Molecular Biology of Atherosclerotic Ischemic Strokes


 and
and
Abstract
:1. Introduction
2. The Role of the Innate Immune System in Neuroinflammation
2.1. NLRP3 Inflammasome
2.2. DICKKOPFF-3 (DKK-3)
2.3. DECTIN-1/SYK
2.4. CXCL4-CCL5 Heterodimer
2.5. Microglial IRF5-IRF4 Regulatory Axis
2.6. The Role of CD200-CD200R Interaction in Neuroinflammation after Stroke
2.7. The Role of Astrocytes Activation in Neuroinflammation
3. The Role of the Adaptative Immune System in Neuroinflammation
4. Relationship between Atrial Fibrillation and Neuroinflammation
5. Neuroinflammation and BBB Dysfunction
6. miRNAs and Neuroinflammation
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| A-FABP | Adipocyte fatty acid-binding protein |
| AF | Atrial fibrillation |
| ApoE | Apolipoprotein E |
| ApoC3 | Apolipoprotein C-III |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| BBB | Blood–brain barrier |
| BTRC | Beta-transducin repeat containing |
| CCL1 | Chemokine (C-C motif) ligand 1 |
| CCL5 | Chemokine (C-C motif) ligand 5 |
| CCL20 | Chemokine (C-C motif) ligand 20 |
| CCR5 | C-C chemokine receptor type 5 |
| CLEC2 | C-type lectin-like receptor 2 |
| CLEC9A | C-type lectin-like receptor 9A |
| COX-2 | Cyclooxygenase-2 |
| CSF-3 | Colony-stimulating factor 3 |
| CX3CL1 | Chemokine (C-X3-C motif) ligand 1 |
| CXCL4 | Chemokine (C-X-C motif) ligand 4 |
| CXCL7 | Chemokine (C-X-C motif) ligand 7 |
| CXCL8 | Chemokine (C-X-C motif) ligand 8 |
| DAMPs | Damage-associated molecular patterns |
| DLL-4 | Delta-like ligand 4 |
| DKK-3 | Dickkopf WNT Signaling Pathway Inhibitor 3 |
| FOXP3 | Forkhead box P3 |
| GABRA 1 | Alpha receptor GABA 1 |
| Gal-3 | Galectin-3 |
| GLT-1 | Glial glutamate transporter |
| GSK3 β | Glycogen synthase kinase 3 beta |
| hemITAMs | Hemi-immunoreceptor tyrosine-based activation motifs |
| hsCRP | High-sensitivity C-reactive protein |
| HSP-72 | Heat shock protein 72 |
| IL-1α | Interleukin 1 alpha |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin |
| IL-7 | Interleukin 7 |
| IL-10 | Interleukin 10 |
| IL-17 | Interleukin 17 |
| IL-18 | Interleukin 18 |
| IL-20 | Interleukin 20 |
| IL-23 | Interleukin 23 |
| iNOS | Inducible nitric oxide synthase |
| IRF4 | Interferon regulatory factor 4 |
| IRF5 | Interferon regulatory factor 5 |
| IRS-1 | Insulin receptor substrate 1 |
| ITAMs | Immunoreceptor tyrosine-based activation motifs |
| JNK | c-Jun N-terminal kinase |
| KIRs | Killer immunoglobulin-like receptors |
| LAM | Laminarin |
| LRR | Leucine-rich repeats |
| Ly6C | Ly6Chi monocytes |
| MCAO | Middle cerebral artery occlusion |
| MiMΦs | Microglia-derived macrophages |
| miRNAs | MicroRNAs |
| MoMΦs | Monocyte-derived macrophages |
| MPO | Myeloperoxidase |
| MMP-9 | Matrix metalloproteinase 9 |
| NADPH oxidase | Nicotinamide adenine dinucleotide phosphate oxidase |
| NAIP | Neuronal apoptosis inhibitory protein |
| NLR | NOD-like receptor |
| NLR | Neutrophil-to-lymphocyte ratio |
| NLRP3 | NLR family pyrin domain containing 3 |
| NFIA | Nuclear factor IA |
| PIC | Piceatannol |
| PIIINP | Amino-terminal peptide of type III procollagen |
| PGC-1a | Peroxisome proliferator-activated receptor γ coactivator 1 |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| PYCARD | PYD And CARD Domain Containing |
| PYD | Pyrin domain |
| ROS | Reactive oxygen species |
| siRNA | Small interfering RNA |
| SYK | Spleen tyrosine kinase |
| TGF-β | Transforming growth factor-beta |
| TNF-α | Tumor Necrosis Factor-alpha |
| TOP1 | DNA topoisomerase I |
| Tregs | Regulatory T cells |
| TXNIP | Thioredoxin-interacting protein |
| VDUP-1 | Vitamin D3-upregulated protein-1 |
| VEGF | Vascular endothelial growth factor |
| vWF | von Willebrand factor |
| Wnt | Wingless-related integration site |
References
- WHO. The Atlas of Heart Disease and Stroke. Available online: https://www.who.int/cardiovascular_diseases/resources/atlas/en/ (accessed on 19 November 2020).
- Lynch, J.R.; Blessing, R.; White, W.D.; Grocott, H.P.; Newman, M.F.; Laskowitz, D.T. Novel diagnostic test for acute stroke. Stroke 2004, 35, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisel, C.; Schwab, J.M.; Prass, K.; Meisel, A.; Dirnagl, U. Central nervous system injury-induced immune deficiency syndrome. Nat. Rev. Neurosci. 2005, 6, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Urra, X.; Cervera, A.; Villamor, N.; Planas, A.M.; Chamorro, A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience 2009, 158, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Weiner, H.L.; Selkoe, D.J. Inflammation and therapeutic vaccination in CNS diseases. Nature 2002, 420, 879–884. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Maida, C.; Pinto, A. Inflammation and Inflammatory Cell Recruitment in Acute Cerebrovascular Diseases. Available online: https://www.eurekaselect.com/130447/article (accessed on 19 November 2020).
