalamus. Front. Neurosci. 2015, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, A.; Lee, S.; Elmquist, J.K.; Gautron, L. Leptin and brain-adipose crosstalks. Nat. Rev. Neurosci. 2018, 19, 153–165. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Munzberg, H.; Leinninger, G.M.; Leshan, R.L. The geometry of leptin action in the brain: More complicated than a simple ARC. Cell Metab. 2009, 9, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmouni, K.; Haynes, W.G. Leptin signaling pathways in the central nervous system: Interactions between neuropeptide Y and melanocortins. Bioessays 2001, 23, 1095–1099. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef]
- Lee, J.; Hong, S.W.; Rhee, E.J.; Lee, W.Y. GLP-1 Receptor Agonist and Non-Alcoholic Fatty Liver Disease. Diabetes Metab. J. 2012, 36, 262–267. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Yang, Q.; Huesman, S.; Rider, T.; Lo, C.C. Lipid transport in cholecystokinin knockout mice. Physiol. Behav. 2015, 151, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [Green Version]
- Goforth, P.B.; Leinninger, G.M.; Patterson, C.M.; Satin, L.S.; Myers, M.G., Jr. Leptin acts via lateral hypothalamic area neurotensin neurons to inhibit orexin neurons by multiple GABA-independent mechanisms. J. Neurosci. 2014, 34, 11405–11415. [Google Scholar] [CrossRef]
- Morello, G.; Imperatore, R.; Palomba, L.; Finelli, C.; Labruna, G.; Pasanisi, F.; Sacchetti, L.; Buono, L.; Piscitelli, F.; Orlando, P.; et al. Orexin-A represses satiety-inducing POMC neurons and contributes to obesity via stimulation of endocannabinoid signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 4759–4764. [Google Scholar] [CrossRef] [Green Version]
- Milbank, E.; Lopez, M. Orexins/Hypocretins: Key Regulators of Energy Homeostasis. Front. Endocrinol. 2019, 10, 830. [Google Scholar] [CrossRef]
- Imbernon, M.; Beiroa, D.; Vazquez, M.J.; Morgan, D.A.; Veyrat-Durebex, C.; Porteiro, B.; Diaz-Arteaga, A.; Senra, A.; Busquets, S.; Velasquez, D.A.; et al. Central melanin-concentrating hormone influences liver and adipose metabolism via specific hypothalamic nuclei and efferent autonomic/JNK1 pathways. Gastroenterology 2013, 144, 636–649.e636. [Google Scholar] [CrossRef] [Green Version]
- Manceau, R.; Majeur, D.; Alquier, T. Neuronal control of peripheral nutrient partitioning. Diabetologia 2020, 63, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Formoso, G.; Perez-Sieira, S.; Gonzalez-Touceda, D.; Dieguez, C.; Tovar, S. Leptin, 20 years of searching for glucose homeostasis. Life Sci. 2015, 140, 4–9. [Google Scholar] [CrossRef]
- Morton, G.J.; Schwartz, M.W. Leptin and the central nervous system control of glucose metabolism. Physiol. Rev. 2011, 91, 389–411. [Google Scholar] [CrossRef] [Green Version]
- Li, R.J.W.; Zhang, S.Y.; Lam, T.K.T. Interaction of glucose sensing and leptin action in the brain. Mol. Metab. 2020, 39, 101011. [Google Scholar] [CrossRef]
- Amitani, M.; Asakawa, A.; Amitani, H.; Inui, A. The role of leptin in the control of insulin-glucose axis. Front. Neurosci. 2013, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Denroche, H.C.; Huynh, F.K.; Kieffer, T.J. The role of leptin in glucose homeostasis. J. Diabetes Investig. 2012, 3, 115–129. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A. Leptin transport across the blood-brain barrier: Implications for the cause and treatment of obesity. Curr. Pharm. Des. 2001, 7, 125–133. [Google Scholar] [CrossRef]
- Yarandi, S.S.; Hebbar, G.; Sauer, C.G.; Cole, C.R.; Ziegler, T.R. Diverse roles of leptin in the gastrointestinal tract: Modulation of motility, absorption, growth, and inflammation. Nutrition 2011, 27, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Minokoshi, Y.; Kim, Y.B.; Peroni, O.D.; Fryer, L.G.; Muller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef]
- Kieffer, T.J.; Habener, J.F. The adipoinsular axis: Effects of leptin on pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E1–E14. [Google Scholar] [CrossRef]
- Huang, W.; Dedousis, N.; Bandi, A.; Lopaschuk, G.D.; O’Doherty, R.M. Liver triglyceride secretion and lipid oxidative metabolism are rapidly altered by leptin in vivo. Endocrinology 2006, 147, 1480–1487. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.P.; Tall, A.R. Transcriptional Profiling Reveals Global Defects in Energy Metabolism, Lipoprotein, and Bile Acid Synthesis and Transport with Reversal by Leptin Treatment in Ob/ob Mouse Liver. J. Biol. Chem. 2001, 276, 49066–49076. [Google Scholar] [CrossRef] [Green Version]
- Kakuma, T.; Lee, Y.; Higa, M.; Wang, Z.W.; Pan, W.; Shimomura, I.; Unger, R.H. Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc. Natl. Acad. Sci. USA 2000, 97, 8536–8541. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Wirtz, M.; Parker, N.; Hogan, M.; Strahler, J.; Michailidis, G.; Schmidt, S.; Vidal-Puig, A.; Diano, S.; Andrews, P.; et al. Leptin-mediated changes in hepatic mitochondrial metabolism, structure, and protein levels. Proc. Natl. Acad. Sci. USA 2009, 106, 13100–13105. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, L.; Ebihara, K.; Kusakabe, T.; Aotani, D.; Yamamoto-Kataoka, S.; Sakai, T.; Aizawa-Abe, M.; Yamamoto, Y.; Fujikura, J.; Hayashi, T.; et al. Leptin activates hepatic 5′-AMP-activated protein kinase through sympathetic nervous system and α1-adrenergic receptor: A potential mechanism for improvement of fatty liver in lipodystrophy by leptin. J. Biol. Chem. 2012, 287, 40441–40447. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; Galgani, M.; De Rosa, V.; Carbone, F.; La Rocca, C.; Ranucci, G.; Iorio, R.; Matarese, G. Leptin: The Prototypic Adipocytokine and its Role in NAFLD. Curr. Pharm. Des. 2010, 16, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wang, M.Y.; Kakuma, T.; Wang, Z.W.; Babcock, E.; McCorkle, K.; Higa, M.; Zhou, Y.T.; Unger, R.H. Liporegulation in diet-induced obesity. The antisteatotic role of hyperleptinemia. J. Biol. Chem. 2001, 276, 5629–5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Li, J.; Xiang, W.; Cui, Y.; Xie, B.; Wang, X.; Xu, Z.; Gan, L. Metformin increases hepatic leptin receptor and decreases steatosis in mice. J. Endocrinol. 2016, 230, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Wang, Y.; Cline, G.W.; Rabin-Court, A.; Song, J.D.; Dufour, S.; Zhang, X.M.; Petersen, K.F.; Shulman, G.I. Leptin Mediates a Glucose-Fatty Acid Cycle to Maintain Glucose Homeostasis in Starvation. Cell 2018, 172, 234–248.e217. [Google Scholar] [CrossRef]