- Mayer, A.M.S.; Clifford, J.A.; Aldulescu, M.; Frenkel, J.A.; Holland, M.A.; Hall, M.L.; Glaser, K.B.; Berry, J. Cyanobacterial Microcystis aeruginosa lipopolysaccharide elicits release of superoxide anion, thromboxane B₂, cytokines, chemokines, and matrix metalloproteinase-9 by rat microglia. Toxicol. Sci. 2011, 121, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Na, K.-S.; Jung, H.-Y.; Kim, Y.-K. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 277–286. [Google Scholar] [CrossRef]
- Ip, C.W.; Kroner, A.; Groh, J.; Huber, M.; Klein, D.; Spahn, I.; Diem, R.; Williams, S.K.; Nave, K.-A.; Edgar, J.M.; et al. Neuroinflammation by Cytotoxic T-Lymphocytes Impairs Retrograde Axonal Transport in an Oligodendrocyte Mutant Mouse. PLoS ONE 2012, 7, e42554. [Google Scholar] [CrossRef]
- Pétrilli, V.; Dostert, C.; A Muruve, D.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef]
- Tureyen, K.; Bowen, K.; Liang, J.; Dempsey, R.J.; Vemuganti, R. Exacerbated brain damage, edema and inflammation in type-2 diabetic mice subjected to focal ischemia. J. Neurochem. 2011, 116, 499–507. [Google Scholar] [CrossRef]
- Cheng, W.; Yang, Y.; Zhang, X.; Guo, J.; Gong, J.; Gong, F.; She, Z.; Huang, Z.; Xia, H.; Li, H. Dickkopf-3 Ablation Attenuates the Development of Atherosclerosis in ApoE-Deficient Mice. J. Am. Heart Assoc. 2017, 6, e004690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaghan, A.; Kioschis, P.; Wu, W.; Zuniga, A.; Bock, D.; Poustka, A.; Delius, H.; Niehrs, C. Dickkopf genes are co-ordinately expressed in mesodermal lineages. Mech. Dev. 1999, 87, 45–56. [Google Scholar] [CrossRef]
- Ye, X.; Hao, Q.; Ma, W.-J.; Zhao, Q.-C.; Wang, W.-W.; Yin, H.-H.; Zhang, T.; Wang, M.; Zan, K.; Yang, X.-X.; et al. Dectin-1/Syk signaling triggers neuroinflammation after ischemic stroke in mice. J. Neuroinflamm. 2020, 17, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenen, R.R.; Von Hundelshausen, P.; Nesmelova, I.V.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.K.; Piccinini, A.M.; Paludan, S.R.; Kowalska, M.A.; et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 2009, 15, 97–103. [Google Scholar] [CrossRef]
- Terao, S.; Yilmaz, G.; Stokes, K.Y.; Russell, J.; Ishikawa, M.; Kawase, T.; Granger, D.N. Blood Cell-Derived RANTES Mediates Cerebral Microvascular Dysfunction, Inflammation, and Tissue Injury After Focal Ischemia–Reperfusion. Stroke 2008, 39, 2560–2570. [Google Scholar] [CrossRef] [Green Version]
- Günthner, R.; Anders, H.-J. Interferon-Regulatory Factors Determine Macrophage Phenotype Polarization. Mediat. Inflamm. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Al Mamun, A.; Chauhan, A.; Yu, H.; Xu, Y.; Sharmeen, R.; Liu, F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Eur. J. Neurosci. 2018, 47, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Ritzel, R.M.; Patel, A.R.; Grenier, J.M.; Crapser, J.; Verma, R.; Jellison, E.R.; McCullough, L.D. Functional differences between microglia and monocytes after ischemic stroke. J. Neuroinflamm. 2015, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mitani, A.; Tanaka, K. Functional changes of glial glutamate transporter GLT-1 during ischemia: An in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J. Neurosci. 2003, 23, 7176–7182. [Google Scholar] [CrossRef] [Green Version]
- Pekny, M.; Wilhelmsson, U.; Pekna, M. The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 2014, 565, 30–38. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Zhang, S.; Wu, J.; Chen, L.; Wang, Y. Data from: Upregulation of NLRP3 inflammasome in the tears and ocular surface of dry eye patients. PLoS ONE 2015, 10, e0126277. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.P.-Y.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Dostert, C.; Pétrilli, V.; Bruggen, R.V.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation Through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Dong, Q.; Song, Z.; Shen, F.; Shi, J.; Li, Y. NLRP3 inflammasome: A promising target in ischemic stroke. Inflamm. Res. 2017, 66, 17–24. [Google Scholar] [CrossRef]
- Kousathana, F.; Georgitsi, M.; Lambadiari, V.; Giamarellos-Bourboulis, E.J.; Dimitriadis, G.; Mouktaroudi, M. Defective production of interleukin-1 beta in patients with type 2 diabetes mellitus: Restoration by proper glycemic control. Cytokine 2017, 90, 177–184. [Google Scholar] [CrossRef]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Available online: https://www.hindawi.com/journals/omcl/2016/2183026/ (accessed on 19 November 2020).
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, Y.; Matsuzaki, K.; Kuwayama, K.; Kitazato, K.; Mure, H.; Kageji, T.; Nagahiro, S. REIC/Dkk-3 induces cell death in human malignant glioma. Neuro-Oncology 2008, 10, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.-J.; Jo, M.; Rho, S.B.; Park, K.; Yoo, Y.-N.; Park, J.; Chae, M.; Zhang, W.; Lee, J.-H. Dkk3, downregulated in cervical cancer, functions as a negative regulator of beta-catenin. Int. J. Cancer 2009, 124, 287–297. [Google Scholar] [CrossRef]
- Peifer, M.; Polakis, P. Wnt signaling in oncogenesis and embryogenesis—A look outside the nucleus. Science 2000, 287, 1606–1609. [Google Scholar] [CrossRef]
- Yu, B.; Kiechl, S.; Qi, D.; Wang, X.; Song, Y.; Weger, S.; Mayr, A.; Le Bras, A.; Karamariti, E.; Zhang, Z.; et al. A Cytokine-Like Protein Dickkopf-Related Protein 3 Is Atheroprotective. Circulation 2017, 136, 1022–1036. [Google Scholar] [CrossRef] [Green Version]
- Caricasole, A.; Ferraro, T.; Iacovelli, L.; Barletta, E.; Caruso, A.; Melchiorri, D.; Terstappen, G.C.; Nicoletti, F. Functional characterization of WNT7A signaling in PC12 cells: Interaction with A FZD5 x LRP6 receptor complex and modulation by Dickkopf proteins. J. Biol. Chem. 2003, 278, 37024–37031. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.-R.; Lee, E.-J.; Seo, S.H.; Lee, S.-H.; Rho, S.B. Dickkopf-3 (DKK-3) obstructs VEGFR-2/Akt/mTOR signaling cascade by interacting of β2-microglobulin (β2M) in ovarian tumorigenesis. Cell Signal. 2015, 27, 2150–2159. [Google Scholar] [CrossRef]
- Chen, T.; Karamariti, E.; Hong, X.; Deng, J.; Wu, Y.; Gu, W.; Simpson, R.; Wong, M.M.; Yu, B.; Hu, Y.; et al. DKK3 (Dikkopf-3) Transdifferentiates Fibroblasts Into Functional Endothelial Cells—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 765–773. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhang, Y.; Xu, T.; Zhao, Q.; Wang, D.; Chen, C.-S.; Tong, W.; Liu, C.; Xu, T.; Ju, Z.; et al. Effects of immediate blood pressure reduction on death and major disability in patients with acute ischemic stroke: The CATIS randomized clinical trial. JAMA 2014, 311, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Abulafia, D.P.; de Rivero Vaccari, J.P.; Lozano, J.D.; Lotocki, G.; Keane, R.W.; Dietrich, W.D. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 2009, 29, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.E.; Kim, G.S.; Chan, P.H. Neuroprotection by interleukin-6 is mediated by signal transducer and activator of transcription 3 and antioxidative signaling in ischemic stroke. Stroke 2011, 42, 3574–3579. [Google Scholar] [CrossRef] [Green Version]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.-C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef] [Green Version]
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 2012, 18, 911–917. [Google Scholar] [CrossRef]
- Willment, J.A.; Marshall, A.S.J.; Reid, D.M.; Williams, D.L.; Wong, S.Y.C.; Gordon, S.; Brown, G.D. The human beta-glucan receptor is widely expressed and functionally equivalent to murine Dectin-1 on primary cells. Eur. J. Immunol. 2005, 35, 1539–1547. [Google Scholar] [CrossRef]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.-K.; Lu, X.; Li, X.; Sun, Q.-Y.; Su, X.; Song, Y.; Sun, H.-M.; Shi, Y. Dectin-1 is inducible and plays a crucial role in Aspergillus-induced innate immune responses in human bronchial epithelial cells. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, M.; Schrettl, M.; Alcazar-Fuoli, L.; Cairns, T.C.; Muñoz, A.; Walker, L.A.; Herbst, S.; Safari, M.; Cheverton, A.M.; Chen, D.; et al. The pH-responsive PacC transcription factor of Aspergillus fumigatus governs epithelial entry and tissue invasion during pulmonary aspergillosis. PLoS Pathog. 2014, 10, e1004413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, D.M.; Montoya, M.; Taylor, P.R.; Borrow, P.; Gordon, S.; Brown, G.D.; Wong, S.Y.C. Expression of the beta-glucan receptor, Dectin-1, on murine leukocytes in situ correlates with its function in pathogen recognition and reveals potential roles in leukocyte interactions. J. Leukoc. Biol. 2004, 76, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Susanti, H.E.; Römmele, C.; Gröbmayr, R.; Günthner, R.; Anders, H.-J. Quantitative expression of C-type lectin receptors in humans and mice. Int. J. Mol. Sci. 2012, 13, 10113–10131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, O.; Poeck, H.; Bscheider, M.; Dostert, C.; Hannesschläger, N.; Endres, S.; Hartmann, G.; Tardivel, A.; Schweighoffer, E.; Tybulewicz, V.; et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009, 459, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Saïd-Sadier, N.; Padilla, E.; Langsley, G.; Ojcius, D.M. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS ONE 2010, 5, e10008. [Google Scholar] [CrossRef] [Green Version]
- Kankkunen, P.; Teirilä, L.; Rintahaka, J.; Alenius, H.; Wolff, H.; Matikainen, S. (1,3)-beta-glucans activate both dectin-1 and NLRP3 inflammasome in human macrophages. J. Immunol. 2010, 184, 6335–6342. [Google Scholar] [CrossRef] [Green Version]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An Essential Role for the NLRP3 Inflammasome in Host Defense against the Human Fungal Pathogen Candida albicans. Available online: https://pubmed.ncbi.nlm.nih.gov/19454352/ (accessed on 20 November 2020).
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; van der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B.H. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012, 13, 246–254. [Google Scholar] [CrossRef]
- Gensel, J.C.; Wang, Y.; Guan, Z.; Beckwith, K.A.; Braun, K.J.; Wei, P.; McTigue, D.M.; Popovich, P.G. Toll-Like Receptors and Dectin-1, a C-Type Lectin Receptor, Trigger Divergent Functions in CNS Macrophages. Available online: https://pubmed.ncbi.nlm.nih.gov/26156997/ (accessed on 20 November 2020).