- Zhao, S.; Zhu, Y.; Schultz, R.D.; Li, N.; He, Z.; Zhang, Z.; Caron, A.; Zhu, Q.; Sun, K.; Xiong, W.; et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab. 2019, 30, 706–719.e706. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Ortmeyer, H.K.; Stefanovic-Racic, M.; O’Doherty, R.M. Leptin augments the acute suppressive effects of insulin on hepatic very low-density lipoprotein production in rats. Endocrinology 2009, 150, 2169–2174. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, N.; Ogawa, Y.; Masuzaki, H.; Ebihara, K.; Aizawa-Abe, M.; Satoh, N.; Ishikawa, E.; Fujisawa, Y.; Kosaki, A.; Yamada, K.; et al. Decreased triglyceride-rich lipoproteins in transgenic skinny mice overexpressing leptin. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E334–E339. [Google Scholar] [CrossRef]
- Hackl, M.T.; Fürnsinn, C.; Schuh, C.M.; Krssak, M.; Carli, F.; Guerra, S.; Freudenthaler, A.; Baumgartner-Parzer, S.; Helbich, T.H.; Luger, A.; et al. Brain leptin reduces liver lipids by increasing hepatic triglyceride secretion and lowering lipogenesis. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Scheja, L.; Heeren, J. Metabolic interplay between white, beige, brown adipocytes and the liver. J. Hepatol. 2016, 64, 1176–1186. [Google Scholar] [CrossRef] [Green Version]
- Olefsky, J.M. Fat talks, liver and muscle listen. Cell 2008, 134, 914–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renquist, B.J.; Murphy, J.G.; Larson, E.A.; Olsen, D.; Klein, R.F.; Ellacott, K.L.J.; Cone, R.D. Melanocortin-3 receptor regulates the normal fasting response. Proc. Natl. Acad. Sci. USA 2012, 109, 1489–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patni, N.; Garg, A. Congenital generalized lipodystrophies—New insights into metabolic dysfunction. Nat. Rev. Endocrinol. 2015, 11, 522. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [Green Version]
- Bosy-Westphal, A.; Braun, W.; Albrecht, V.; Muller, M.J. Determinants of ectopic liver fat in metabolic disease. Eur. J. Clin. Nutr. 2019, 73, 209–214. [Google Scholar] [CrossRef]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim. 2015, 1, 15080. [Google Scholar] [CrossRef]
- WGO Practice Guideline—NAFLD & NASH. 2012. Available online: https://www.worldgastroenterology.org/ (accessed on 27 September 2020).
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 2013, 59, 1174–1197. [Google Scholar] [CrossRef]
- Solga, S.F.; Diehl, A.M. Non-alcoholic fatty liver disease: Lumen-liver interactions and possible role for probiotics. J. Hepatol. 2003, 38, 681–687. [Google Scholar] [CrossRef]
- Chalasani, N.; Szabo, G. Preface. Alcoholic and Non-Alcoholic Fatty Liver Disease: Bench to Bedside; Springer International Publishing: Berlin/Heidelberg, Germany, 2015. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Trak-Smayra, V.; Paradis, V.; Massart, J.; Nasser, S.; Jebara, V.; Fromenty, B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int. J. Exp. Pathol. 2011, 92, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Asilmaz, E.; Cohen, P.; Miyazaki, M.; Dobrzyn, P.; Ueki, K.; Fayzikhodjaeva, G.; Soukas, A.A.; Kahn, C.R.; Ntambi, J.M.; Socci, N.D.; et al. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J. Clin. Investig. 2004, 113, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Denechaud, P.D.; Dentin, R.; Girard, J.; Postic, C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Lett. 2008, 582, 68–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef] [Green Version]
- Fishman, S.; Muzumdar, R.H.; Atzmon, G.; Ma, X.; Yang, X.; Einstein, F.H.; Barzilai, N. Resistance to leptin action is the major determinant of hepatic triglyceride accumulation in vivo. FASEB J. 2007, 21, 53–60. [Google Scholar] [CrossRef]
- Qiu, L.; Lin, J.; Xu, F.; Gao, Y.; Zhang, C.; Liu, Y.; Luo, Y.; Yang, J.Y. Inhibition of aldose reductase activates hepatic peroxisome proliferator-activated receptor-alpha and ameliorates hepatosteatosis in diabetic db/db mice. Exp. Diabetes Res. 2012, 2012, 789730. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Chen, J.; Wang, J.; Yao, J.; Huang, Y.; Zhang, G.; Bao, Z. Circadian Clock Genes in the Metabolism of Non-alcoholic Fatty Liver Disease. Front. Physiol. 2019, 10, 423. [Google Scholar] [CrossRef] [Green Version]
- Mazzoccoli, G.; De Cosmo, S.; Mazza, T. The Biological Clock: A Pivotal Hub in Non-alcoholic Fatty Liver Disease Pathogenesis. Front. Physiol. 2018, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Elkin, K.; Yip, J.; Guan, L.; Han, W.; Ding, Y. From circadian clocks to non-alcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Dibner, C.; Gachon, F. Circadian Dysfunction and Obesity: Is Leptin the Missing Link? Cell Metab. 2015, 22, 359–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in NAFLD - Revisited After a Decade. Hepatology 2020. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Thematic review series: Lipotoxicity: Many roads to cell dysfunction and cell death lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef] [Green Version]
- Perez-Perez, A.; Sanchez-Jimenez, F.; Vilarino-Garcia, T.; Sanchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef]
- Ikejima, K.; Honda, H.; Yoshikawa, M.; Hirose, M.; Kitamura, T.; Takei, Y.; Sato, N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology 2001, 34, 288–297. [Google Scholar] [CrossRef]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, I.A.; Farrell, G.C.; Schriemer, R.; Robertson, G.R. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol. 2002, 37, 206–213. [Google Scholar] [CrossRef]
- Yan, C.; Yang, Q.; Shen, H.M.; Spitsbergen, J.M.; Gong, Z. Chronically high level of tgfb1a induction causes both hepatocellular carcinoma and cholangiocarcinoma via a dominant Erk pathway in zebrafish. Oncotarget 2017, 8, 77096–77109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikejima, K.; Takei, Y.; Honda, H.; Hirose, M.; Yoshikawa, M.; Zhang, Y.J.; Lang, T.; Fukuda, T.; Yamashina, S.; Kitamura, T.; et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology 2002, 122, 1399–1410. [Google Scholar] [CrossRef]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palu, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [Green Version]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC—Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Ganini, D.; Tokar, E.J.; Kumar, A.; Das, S.; Corbett, J.; Kadiiska, M.B.; Waalkes, M.P.; Diehl, A.M.; Mason, R.P. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. J. Hepatol. 2013, 58, 778–784. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Sakaida, I.; Uchida, K.; Terai, S.; Okita, K. Leptin enhances TNF-α production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sci. 2005, 77, 1502–1515. [Google Scholar] [CrossRef]