- Baldwin, K.T.; Carbajal, K.S.; Segal, B.M.; Giger, R.J. Neuroinflammation Triggered by β-glucan/dectin-1 Signaling Enables CNS Axon Regeneration. Available online: https://pubmed.ncbi.nlm.nih.gov/25675510/ (accessed on 20 November 2020).
- Turner, M.; Schweighoffer, E.; Colucci, F.; Di Santo, J.P.; Tybulewicz, V.L. Tyrosine kinase SYK: Essential functions for immunoreceptor signalling. Immunol. Today 2000, 21, 148–154. [Google Scholar] [CrossRef]
- Riccaboni, M.; Bianchi, I.; Petrillo, P. Spleen Tyrosine Kinases: Biology, Therapeutic Targets and Drugs. Available online: https://pubmed.ncbi.nlm.nih.gov/20553955/ (accessed on 20 November 2020).
- Duta, F.; Ulanova, M.; Seidel, D.; Puttagunta, L.; Musat-Marcu, S.; Harrod, K.S.; Schreiber, A.D.; Steinhoff, U.; Befus, A.D. Differential Expression of Spleen Tyrosine Kinase Syk Isoforms in Tissues: Effects of the Microbial Flora. Available online: https://pubmed.ncbi.nlm.nih.gov/16708245/ (accessed on 20 November 2020).
- Friedrich, V.; Flores, R.; Muller, A.; Bi, W.; Peerschke, E.I.; Sehba, F.A. Reduction of neutrophil activity decreases early microvascular injury after subarachnoid haemorrhage. J. Neuroinflamm. 2011, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kawabori, M.; Yenari, M.A. Innate inflammatory responses in stroke: Mechanisms and potential therapeutic targets. Curr. Med. Chem 2014, 21, 2076–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ystgaard, M.B.; Sejersted, Y.; Løberg, E.M.; Lien, E.; Yndestad, A.; Saugstad, O.D. Early Upregulation of NLRP3 in the Brain of Neonatal Mice Exposed to Hypoxia-Ischemia: No Early Neuroprotective Effects of NLRP3 Deficiency. Neonatology 2015, 108, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Pamuk, O.N.; Lapchak, P.H.; Rani, P.; Pine, P.; Dalle Lucca, J.J.; Tsokos, G.C. Spleen tyrosine kinase inhibition prevents tissue damage after ischemia-reperfusion. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G391–G399. [Google Scholar] [CrossRef]
- Huysamen, C.; Willment, J.A.; Dennehy, K.M.; Brown, G.D. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol. Chem. 2008, 283, 16693–16701. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, S.; Ishikawa, E.; Sakuma, M.; Hara, H.; Ogata, K.; Saito, T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 2008, 9, 1179–1188. [Google Scholar] [CrossRef]
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martínez, D.; Hernanz-Falcón, P.; Rosewell, I.; e Sousa, C.R. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009, 458, 899–903. [Google Scholar] [CrossRef]
- Yang, W.S.; Lee, J.; Kim, T.W.; Kim, J.H.; Lee, S.; Rhee, M.H.; Hong, S.; Cho, J.Y. Src/NF-κB-targeted inhibition of LPS-induced macrophage activation and dextran sodium sulphate-induced colitis by Archidendron clypearia methanol extract. J. Ethnopharmacol. 2012, 142, 287–293. [Google Scholar] [CrossRef]
- Yu, T.; Lee, S.; Yang, W.S.; Jang, H.-J.; Lee, Y.J.; Kim, T.W.; Kim, S.Y.; Lee, J.; Cho, J.Y. The ability of an ethanol extract of Cinnamomum cassia to inhibit Src and spleen tyrosine kinase activity contributes to its anti-inflammatory action. J. Ethnopharmacol. 2012, 139, 566–573. [Google Scholar] [CrossRef]
- Jeong, D.; Yang, W.S.; Yang, Y.; Nam, G.; Kim, J.H.; Yoon, D.H.; Noh, H.J.; Lee, S.; Kim, T.W.; Sung, G.-H.; et al. In vitro and in vivo anti-inflammatory effect of Rhodomyrtus tomentosa methanol extract. J. Ethnopharmacol. 2013, 146, 205–213. [Google Scholar] [CrossRef]
- Yoon, J.Y.; Jeong, H.Y.; Kim, S.H.; Kim, H.G.; Nam, G.; Kim, J.P.; Yoon, D.H.; Hwang, H.; Kimc, T.W.; Hong, S.; et al. Methanol extract of Evodia lepta displays Syk/Src-targeted anti-inflammatory activity. J. Ethnopharmacol. 2013, 148, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli-Badenier, M.; Braunersreuther, V.; Viviani, G.L.; Dallegri, F.; Quercioli, A.; Veneselli, E.; Mach, F.; Montecucco, F. CC and CXC chemokines are pivotal mediators of cerebral injury in ischaemic stroke. Thromb. Haemost. 2011, 105, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Di Raimondo, D.; di Sciacca, R.; Pinto, A.; Licata, G. Inflammatory cytokines in acute ischemic stroke. Curr. Pharm. Des. 2008, 14, 3574–3589. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M.; Baggiolini, M.; Charo, I.F.; Hébert, C.A.; Horuk, R.; Matsushima, K.; Miller, L.H.; Oppenheim, J.J.; Power, C.A. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharm. Rev. 2000, 52, 145–176. [Google Scholar]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of the CXC chemokines platelet factor-4 (CXCL4/PF-4) and its variant (CXCL4L1/PF-4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev. 2011, 22, 1–18. [Google Scholar] [CrossRef]