- Metlakunta, A.; Huang, W.; Stefanovic-Racic, M.; Dedousis, N.; Sipula, I.; O’Doherty, R.M. Kupffer cells facilitate the acute effects of leptin on hepatic lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E11–E18. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Leclercq, I.; Brymora, J.M.; Xu, N.; Ramezani-Moghadam, M.; London, R.M.; Brigstock, D.; George, J. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 2009, 137, 713–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Zheng, S.; Chen, A. Curcumin eliminates leptin’s effects on hepatic stellate cell activation via interrupting leptin signaling. Endocrinology 2009, 150, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Tsai, T.H.; Huang, Y.T.; Lee, T.Y.; Chan, C.C.; Lee, K.C.; Lin, H.C. Hepatic endothelin-1 and endocannabinoids-dependent effects of hyperleptinemia in nonalcoholic steatohepatitis-cirrhotic rats. Hepatology 2012, 55, 1540–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Minicis, S.; Seki, E.; Oesterreicher, C.; Schnabl, B.; Schwabe, R.F.; Brenner, D.A. Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology 2008, 48, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Ikeda, K.; Rockey, D.C.; Friedman, S.L.; Anania, F.A. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology 2002, 35, 762–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourhoseini, S.; Seth, R.K.; Das, S.; Dattaroy, D.; Kadiiska, M.B.; Xie, G.; Michelotti, G.A.; Nagarkatti, M.; Diehl, A.M.; Chatterjee, S. Upregulation of miR21 and repression of Grhl3 by leptin mediates sinusoidal endothelial injury in experimental nonalcoholic steatohepatitis. PLoS ONE 2015, 10, e0116780. [Google Scholar] [CrossRef]
- Boutari, C.; Perakakis, N.; Mantzoros, C.S. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol. Metab. 2018, 33, 33–43. [Google Scholar] [CrossRef]
- Dattaroy, D.; Pourhoseini, S.; Das, S.; Alhasson, F.; Seth, R.K.; Nagarkatti, M.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G298–G312. [Google Scholar] [CrossRef]
- Loyer, X.; Paradis, V.; Henique, C.; Vion, A.C.; Colnot, N.; Guerin, C.L.; Devue, C.; On, S.; Scetbun, J.; Romain, M.; et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARalpha expression. Gut 2016, 65, 1882–1894. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Yang, Y.L.; Wang, P.W.; Wang, F.S.; Huang, Y.H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; Correia de Sousa, M.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Landrier, J.F.; Derghal, A.; Mounien, L. MicroRNAs in Obesity and Related Metabolic Disorders. Cells 2019, 8, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, X.; Cheng, F.; Ji, L.; Zhu, X.; Cao, Q.; Zhang, Y.; Jia, X.; Zhou, Q.; Guan, W.; Zhou, Y. Leptin reduces microRNA-122 level in hepatic stellate cells in vitro and in vivo. Mol. Immunol. 2017, 92, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Zhu, X.; Zhai, X.; Ji, L.; Cheng, F.; Zhu, Y.; Yu, P.; Zhou, Y. Leptin suppresses microRNA-122 promoter activity by phosphorylation of foxO1 in hepatic stellate cell contributing to leptin promotion of mouse liver fibrosis. Toxicol. Appl. Pharmacol. 2018, 339, 143–150. [Google Scholar] [CrossRef]
- Li, Z.; Ji, L.; Su, S.; Zhu, X.; Cheng, F.; Jia, X.; Zhou, Q.; Zhou, Y. Leptin up-regulates microRNA-27a/b-3p level in hepatic stellate cells. Exp. Cell Res. 2018, 366, 63–70. [Google Scholar] [CrossRef]
- Ji, J.; Zhang, J.; Huang, G.; Qian, J.; Wang, X.; Mei, S. Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett. 2009, 583, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Guan, W.; Qiao, H.; Cheng, Y.; Li, Z.; Zhai, X.; Zhou, Y. GATA binding protein 2 mediates leptin inhibition of PPARγ1 expression in hepatic stellate cells and contributes to hepatic stellate cell activation. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2367–2377. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.K.; Titus, M.A.; Ding, X.; Floyd, J.; Srinivasan, S.; Sitaraman, S.V.; Anania, F.A. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: Mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 2004, 18, 1612–1614. [Google Scholar] [CrossRef]
- Aleffi, S.; Petrai, I.; Bertolani, C.; Parola, M.; Colombatto, S.; Novo, E.; Vizzutti, F.; Anania, F.A.; Milani, S.; Rombouts, K.; et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 2005, 42, 1339–1348. [Google Scholar] [CrossRef]
- Tang, M.; Potter, J.J.; Mezey, E. Leptin enhances the effect of transforming growth factor β in increasing type I collagen formation. Biochem. Biophys. Res. Commun. 2002, 297, 906–911. [Google Scholar] [CrossRef]
- Cao, Q.; Mak, K.M.; Ren, C.; Lieber, C.S. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells. Respective roles of the JAK/STAT and JAK-mediated H2O2-dependent MAPK pathways. J. Biol. Chem. 2004, 279, 4292–4304. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Mak, K.M.; Lieber, C.S. Leptin represses matrix metalloproteinase-1 gene expression in LX2 human hepatic stellate cells. J. Hepatol. 2007, 46, 124–133. [Google Scholar] [CrossRef]
- Roeb, E. Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol. 2018, 68–69, 463–473. [Google Scholar] [CrossRef] [PubMed]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef]
- La Cava, A. Leptin in inflammation and autoimmunity. Cytokine 2017, 98, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Maya-Monteiro, C.M.; Bozza, P.T. Leptin and mTOR: Partners in metabolism and inflammation. Cell Cycle 2008, 7, 1713–1717. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.E.; Klintworth, H.; Kowdley, K.V. Iron metabolism in Nonalcoholic Fatty Liver Disease. Curr. Gastroenterol. Rep. 2012, 14, 8–16. [Google Scholar] [CrossRef]
- Chung, B.; Matak, P.; McKie, A.T.; Sharp, P. Leptin increases the expression of the iron regulatory hormone hepcidin in HuH7 human hepatoma cells. J. Nutr. 2007, 137, 2366–2370. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kuragano, T.; Kimura, T.; Nanami, M.; Hasuike, Y.; Nakanishi, T. Interplay of adipocyte and hepatocyte: Leptin upregulates hepcidin. Biochem. Biophys. Res. Commun. 2018, 495, 1548–1554. [Google Scholar] [CrossRef]
- Marmur, J.; Beshara, S.; Eggertsen, G.; Onelov, L.; Albiin, N.; Danielsson, O.; Hultcrantz, R.; Stal, P. Hepcidin levels correlate to liver iron content, but not steatohepatitis, in non-alcoholic fatty liver disease. BMC Gastroenterol. 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Auguet, T.; Aragones, G.; Berlanga, A.; Martinez, S.; Sabench, F.; Binetti, J.; Aguilar, C.; Porras, J.A.; Molina, A.; Del Castillo, D.; et al. Hepcidin in morbidly obese women with non-alcoholic fatty liver disease. PLoS ONE 2017, 12, e0187065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.N.; Lee, K.T.; Ker, C.G. Leptin in hepatocellular carcinoma. World J. Gastroenterol. 2010, 16, 5801–5809. [Google Scholar] [CrossRef] [PubMed]
- Karagozian, R.; Derdák, Z.; Baffy, G. Obesity-associated mechanisms of hepatocarcinogenesis. Metab. Clin. Exp. 2014, 63, 607–617. [Google Scholar] [CrossRef]
- Zoller, H.; Tilg, H. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Metab. Clin. Exp. 2016, 65, 1151–1160. [Google Scholar] [CrossRef]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Wang, J.; Fu, P.P.; Sharma, S.; Nagalingam, A.; Mells, J.; Handy, J.; Page, A.J.; Cohen, C.; Anania, F.A.; et al. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 2010, 52, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Cheung, O.K.; Cheng, A.S. Gender Differences in Adipocyte Metabolism and Liver Cancer Progression. Front. Genet. 2016, 7, 168. [Google Scholar] [CrossRef]
- Manieri, E.; Herrera-Melle, L.; Mora, A.; Tomas-Loba, A.; Leiva-Vega, L.; Fernandez, D.I.; Rodriguez, E.; Moran, L.; Hernandez-Cosido, L.; Torres, J.L.; et al. Adiponectin accounts for gender differences in hepatocellular carcinoma incidence. J. Exp. Med. 2019, 216, 1108–1119. [Google Scholar] [CrossRef]
- Shen, M.; Shi, H. Estradiol and Estrogen Receptor Agonists Oppose Oncogenic Actions of Leptin in HepG2 Cells. PLoS ONE 2016, 11, e0151455. [Google Scholar] [CrossRef]
- Elinav, E.; Abd-Elnabi, A.; Pappo, O.; Bernstein, I.; Klein, A.; Engelhardt, D.; Rabbani, E.; Ilan, Y. Suppression of hepatocellular carcinoma growth in mice via leptin, is associated with inhibition of tumor cell growth and natural killer cell activation. J. Hepatol. 2006, 44, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Hu, Y.; Dong, F.; Xu, X.; Hu, A.; Gao, G. Hepatoma cell-derived leptin downregulates the immunosuppressive function of regulatory T-cells to enhance the anti-tumor activity of CD8 + T-cells. Immunol. Cell Biol. 2016, 94, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.J.; Lau, K.N.; Johnson, S.; Martinie, J.B.; Iannitti, D.A.; McKillop, I.H.; Sindram, D. Leptin inhibits hepatocellular carcinoma proliferation via p38-MAPK-dependent signalling. HPB 2011, 13, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.; Wang, S.H.; Chen, Y.R.; Li, Z.L.; Chin, Y.T.; Yang, Y.S.H.; Wu, Y.H.; Su, K.W.; Chu, H.R.; Chiu, H.C.; et al. Leptin-derived peptides block leptin-induced proliferation by reducing expression of pro-inflammatory genes in hepatocellular carcinoma cells. Food Chem. Toxicol. 2019, 133, 110808. [Google Scholar] [CrossRef]
- Bergmann, J.; Muller, M.; Baumann, N.; Reichert, M.; Heneweer, C.; Bolik, J.; Lucke, K.; Gruber, S.; Carambia, A.; Boretius, S.; et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017, 65, 89–103. [Google Scholar] [CrossRef]
- Newman, G.; Gonzalez-Perez, R.R. Leptin-cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Mittenbuhler, M.J.; Sprenger, H.G.; Gruber, S.; Wunderlich, C.M.; Kern, L.; Bruning, J.C.; Wunderlich, F.T. Hepatic leptin receptor expression can partially compensate for IL-6Ralpha deficiency in DEN-induced hepatocellular carcinoma. Mol. Metab. 2018, 17, 122–133. [Google Scholar] [CrossRef]
- Zhou, J.; Lei, W.; Shen, L.; Luo, H.S.; Shen, Z.X. Primary study of leptin and human hepatocellular carcinoma in vitro. World J. Gastroenterol. 2008, 14, 2900–2904. [Google Scholar] [CrossRef]
- Saxena, N.K.; Sharma, D.; Ding, X.; Lin, S.; Marra, F.; Merlin, D.; Anania, F.A. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 2497–2507. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chang, Y.C.; Liu, C.L.; Liu, T.P.; Chang, K.J.; Guo, I.C. Leptin induces proliferation and anti-apoptosis in human hepatocarcinoma cells by up-regulating cyclin D1 and down-regulating Bax via a Janus kinase 2-linked pathway. Endocr. Relat. Cancer 2007, 14, 513–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fava, G.; Alpini, G.; Rychlicki, C.; Saccomanno, S.; DeMorrow, S.; Trozzi, L.; Candelaresi, C.; Venter, J.; Di Sario, A.; Marzioni, M.; et al. Leptin enhances cholangiocarcinoma cell growth. Cancer Res. 2008, 68, 6752–6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, K.; Yang, H.; Xia, M.; Ara, A.I.; Mato, J.M.; Lu, S.C. Leptin’s mitogenic effect in human liver cancer cells requires induction of both methionine adenosyltransferase 2A and 2β. Hepatology 2008, 47, 521–531. [Google Scholar] [CrossRef] [Green Version]
- VanSaun, M.N.; Mendonsa, A.M.; Lee Gorden, D. Hepatocellular Proliferation Correlates with Inflammatory Cell and Cytokine Changes in a Murine Model of Nonalchoholic Fatty Liver Disease. PLoS ONE 2013, 8, e73054. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Q.; Lin, H.Z.; Hwang, J.; Diehl, A.M.; Chacko, V.P. Hepatic hyperplasia in noncirrhotic fatty livers: Is obesity-related hepatic steatosis a premalignant condition? Cancer Res. 2001, 61, 5016–5023. [Google Scholar]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Kitade, M.; Yoshiji, H.; Kojima, H.; Ikenaka, Y.; Noguchi, R.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Asada, K.; et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology 2006, 44, 983–991. [Google Scholar] [CrossRef]