- Daly, T.J.; LaRosa, G.J.; Dolich, S.; Maione, T.E.; Cooper, S.; Broxmeyer, H.E. High Activity Suppression of Myeloid Progenitor Proliferation by Chimeric Mutants of Interleukin 8 and Platelet Factor 4. J. Biol. Chem. 1995, 270, 23282–23292. [Google Scholar] [CrossRef] [Green Version]
- Gengrinovitch, S.; Greenberg, S.M.; Cohen, T.; Gitay-Goren, H.; Rockwell, P.; Maione, T.E.; Levi, B.Z.; Neufeld, G. Platelet factor-4 inhibits the mitogenic activity of VEGF121 and VEGF165 using several concurrent mechanisms. J. Biol. Chem. 1995, 270, 15059–15065. [Google Scholar] [CrossRef] [Green Version]
- Crisi, G.M.; Katz, I.R.; Zucker, M.B.; Thorbecke, G.J. Induction of inhibitory activity for B cell differentiation in human CD8 T cells with pokeweed mitogen, dimaprit, and cAMP upregulating agents: Countersuppressive effect of platelet factor 4. Cell Immunol. 1996, 172, 205–216. [Google Scholar] [CrossRef]
- Katayama, H.; Yokoyama, A.; Kohno, N.; Sakai, K.; Hiwada, K.; Yamada, H.; Hirai, K. Production of eosinophilic chemokines by normal pleural mesothelial cells. Am. J. Respir. Cell Mol. Biol. 2002, 26, 398–403. [Google Scholar] [CrossRef]
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 2014, 292376. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Xiong, X.; Zhang, Y.; Yan, D.; Jian, Z.; Xu, B.; Zhao, H. MKEY, a Peptide Inhibitor of CXCL4-CCL5 Heterodimer Formation, Protects Against Stroke in Mice. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Rayasam, A.; Hsu, M.; Kijak, J.A.; Kissel, L.; Hernandez, G.; Sandor, M.; Fabry, Z. Immune responses in stroke: How the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology 2018, 154, 363–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.R.; Ritzel, R.; McCullough, L.D.; Liu, F. Microglia and ischemic stroke: A double-edged sword. Int. J. Physiol. Pathophysiol. Pharm. 2013, 5, 73–90. [Google Scholar]
- Garcia-Bonilla, L.; Racchumi, G.; Murphy, M.; Anrather, J.; Iadecola, C. Endothelial CD36 Contributes to Postischemic Brain Injury by Promoting Neutrophil Activation via CSF3. J. Neurosci. 2015, 35, 14783–14793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wang, H.; Sun, G.; Zhang, J.; Edwards, N.J.; Aronowski, J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. J. Neurosci. 2015, 35, 11281–11291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.-N.; Shi, S.X.-Y.; Li, Z.; Li, M.; Wood, K.; Gonzales, R.J.; Liu, Q. Depletion of microglia exacerbates postischemic inflammation and brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2224–2236. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, K.E.; DeMars, K.M.; Alexander, J.C.; de Leon, L.G.; Pacheco, S.C.; Graves, C.; Yang, C.; McCrea, A.O.; Frankowski, J.C.; Garrett, T.J.; et al. Targeting resolution of neuroinflammation after ischemic stroke with a lipoxin A4 analog: Protective mechanisms and long-term effects on neurological recovery. Brain Behav. 2017, 7, e00688. [Google Scholar] [CrossRef]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Clarke, D.T.W.; McMillan, N.A.J. Gene delivery: Cell-specific therapy on target. Nat. Nanotechnol. 2014, 9, 568–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.-Y.; Liu, L.; Yang, Q.-W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef]
- Ahn, K.-C.; MacKenzie, E.M.; Learman, C.R.; Hall, T.C.; Weaver, C.L.; Dunbar, G.L.; Song, M.-S. Inhibition of p53 attenuates ischemic stress-induced activation of astrocytes. Neuroreport 2015, 26, 862–869. [Google Scholar] [CrossRef]
- Choudhury, G.R.; Ryou, M.-G.; Poteet, E.; Wen, Y.; He, R.; Sun, F.; Yuan, F.; Jin, K.; Yang, S.-H. Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res. 2014, 1551, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Grande, B.; Swana, M.; Nguyen, L.; Englezou, P.; Maysami, S.; Allan, S.M.; Rothwell, N.J.; Garlanda, C.; Denes, A.; Pinteaux, E. The acute-phase protein PTX3 is an essential mediator of glial scar formation and resolution of brain edema after ischemic injury. J. Cereb. Blood Flow Metab. 2014, 34, 480–488. [Google Scholar] [CrossRef]