- Ribatti, D.; Belloni, A.S.; Nico, B.; Di Comite, M.; Crivellato, E.; Vacca, A. Leptin-leptin receptor are involved in angiogenesis in human hepatocellular carcinoma. Peptides 2008, 29, 1596–1602. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Machida, K. Existence of cancer stem cells in hepatocellular carcinoma: Myth or reality? Hepatol. Int. 2017, 11, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Uthaya Kumar, D.B.; Chen, C.L.; Liu, J.C.; Feldman, D.E.; Sher, L.S.; French, S.; DiNorcia, J.; French, S.W.; Naini, B.V.; Junrungsee, S.; et al. TLR4 Signaling via NANOG Cooperates With STAT3 to Activate Twist1 and Promote Formation of Tumor-Initiating Stem-Like Cells in Livers of Mice. Gastroenterology 2016, 150, 707–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.V.; Coutinho, J.; Carepa, F.; Costa, A.; Proença, H.; Cortez-Pinto, H. How adiponectin, leptin, and ghrelin orchestrate together and correlate with the severity of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Hossain, I.A.; Akter, S.; Rahman, M.K.; Ali, L. Gender specific association of serum leptin and insulinemic indices with nonalcoholic fatty liver disease in prediabetic subjects. PLoS ONE 2015, 10, e0142165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, B.Q.; Lu, L.L.; Yuan, C.; Xin, Y.N.; Xuan, S.Y. Leptin Receptor Gene Polymorphisms and the Risk of Non-Alcoholic Fatty Liver Disease and Coronary Atherosclerosis in the Chinese Han Population. Hepat. Mon. 2016, 16, e35055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Zheng, M.; Zou, T.; Liu, W.; Gu, X.; Zhang, X.; Cheng, X. The LEPR K109R and Q223R might contribute to the risk of NAFLD: A meta-analysis. Curr. Mol. Med. 2018, 18, 91–99. [Google Scholar] [CrossRef]
- Wong, V.W.S.; Wong, G.L.H.; Choi, P.C.L.; Chan, A.W.H.; Li, M.K.P.; Chan, H.Y.; Chim, A.M.L.; Yu, J.; Sung, J.J.Y.; Chan, H.L.Y. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Lotan, R.; Shlomai, A.; Webb, M.; Harrari, G.; Buch, A.; Nitzan Kaluski, D.; Halpern, Z.; Oren, R. Predictors for incidence and remission of NAFLD in the general population during a seven-year prospective follow-up. J. Hepatol. 2012, 56, 1145–1151. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Anastasilakis, A.D.; Geladari, E.V.; Mantzoros, C.S. Irisin in patients with nonalcoholic fatty liver disease. Metab. Clin. Exp. 2014, 63, 207–217. [Google Scholar] [CrossRef]
- Haghgoo, S.M.; Sharafi, H.; Alavian, S.M. Serum cytokines, adipokines and ferritin for non-invasive assessment of liver fibrosis in chronic liver disease: A systematic review. Clin. Chem. Lab. Med. 2019, 57, 577–610. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, S.; Farrell, G.; Frost, L.; Kriketos, A.; Lin, R.; Fung, C.; Liddle, C.; Samarasinghe, D.; George, J. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: A manifestation of lipotoxicity? Hepatology 2002, 36, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.M.; Hakkarainen, A.; Lehtimaki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Jarvinen, H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadarlis, A.; Antza, C.; Bakaloudi, D.R.; Doundoulakis, I.; Kalopitas, G.; Samara, M.; Dardavessis, T.; Maris, T.; Chourdakis, M. Systematic review with meta-analysis: The effect of vitamin E supplementation in adult patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Saryusz-Wolska, M.; Szymańska-Garbacz, E.; Jabłkowski, M.; Białkowska, J.; Pawłowski, M.; Kwiecińska, E.; Omulecka, A.; Borkowska, A.; Ignaczak, A.; Loba, J.; et al. Rosiglitazone treatment in nondiabetic subjects with nonalcoholic fatty liver disease. Pol. Arch. Med. Wewn. 2011, 121, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S.; Polymerou, V.; Katsinelos, P. Effects of combined low-dose spironolactone plus vitamin E vs vitamin E monotherapy on insulin resistance, non-invasive indices of steatosis and fibrosis, and adipokine levels in non-alcoholic fatty liver disease: A randomized controlled trial. Diabetes Obes. Metab. 2017, 19, 1805–1809. [Google Scholar] [CrossRef] [PubMed]
- Navekar, R.; Rafraf, M.; Ghaffari, A.; Asghari-Jafarabadi, M.; Khoshbaten, M. Turmeric Supplementation Improves Serum Glucose Indices and Leptin Levels in Patients with Nonalcoholic Fatty Liver Diseases. J. Am. Coll. Nutr. 2017, 36, 261–267. [Google Scholar] [CrossRef]
- Baykal, A.P.; Parks, E.J.; Shamburek, R.; Syed-Abdul, M.M.; Chacko, S.K.; Cochran, E.; Startzell, M.; Gharib, A.M.; Ouwerkerk, R.; Abd-Elmoniem, K.Z.; et al. Leptin decreases de novo lipogenesis in patients with lipodystrophy. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Javor, E.D.; Ghany, M.G.; Cochran, E.K.; Oral, E.A.; DePaoli, A.M.; Premkumar, A.; Kleiner, D.E.; Gorden, P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology 2005, 41, 753–760. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Mantzoros, C.S. Leptin in Health and Disease: Facts and Expectations at its Twentieth Anniversary. Metab. Clin. Exp. 2015, 64, 5–12. [Google Scholar] [CrossRef]
- Duan, X.F.; Tang, P.; Li, Q.; Yu, Z.T. Obesity, adipokines and hepatocellular carcinoma. Int. J. Cancer 2013, 133, 1776–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelwahab, K.; Abdelmaaboud, M.; Magdy, M.; Abdelhalim, M.A. Diagnostic value of serum leptin as a tumor marker in hepatocellular carcinoma. QJM Int. J. Med. 2020, 113. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Lin, S.Y. Leptin in relation to hepatocellular carcinoma in patients with liver cirrhosis. Horm. Res. 2003, 60, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Takai, K.; Imai, K.; Shimizu, M.; Naiki, T.; Nagaki, M.; Moriwaki, H. Increased levels of serum leptin are a risk factor for the recurrence of stage I/II hepatocellular carcinoma after curative treatment. J. Clin. Biochem. Nutr. 2011, 49, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.N.; Chuang, S.C.; Yeh, Y.T.; Yang, S.F.; Chai, C.Y.; Chen, W.T.; Kuo, K.K.; Chen, J.S.; Lee, K.T. Potential prognostic value of leptin receptor in hepatocellular carcinoma. J. Clin. Pathol. 2006, 59, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- Rocco, A.; Compare, D.; Angrisani, D.; Zamparelli, M.S.; Nardone, G.; Rocco, A.; Compare, D.; Angrisani, D. Alcoholic disease: Liver and beyond. World J. Gastroenterol. 2014, 20, 14652–14659. [Google Scholar] [CrossRef]
- Lamas-Paz, A.; Hao, F.; Nelson, L.J.; Vazquez, M.T.; Canals, S.; Gomez Del Moral, M.; Martinez-Naves, E.; Nevzorova, Y.A.; Cubero, F.J. Alcoholic liver disease: Utility of animal models. World J. Gastroenterol. 2018, 24, 5063–5075. [Google Scholar] [CrossRef]
- Balasubramaniyan, V.; Manju, V.; Nalini, N. Effect of leptin administration on plasma and tissue lipids in alcohol induced liver. Hum. Exp. Toxicol. 2003, 22, 149–154. [Google Scholar] [CrossRef]
- Otaka, M.; Konishi, N.; Odashima, M.; Jin, M.; Wada, I.; Matsuhashi, T.; Ohba, R.; Watanabe, S. Effect of alcohol consumption on leptin level in serum, adipose tissue, and gastric mucosa. Dig. Dis. Sci. 2007, 52, 3066–3069. [Google Scholar] [CrossRef]
- Tomita, K.; Azuma, T.; Kitamura, N.; Tamiya, G.; Ando, S.; Nagata, H.; Kato, S.; Inokuchi, S.; Nishimura, T.; Ishii, H.; et al. Leptin deficiency enhances sensitivity of rats to alcoholic steatohepatitis through suppression of metallothionein. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1078–G1085. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Sun, X.; Li, Q.; Zhao, Y.; Zhong, W.; Sun, X.; Jia, W.; McClain, C.J.; Zhou, Z. Leptin deficiency contributes to the pathogenesis of alcoholic fatty liver disease in mice. Am. J. Pathol. 2012, 181, 1279–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhong, W.; Sun, Q.; Sun, X.; Zhou, Z. Adipose-specific lipin1 overexpression in mice protects against alcohol-induced liver injury. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniyan, V.; Murugaiyan, G.; Shukla, R.; Bhonde, R.R.; Nalini, N. Leptin downregulates ethanol-induced secretion of proinflammatory cytokines and growth factor. Cytokine 2007, 37, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniyan, V.; Shukla, R.; Murugaiyan, G.; Bhonde, R.R.; Nalini, N. Mouse recombinant leptin protects human hepatoma HepG2 against apoptosis, TNF-alpha response and oxidative stress induced by the hepatotoxin-ethanol. Biochim. Biophys. Acta 2007, 1770, 1136–1144. [Google Scholar] [CrossRef]
- Addolorato, G.; Capristo, E.; Marini, M.; Santini, P.; Scognamiglio, U.; Attilia, M.L.; Messineo, D.; Sasso, G.F.; Gasbarrini, G.; Ceccanti, M. Body composition changes induced by chronic ethanol abuse: Evaluation by dual energy X-ray absorptiometry. Am. J. Gastroenterol. 2000, 95, 2323–2327. [Google Scholar] [CrossRef]
- Greco, A.V.; Mingrone, G.; Favuzzi, A.; Capristo, E.; Gniuli, D.; Addolorato, G.; Brunani, A.; Cavagnin, F.; Gasbarrini, G. Serum leptin levels in post-hepatitis liver cirrhosis. J. Hepatol. 2000, 33, 38–42. [Google Scholar] [CrossRef]
- Campillo, B.; Sherman, E.; Richardet, J.P.; Bories, P.N. Serum leptin levels in alcoholic liver cirrhosis: Relationship with gender, nutritional status, liver function and energy metabolism. Eur. J. Clin. Nutr. 2001, 55, 980–988. [Google Scholar] [CrossRef]
- Henriksen, J.H.; Holst, J.J.; Moller, S.; Brinch, K.; Bendtsen, F. Increased Circulating Leptin in Alcoholic Cirrhosis: Relation to Release and Disposal. Hepatology 1999, 29, 1818–1824. [Google Scholar] [CrossRef]
- McCullough, A.J.; Bugianesi, E.; Marchesini, G.; Kalhan, S.C. Gender-Dependent Alterations in Serum Leptin in Alcoholic Cirrhosis. Gastroenterology 1998, 115, 947–953. [Google Scholar] [CrossRef]
- Naveau, S.; Perlemuter, G.; Chaillet, M.; Raynard, B.; Balian, A.; Portier, A.; Galanaud, P.; Emilie, D.; Chaput, J.-C. Serum Leptin in Patients with Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2006, 30, 1422–1428. [Google Scholar] [CrossRef]
- Nicolas, J.M.; Fernandez-Sola, J.; Fatjo, F.; Casamitjana, R.; Bataller, R.; Sacanella, E.; Tobias, E.; Badia, E.; Estruch, R. Increased circulating leptin levels in chronic alcoholism. Alcohol. Clin. Exp. Res. 2001, 25, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Proskynitopoulos, P.J.; Rhein, M.; Jackel, E.; Manns, M.P.; Frieling, H.; Bleich, S.; Thum, T.; Hillemacher, T.; Glahn, A. Leptin Expression and Gene Methylation Patterns in Alcohol-Dependent Patients with Ethyltoxic Cirrhosis-Normalization After Liver Transplantation and Implications for Future Research. Alcohol Alcohol. 2018, 53, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Kosuta, I.; Mrzljak, A.; Kolaric, B.; Vucic Lovrencic, M. Leptin as a Key Player in Insulin Resistance of Liver Cirrhosis? A Cross-Sectional Study in Liver Transplant Candidates. J. Clin. Med. 2020, 9, 560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begriche, K.; Massart, J.; Robin, M.-A.; Borgne-sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-Induced Hepatotoxicity: A Comprehensive Update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Polat, M.; Cerrah, S.; Albayrak, B.; Ipek, S.; Arabul, M.; Aslan, F.; Yilmaz, O. Assessing the Effect of Leptin on Liver Damage in Case of Hepatic Injury Associated with Paracetamol Poisoning. Gastroenterol. Res. Pract. 2015, 2015, 357360. [Google Scholar] [CrossRef]
- Watson, A.M.; Poloyac, S.M.; Howard, G.; Blouin, R.A. Effect of leptin on cytochrome P-450, conjugation, and antioxidant enzymes in the ob/ob mouse. Drug Metab. Dispos. 1999, 27, 695–700. [Google Scholar]
- Larsen, M.C.; Bushkofsky, J.R.; Gorman, T.; Adhami, V.; Mukhtar, H.; Wang, S.; Reeder, S.B.; Sheibani, N.; Jefcoate, C.R. Cytochrome P450 1B1: An unexpected modulator of liver fatty acid homeostasis. Arch. Biochem. Biophys. 2015, 571, 21–39. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.N.; Fan, S.; Weng, C.F. Down-regulation of TGFbeta1 and leptin ameliorates thioacetamide-induced liver injury in lipopolysaccharide-primed rats. J. Endotoxin Res. 2007, 13, 176–188. [Google Scholar] [CrossRef]
- Honda, H.; Ikejima, K.; Hirose, M.; Yoshikawa, M.; Lang, T.; Enomoto, N.; Kitamura, T.; Takei, Y.; Sato, N. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology 2002, 36, 12–21. [Google Scholar] [CrossRef]
- Dai, K.; Qi, J.Y.; Tian, D.Y. Leptin administration exacerbates thioacetamide-induced liver fibrosis in mice. World J. Gastroenterol. 2005, 11, 4822–4826. [Google Scholar] [CrossRef]
- Adachi, A.; Miura, T. Animal model studies on viral infections. Front. Microbiol. 2014, 5, 672. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Hamesch, K.; Weiskirchen, R.; Tacke, F. The concanavalin A model of acute hepatitis in mice. Lab. Anim. 2015, 49, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, B.; Lear-Kaul, K.C.; Faggioni, R.; Fantuzzi, G. Leptin deficiency, not obesity, protects mice from Con A-induced hepatitis. Eur. J. Immunol. 2002, 32, 552–560. [Google Scholar] [CrossRef]
- Faggioni, R.; Jones-Carson, J.; Reed, D.A.; Dinarello, C.A.; Feingold, K.R.; Grunfeld, C.; Fantuzzi, G. Leptin-deficient (ob/ob) mice are protected from T cell-mediated hepatotoxicity: Role of tumor necrosis factor alpha and IL-18. Proc. Natl. Acad. Sci. USA 2000, 97, 2367–2372. [Google Scholar] [CrossRef] [Green Version]