- Xu, L.; Emery, J.F.; Ouyang, Y.-B.; Voloboueva, L.A.; Giffard, R.G. Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia 2010, 58, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Qiu, Y.; Lin, Y.; Miao, Z.; Zhang, J.; Lu, X. Protection by pyruvate against glutamate neurotoxicity is mediated by astrocytes through a glutathione-dependent mechanism. Mol. Biol. Rep. 2011, 38, 3235–3242. [Google Scholar] [CrossRef]
- Ouyang, Y.-B.; Xu, L.; Yue, S.; Giffard, R. Neuroprotection by astrocytes in brain ischemia: Importance of microRNAs. Neurosci. Lett. 2013, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shichita, T.; Sugiyama, Y.; Ooboshi, H.; Sugimori, H.; Nakagawa, R.; Takada, I.; Iwaki, T.; Okada, Y.; Iida, M.; Cua, D.J.; et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat. Med. 2009, 15, 946–950. [Google Scholar] [CrossRef]
- Gelderblom, M.; Weymar, A.; Bernreuther, C.; Velden, J.; Arunachalam, P.; Steinbach, K.; Orthey, E.; Arumugam, T.V.; Leypoldt, F.; Simova, O.; et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012, 120, 3793–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesz, A.; Zhou, W.; Mracskó, É.; Karcher, S.; Bauer, H.; Schwarting, S.; Sun, L.; Bruder, D.; Stegemann, S.; Cerwenka, A.; et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain 2011, 134, 704–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef]
- Liesz, A.; Zhou, W.; Na, S.-Y.; Hämmerling, G.J.; Garbi, N.; Karcher, S.; Mracsko, E.; Backs, J.; Rivest, S.; Veltkamp, R. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J. Neurosci. 2013, 33, 17350–17362. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Pecoraro, R.; Casuccio, A.; Di Raimondo, D.; Buttà, C.; Clemente, G.; Della Corte, V.; Guggino, G.; Arnao, V.; Maida, C.; et al. Peripheral Frequency of CD4+ CD28− Cells in Acute Ischemic Stroke. Medicine 2015, 94, e813. [Google Scholar] [CrossRef]
- Nakajima, T.; Goek, O.; Zhang, X.; Kopecky, S.L.; Frye, R.L.; Goronzy, J.J.; Weyand, C.M. De novo expression of killer immunoglobulin-like receptors and signaling proteins regulates the cytotoxic function of CD4 T cells in acute coronary syndromes. Circ. Res. 2003, 93, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Casuccio, A.; Di Bona, D.; Aiello, A.; Accardi, G.; Arnao, V.; Clemente, G.; Corte, V.D.; et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute ischemic stroke. J. Neuroinflamm. 2019, 16, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurses, K.M.; Yalcin, M.U.; Kocyigit, D.; Canpinar, H.; Evranos, B.; Yorgun, H.; Sahiner, M.L.; Kaya, E.B.; Ozer, N.; Tokgozoglu, L.; et al. Effects of persistent atrial fibrillation on serum galectin-3 levels. Am. J. Cardiol. 2015, 115, 647–651. [Google Scholar] [CrossRef]
- Li, M.; Yang, G.; Xie, B.; Babu, K.; Huang, C. Changes in matrix metalloproteinase-9 levels during progression of atrial fibrillation. J. Int. Med. Res. 2014, 42, 224–230. [Google Scholar] [CrossRef]
- Jensen, L.T. The aminoterminal propeptide of type III procollagen. Studies on physiology and pathophysiology. Dan. Med. Bull. 1997, 44, 70–78. [Google Scholar]
- Naya, T.; Yukiiri, K.; Hosomi, N.; Takahashi, T.; Ohkita, H.; Mukai, M.; Koziol, J.A.; Kohno, M. Brain natriuretic peptide as a surrogate marker for cardioembolic stroke with paroxysmal atrial fibrillation. Cereb. Dis. 2008, 26, 434–440. [Google Scholar] [CrossRef]
- Rodriguez-Yañez, M.; Arias-Rivas, S.; Santamaría, M.; Sobrino, T.; Castillo, J. High pro-BNP levels predict the occurrence of atrial fibrillation after cryptogenic stroke. Neurology 2013, 81. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Inflammation in ischemic stroke subtypes. Curr. Pharm. Des. 2012, 18, 4289–4310. [Google Scholar] [CrossRef]
- Gökhan, S.; Ozhasenekler, A.; Mansur Durgun, H.; Akil, E.; Ustündag, M.; Orak, M. Neutrophil lymphocyte ratios in stroke subtypes and transient ischemic attack. Eur. Rev. Med. Pharm. Sci. 2013, 17, 653–657. [Google Scholar]
- Zhao, L.; Dai, Q.; Chen, X.; Li, S.; Shi, R.; Yu, S.; Yang, F.; Xiong, Y.; Zhang, R. Neutrophil-to-Lymphocyte Ratio Predicts Length of Stay and Acute Hospital Cost in Patients with Acute Ischemic Stroke. J. Stroke Cereb. Dis. 2016, 25, 739–744. [Google Scholar] [CrossRef]
- Nakase, T.; Yamazaki, T.; Ogura, N.; Suzuki, A.; Nagata, K. The impact of inflammation on the pathogenesis and prognosis of ischemic stroke. J. Neurol. Sci. 2008, 271, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Licata, G.; Tuttolomondo, A.; Di Raimondo, D.; Corrao, S.; Di Sciacca, R.; Pinto, A. Immuno-inflammatory activation in acute cardio-embolic strokes in comparison with other subtypes of ischaemic stroke. Thromb. Haemost. 2009, 101, 929–937. [Google Scholar] [PubMed] [Green Version]