- Maurya, R.; Bhattacharya, P.; Dey, R.; Nakhasi, H.L. Leptin Functions in Infectious Diseases. Front. Immunol. 2018, 9, 2741. [Google Scholar] [CrossRef] [Green Version]
- Alti, D.; Sambamurthy, C.; Kalangi, S.K. Emergence of Leptin in Infection and Immunity: Scope and Challenges in Vaccines Formulation. Front. Cell Infect. Microbiol. 2018, 8, 147. [Google Scholar] [CrossRef]
- Manolakopoulos, S.; Bethanis, S.; Liapi, C.; Stripeli, F.; Sklavos, P.; Margeli, A.; Christidou, A.; Katsanika, A.; Vogiatzakis, E.; Tzourmakliotis, D.; et al. An assessment of serum leptin levels in patients with chronic viral hepatitis: A prospective study. BMC Gastroenterol. 2007, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Pavlidis, C.; Panoutsopoulos, G.I.; Tiniakos, D.; Koutsounas, S.; Vlachogiannakos, J. Serum leptin and ghrelin in chronic hepatitis C patients with steatosis. World J. Hepatol. 2011, 17, 5097–5104. [Google Scholar] [CrossRef]
- Romero-go, M.; Castellano-Megias, V.M.; Grande, L.; Irles, A.; Cruz, M.; Nogales, C.; Alco, J.C. Serum Leptin Levels Correlate With Hepatic Steatosis in Chronic Hepatitis C. Am. J. Gastroenterol. 2003, 98, 1135–1141. [Google Scholar] [CrossRef]
- Testa, R.; Franceschini, R.; Giannini, E.; Cataldi, A.; Botta, F.; Fasoli, A.; Tenerelli, P.; Rolandi, E.; Barreca, T. Serum leptin levels in patients with viral chronic hepatitis or liver cirrhosis. J. Hepatol. 2000, 33, 33–37. [Google Scholar] [CrossRef]
- Korah, T.E.; El-Sayed, S.; Elshafie, M.K.; Hammoda, G.E.; Safan, M.A. Significance of serum leptin and adiponectin levels in Egyptian patients with chronic hepatitis C virus associated hepatic steatosis and fibrosis. World J. Hepatol. 2013, 5, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-W.; Zhang, N.; Han, Q.-Y.; Zeng, J.-T.; Chu, Y.-L.; Qiu, J.-M.; Wang, Y.-W.; Ma, L.-T.; Wang, X.-Q. Correlation of serum leptin levels with anthropometric and metabolic parameters and biochemical liver function in Chinese patients with chronic hepatitis C virus infection. World J. Gastroenterol. 2005, 11, 3357–3362. [Google Scholar] [CrossRef] [PubMed]
- Tiftikci, A.; Atug, O.; Yilmaz, Y.; Eren, F.; Ozdemir, T.; Yapali, S. Serum Levels of Adipokines in Patients with Chronic HCV Infection: Relationship with Steatosis and Fibrosis. Arch. Med. Res. 2009, 40, 294–298. [Google Scholar] [CrossRef]
- Cua, I.H.Y.; Hui, J.M.; Bandara, P.; Kench, J.G.; Farrell, G.C.; McCaughan, G.W.; George, J. Insulin Resistance and Liver Injury in Hepatitis C Is Not Associated with Virus-Specific Changes in Adipocytokines. Hepatology 2007, 46, 66–73. [Google Scholar] [CrossRef]
- Giannini, E.; Ceppa, P.; Botta, F.; Mastracci, L.; Romagnoli, P.; Comino, I.; Pasini, A.; Risso, D.; Lantieri, P.B.; Icardi, G.; et al. Leptin Has No Role in Determining Severity of Steatosis and Fibrosis in Patients With Chronic Hepatitis C. Am. J. Gastroenterol. 2000, 95, 3211–3217. [Google Scholar] [CrossRef]
- González-reimers, E.; López-prieto, J.; Quintero-platt, G.; Pelazas-gonzález, R.; Alemán-valls, M.R.; Pérez-hernández, O.; José, M.; Gómez-rodríguez, M.A.; Martín-gonzález, C.; Santolaria-fernández, F.; et al. Adipokines, cytokines and body fat stores in hepatitis C virus liver steatosis. World J. Hepatol. 2016, 8, 74–82. [Google Scholar] [CrossRef]
- Myers, R.P.; Messous, D.; Poynard, T.; Imbert-bismut, F. Association between leptin, metabolic factors and liver histology in patients with chronic hepatitis C. Can. J. Gastroenterol. 2007, 21, 289–294. [Google Scholar] [CrossRef]
- Nkontchou, G.; Bastard, J.-P.; Ziol, M.; Aout, M.; Cosson, E.; Ganne-carrie, N.; Grando-lemaire, V.; Roulot, D.; Capeau, J.; Trinchet, J.-C.; et al. Insulin resistance, serum leptin, and adiponectin levels and outcomes of viral hepatitis C cirrhosis. J. Hepatol. 2010, 53, 827–833. [Google Scholar] [CrossRef]
- Dayakar, A.; Chandrasekaran, S.; Veronica, J.; Maurya, R. Leptin induces the phagocytosis and protective immune response in Leishmania donovani infected THP-1 cell line and human PBMCs. Exp. Parasitol. 2016, 160, 54–59. [Google Scholar] [CrossRef]
- Maurya, R.; Bhattacharya, P.; Ismail, N.; Dagur, P.K.; Joshi, A.B.; Razdan, K.; McCoy, J.P.; Ascher, J.; Dey, R.; Nakhasi, H.L. Differential Role of Leptin as an Immunomodulator in Controlling Visceral Leishmaniasis in Normal and Leptin-Deficient Mice. Am. J. Trop. Med. Hyg. 2016, 95, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fievez, A.; Silva-Freitas, M.L.; Sousa, A.Q.; Santos-Oliveira, J.R.; Da-Cruz, A.M. Lower levels of leptin are associated with severity parameters in visceral leishmaniasis patients. PLoS ONE 2019, 14, e0214413. [Google Scholar] [CrossRef] [PubMed]
- Nagajyothi, F.; Zhao, D.; Machado, F.S.; Weiss, L.M.; Schwartz, G.J.; Desruisseaux, M.S.; Zhao, Y.; Factor, S.M.; Huang, H.; Albanese, C.; et al. Crucial role of the central leptin receptor in murine Trypanosoma cruzi (Brazil strain) infection. J. Infect. Dis. 2010, 202, 1104–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olveda, D.U.; Olveda, R.M.; McManus, D.P.; Cai, P.; Chau, T.N.; Lam, A.K.; Li, Y.; Harn, D.A.; Vinluan, M.L.; Ross, A.G. The chronic enteropathogenic disease schistosomiasis. Int. J. Infect. Dis. 2014, 28, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, J.J.; Rennie-Tankesley, L.; Mezey, E. Influence of leptin in the development of hepatic fibrosis produced in mice by Schistosoma mansoni infection and by chronic carbon tetrachloride administration. J. Hepatol. 2003, 38, 281–288. [Google Scholar] [CrossRef]
- Anthony, B.; Allen, J.T.; Li, Y.S.; McManus, D.P. Hepatic stellate cells and parasite-induced liver fibrosis. Parasit. Vectors 2010, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Potter, J.J.; Mezey, E. Leptin deficiency reduces but does not eliminate the development of hepatic fibrosis in mice infected with Schistosoma mansoni. Liver 2002, 22, 173–177. [Google Scholar] [CrossRef]
- Abu Rmilah, A.; Zhou, W.; Nelson, E.; Lin, L.; Amiot, B.; Nyberg, S.L. Understanding the marvels behind liver regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e340. [Google Scholar] [CrossRef]