- Tuttolomondo, A.; Di Sciacca, R.; Di Raimondo, D.; Serio, A.; D’Aguanno, G.; La Placa, S.; Pecoraro, R.; Arnao, V.; Marino, L.; Monaco, S.; et al. Plasma levels of inflammatory and thrombotic/fibrinolytic markers in acute ischemic strokes: Relationship with TOAST subtype, outcome and infarct site. J. Neuroimmunol. 2009, 215, 84–89. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Yang, Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg. Focus 2007, 22, E4. [Google Scholar] [CrossRef] [Green Version]
- Sumi, N.; Nishioku, T.; Takata, F.; Matsumoto, J.; Watanabe, T.; Shuto, H.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Lipopolysaccharide-activated microglia induce dysfunction of the blood-brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell Mol. Neurobiol. 2010, 30, 247–253. [Google Scholar] [CrossRef]
- da Fonseca, A.C.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R.S. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jin, X.; Liu, K.J.; Liu, W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J. Neurosci 2012, 32, 3044–3057. [Google Scholar] [CrossRef]
- Asahi, M.; Asahi, K.; Jung, J.C.; del Zoppo, G.J.; Fini, M.E.; Lo, E.H. Role for matrix metalloproteinase 9 after focal cerebral ischemia: Effects of gene knockout and enzyme inhibition with BB-94. J. Cereb. Blood Flow Metab. 2000, 20, 1681–1689. [Google Scholar] [CrossRef] [Green Version]
- Rom, S.; Dykstra, H.; Zuluaga-Ramirez, V.; Reichenbach, N.L.; Persidsky, Y. miR-98 and let-7g* protect the blood–brain barrier under neuroinflammatory conditions. J. Cereb. Blood Flow Metab. 2015, 35, 1957–1965. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E. Regulation by glycogen synthase kinase-3 of inflammation and T cells in CNS diseases. Front. Mol. Neurosci. 2011, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, S.H.; Fan, S.; Zhang, M.; Papugani, A.; Reichenbach, N.; Dykstra, H.; Mercer, A.J.; Tuma, R.F.; Persidsky, Y. Inhibition of glycogen synthase kinase 3beta (GSK3beta) decreases inflammatory responses in brain endothelial cells. Am. J. Pathol. 2010, 176, 881–892. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Fan, S.; Dykstra, H.; Rom, S.; Mercer, A.; Reichenbach, N.L.; Gofman, L.; Persidsky, Y. Inhibition of glycogen synthase kinase 3β promotes tight junction stability in brain endothelial cells by half-life extension of occludin and claudin-5. PLoS ONE 2013, 8, e55972. [Google Scholar] [CrossRef] [PubMed]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Schober, A.; Zernecke, A. Chemokines in vascular remodeling. Thromb. Haemost. 2007, 97, 730–737. [Google Scholar] [PubMed]
- Stamatovic, S.M.; Shakui, P.; Keep, R.F.; Moore, B.B.; Kunkel, S.L.; Van Rooijen, N.; Andjelkovic, A.V. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2005, 25, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Xu, W.; Nan, S.; Chang, M.; Zhang, Y. MicroRNA-384-5p Promotes Endothelial Progenitor Cell Proliferation and Angiogenesis in Cerebral Ischemic Stroke through the Delta-Likeligand 4-Mediated Notch Signaling Pathway. Cereb. Dis. 2020, 49, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Vanhoutte, P.M. Adiponectin and adipocyte fatty acid binding protein in the pathogenesis of cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1231–H1240. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Hertzel, A.V.; Steen, K.A.; Bernlohr, D.A. Loss of Fatty Acid Binding Protein 4/aP2 Reduces Macrophage Inflammation Through Activation of SIRT3. Mol. Endocrinol. 2016, 30, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, L.B.; Adams, R.; Alberts, M.J.; Appel, L.J.; Brass, L.M.; Bushnell, C.D.; Culebras, A.; Degraba, T.J.; Gorelick, P.B.; Guyton, J.R.; et al. Primary prevention of ischemic stroke: A guideline from the American Heart Association/American Stroke Association Stroke Council: Cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group: The American Academy of Neurology affirms the value of this guideline. Stroke 2006, 37, 1583–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.-L. Association of Serum Levels of Adipocyte Fatty Acid-Binding Protein and High-Sensitivity C Reactive Protein with Severity of Acute Ischemic Stroke. Cell Biochem. Biophys. 2015, 72, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Geng, L.; Zhang, F.; Shu, L.; Wei, L.; Yeung, P.K.K.; Lam, K.S.L.; Chung, S.K.; Chang, J.; Vanhoutte, P.M.; et al. Adipocyte fatty acid-binding protein exacerbates cerebral ischaemia injury by disrupting the blood–brain barrier. Eur. Heart J. 2020, 41, 3169–3180. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Yang, H.; Xiao, B. Role of microRNAs in endothelial inflammation and senescence. Mol. Biol. Rep. 2012, 39, 4509–4518. [Google Scholar] [CrossRef]