- Gilgenkrantz, H.; Collin de l’Hortet, A. Understanding Liver Regeneration: From Mechanisms to Regenerative Medicine. Am. J. Pathol. 2018, 188, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Higgins, G.M.; Anderson, R.M. Experimental pathology of the liver—Restoration of the liver of the white rat following partial surgical removal. Arch. Pathol. Lab. Med. 1931, 12, 186–202. [Google Scholar]
- Ruccoleri, A.L.B.; Allucci, R.A.G.; Ermolec, D.O.R.I.R.G.; Lackshear, P.A.B.; Imeonova, P.E.S.; Hurman, R.O.G.T.; Uster, M.I.I.L. Induction of Early-Immediate Genes by Tumor Necrosis Factor a Contribute to Liver Repair Following Chemical-Induced Hepatotoxicity. Hepatology 1997, 25, 133–141. [Google Scholar] [CrossRef]
- Akhurst, B.; Croager, E.J.; Farley-Roche, C.A.; Ong, J.K.; Dumble, M.L.; Knight, B.; Yeoh, G.C. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology 2001, 34, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Preisegger, K.H.; Factor, V.M.; Fuchsbichler, A.; Stumptner, C.; Denk, H.; Thorgeirsson, S.S. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab. Investig. 1999, 79, 103–109. [Google Scholar] [PubMed]
- Leclercq, I.A.; Vansteenberghe, M.; Lebrun, V.B.; VanHul, N.K.; Abarca-Quinones, J.; Sempoux, C.L.; Picard, C.; Starkel, P.; Horsmans, Y.L. Defective hepatic regeneration after partial hepatectomy in leptin-deficient mice is not rescued by exogenous leptin. Lab. Investig. 2006, 86, 1161–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, I.A.; Field, J.; Farrell, G.C. Leptin-Specific Mechanisms for Impaired Liver Regeneration in ob/ob Mice After Toxic Injury. Gastroenterology 2003, 124, 1451–1464. [Google Scholar] [CrossRef]
- Yang, S.Q.; Lin, H.Z.; Mandal, A.K.; Huang, J.; Diehl, A.M. Disrupted Signaling and Inhibited Regeneration in Obese Mice With Fatty Livers: Implications for Nonalcoholic Fatty Liver Disease Pathophysiology. Hepatology 2001, 34, 694–706. [Google Scholar] [CrossRef]
- Yamauchi, H.; Uetsuka, K.; Okada, T.; Nakayama, H.; Doi, K. Impaired liver regeneration after partial hepatectomy in db/db mice. Exp. Toxicol. Pathol. 2003, 54, 281–286. [Google Scholar] [CrossRef]
- Shirai, M.; Yamauchi, H.; Nakayama, H.; Doi, K.; Uetsuka, K. Expression of epidermal growth factor receptor protein in the liver of db/db mice after partial hepatectomy. Exp. Toxicol. Pathol. 2007, 59, 157–162. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Jin, X.; Zhang, Z.; Jiang, Y.; Koniaris, L.G. Epidermal growth factor receptor restoration rescues the fatty liver regeneration in mice. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E440–E449. [Google Scholar] [CrossRef]
- Picard, C.; Lambotte, L.; Starkel, P.; Sempoux, C.; Saliez, A.; Berge, V.D.; Horsmans, Y. Steatosis is not sufficient to cause an impaired regenerative response after partial hepatectomy in rats. J. Hepatol. 2002, 32, 645–652. [Google Scholar] [CrossRef]
- Selzner, M.; Clavien, P.-A. Failure of Regeneration of the Steatotic Rat Liver: Disruption at Two Different Levels in the Regeneration Pathway. Hepatology 2000, 31, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Shteyer, E.; Liao, Y.; Muglia, L.J.; Hruz, P.W.; Rudnick, D.A. Disruption of Hepatic Adipogenesis Is Associated With Impaired Liver Regeneration in Mice. Hepatology 2004, 40, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Cilekar, M.; Uysal, O.; Bal, C.; Turel, S.; Yilmaz, S. Leptin increases mitotic index and regeneration ratio in hepatectomized rats. Med. Sci. Monit. Basic Res. 2013, 19, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sydor, S.; Gu, Y.; Schlattjan, M.; Bechmann, L.P.; Rauen, U.; Best, J.; Paul, A.; Baba, H.A.; Sowa, J.-P.; Gerken, G.; et al. Steatosis does not impair liver regeneration after partial hepatectomy. Lab. Investig. 2012, 93, 20–30. [Google Scholar] [CrossRef]
- Matsumoto, K.; Miyake, Y.; Umeda, Y.; Matsushita, H. Serial Changes of Serum Growth Factor Levels and Liver Regeneration after Partial Hepatectomy in Healthy Humans. Int. J. Mol. Sci. 2013, 14, 20877–20889. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N.; Committee, E.H.S. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef]
- Wong, M.C.S.; Huang, J.L.W.; George, J.; Huang, J.; Leung, C.; Eslam, M.; Chan, H.L.Y.; Ng, S.C. The changing epidemiology of liver diseases in the Asia-Pacific region. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 57–73. [Google Scholar] [CrossRef]
- Kamdem, S.D.; Moyou-Somo, R.; Brombacher, F.; Nono, J.K. Host Regulators of Liver Fibrosis During Human Schistosomiasis. Front. Immunol. 2018, 9, 2781. [Google Scholar] [CrossRef]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Reitman, M.L. Leptin in the liver: A toxic or beneficial mix? Cell Metab. 2012, 16, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePaoli, A.M. 20 years of leptin: Leptin in common obesity and associated disorders of metabolism. J. Endocrinol. 2014, 223, T71–T81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrysafi, P.; Perakakis, N.; Farr, O.M.; Stefanakis, K.; Peradze, N.; Sala-Vila, A.; Mantzoros, C.S. Leptin alters energy intake and fat mass but not energy expenditure in lean subjects. Nat. Commun. 2020, 11, 5145. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S.; Maratos-Flier, E. Leptin’s Physiologic Role: Does the Emperor of Energy Balance Have No Clothes? Cell Metab. 2017, 26, 24–26. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Uña, M.; López-Mancheño, Y.; Diéguez, C.; Fernández-Rojo, M.A.; Novelle, M.G. Unraveling the Role of Leptin in Liver Function and Its Relationship with Liver Diseases. Int. J. Mol. Sci. 2020, 21, 9368. https://doi.org/10.3390/ijms21249368
Martínez-Uña M, López-Mancheño Y, Diéguez C, Fernández-Rojo MA, Novelle MG. Unraveling the Role of Leptin in Liver Function and Its Relationship with Liver Diseases. International Journal of Molecular Sciences. 2020; 21(24):9368. https://doi.org/10.3390/ijms21249368
Chicago/Turabian StyleMartínez-Uña, Maite, Yaiza López-Mancheño, Carlos Diéguez, Manuel A. Fernández-Rojo, and Marta G. Novelle. 2020. "Unraveling the Role of Leptin in Liver Function and Its Relationship with Liver Diseases" International Journal of Molecular Sciences 21, no. 24: 9368. https://doi.org/10.3390/ijms21249368
APA StyleMartínez-Uña, M., López-Mancheño, Y., Diéguez, C., Fernández-Rojo, M. A., & Novelle, M. G. (2020). Unraveling the Role of Leptin in Liver Function and Its Relationship with Liver Diseases. International Journal of Molecular Sciences, 21(24), 9368. https://doi.org/10.3390/ijms21249368