- Long, G.; Wang, F.; Li, H.; Yin, Z.; Sandip, C.; Lou, Y.; Wang, Y.; Chen, C.; Wang, D.W. Circulating miR-30a, miR-126 and let-7b as biomarker for ischemic stroke in humans. BMC Neurol 2013, 13, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, L.Y.; Chan, C.P.Y.; Leung, Y.K.; Jiang, H.L.; Abrigo, J.M.; Wang, D.F.; Chung, J.S.H.; Rainer, T.H.; Graham, C.A. Comparison of miR-124-3p and miR-16 for early diagnosis of hemorrhagic and ischemic stroke. Clin. Chim. Acta 2014, 433, 139–144. [Google Scholar] [CrossRef]
- Xue, Y.; Wei, Z.; Ding, H.; Wang, Q.; Zhou, Z.; Zheng, S.; Zhang, Y.; Hou, D.; Liu, Y.; Zen, K.; et al. MicroRNA-19b/221/222 induces endothelial cell dysfunction via suppression of PGC-1α in the progression of atherosclerosis. Atherosclerosis 2015, 241, 671–681. [Google Scholar] [CrossRef]
- Hu, S.-L.; Cui, G.-L.; Huang, J.; Jiang, J.-G.; Wang, D.-W. An APOC3 3′UTR variant associated with plasma triglycerides levels and coronary heart disease by creating a functional miR-4271 binding site. Sci. Rep. 2016, 6, 32700. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuolin, M.; Kan, P.; Zhang, B. The Diagnostic Value of Serum miRNA-221-3p, miRNA-382-5p, and miRNA-4271 in Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2017, 26. [Google Scholar] [CrossRef]
- Bao, Y.; Lin, C.; Ren, J.; Liu, J. MicroRNA-384-5p regulates ischemia-induced cardioprotection by targeting phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit delta (PI3K p110δ). Apoptosis 2013, 18, 260–270. [Google Scholar] [CrossRef]
- Wang, B.; Zhong, Y.; Huang, D.; Li, J. Macrophage autophagy regulated by miR-384-5p-mediated control of Beclin-1 plays a role in the development of atherosclerosis. Am. J. Transl. Res. 2016, 8, 606–614. [Google Scholar]
- Ogata, K.; Sumida, K.; Miyata, K.; Kushida, M.; Kuwamura, M.; Yamate, J. Circulating miR-9* and miR-384-5p as potential indicators for trimethyltin-induced neurotoxicity. Toxicol. Pathol. 2015, 43, 198–208. [Google Scholar] [CrossRef]
- Fasanaro, P.; Greco, S.; Ivan, M.; Capogrossi, M.C.; Martelli, F. microRNA: Emerging therapeutic targets in acute ischemic diseases. Pharm. Ther. 2010, 125, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Dong, Z.; Li, H.; Liu, X.; Liu, H.; Dong, R. MicroRNA-137 protects neurons against ischemia/reperfusion injury through regulation of the Notch signaling pathway. Exp. Cell Res. 2017, 352, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009, 137, 1124–1135. [Google Scholar] [CrossRef] [Green Version]
- Diez, H.; Fischer, A.; Winkler, A.; Hu, C.-J.; Hatzopoulos, A.K.; Breier, G.; Gessler, M. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp. Cell Res. 2007, 313, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sharghi-Namini, S.; Tan, E.; Ong, L.-L.S.; Ge, R.; Asada, H.H. Dll4-containing exosomes induce capillary sprout retraction in a 3D microenvironment. Sci. Rep. 2014, 4, 4031. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Li, J.; Li, Y.; Yang, X.; Feng, S.; Han, S.; Li, J. Downregulation of miR-181b in mouse brain following ischemic stroke induces neuroprotection against ischemic injury through targeting heat shock protein A5 and ubiquitin carboxyl-terminal hydrolase isozyme L1. J. Neurosci. Res. 2013, 91, 1349–1362. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Evidence | Findings | References |
|---|---|---|
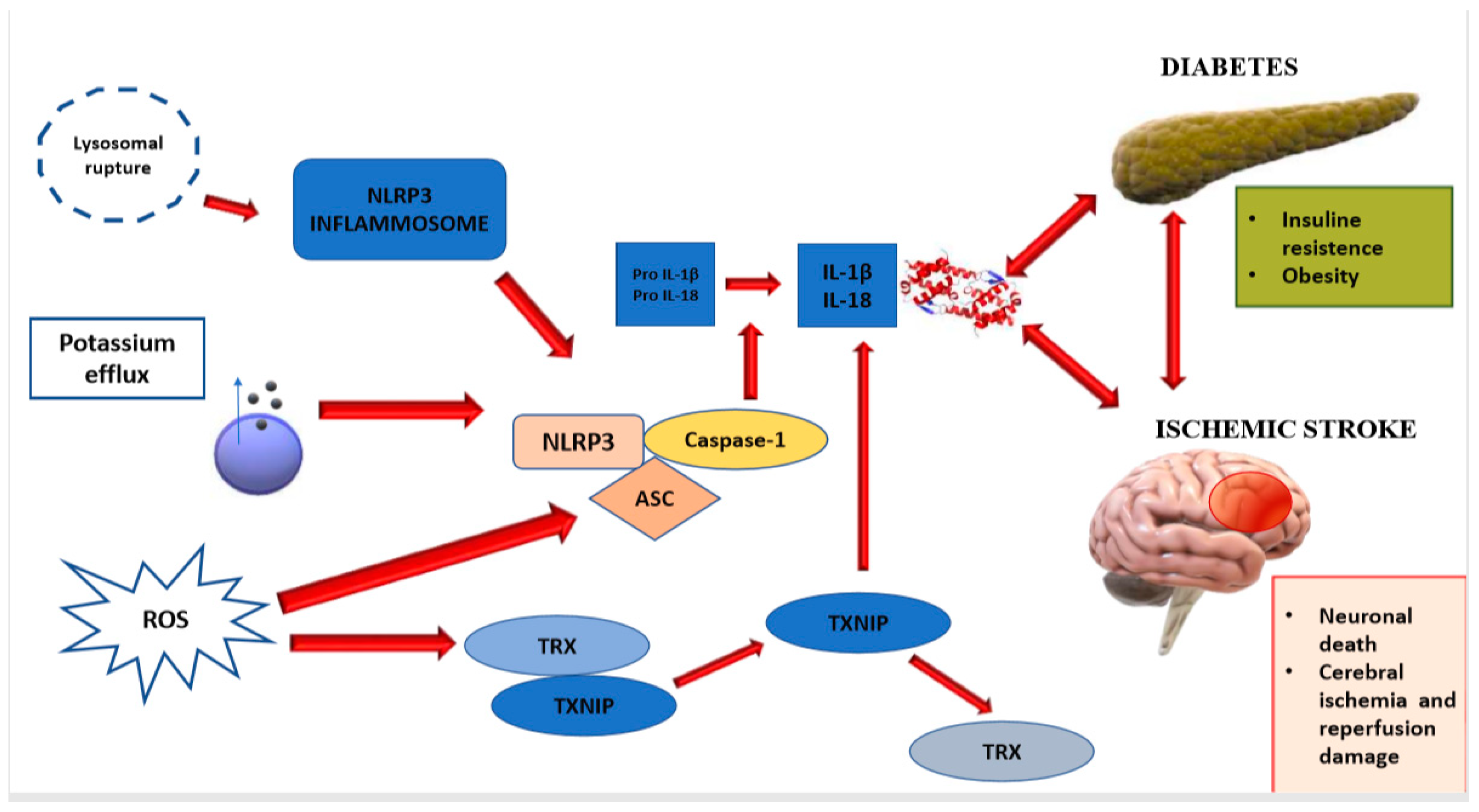
| NLRP3 inflammasome is one of the main contributors to the process of neuroinflammation and consequent brain damage. | NLRP3 inflammasome is responsible for activation of IL-1β, and the release of IL-18, which phosphorylate IRS-1, worsening insulin-resistance and causing neuronal death. | [11,12] |
| High levels or low levels of DKK-3 worsen outcome after an ischemic stroke. | DKK-3 concentrations have an impact on endothelial dysfunction and atherosclerosis. | [13,14] |
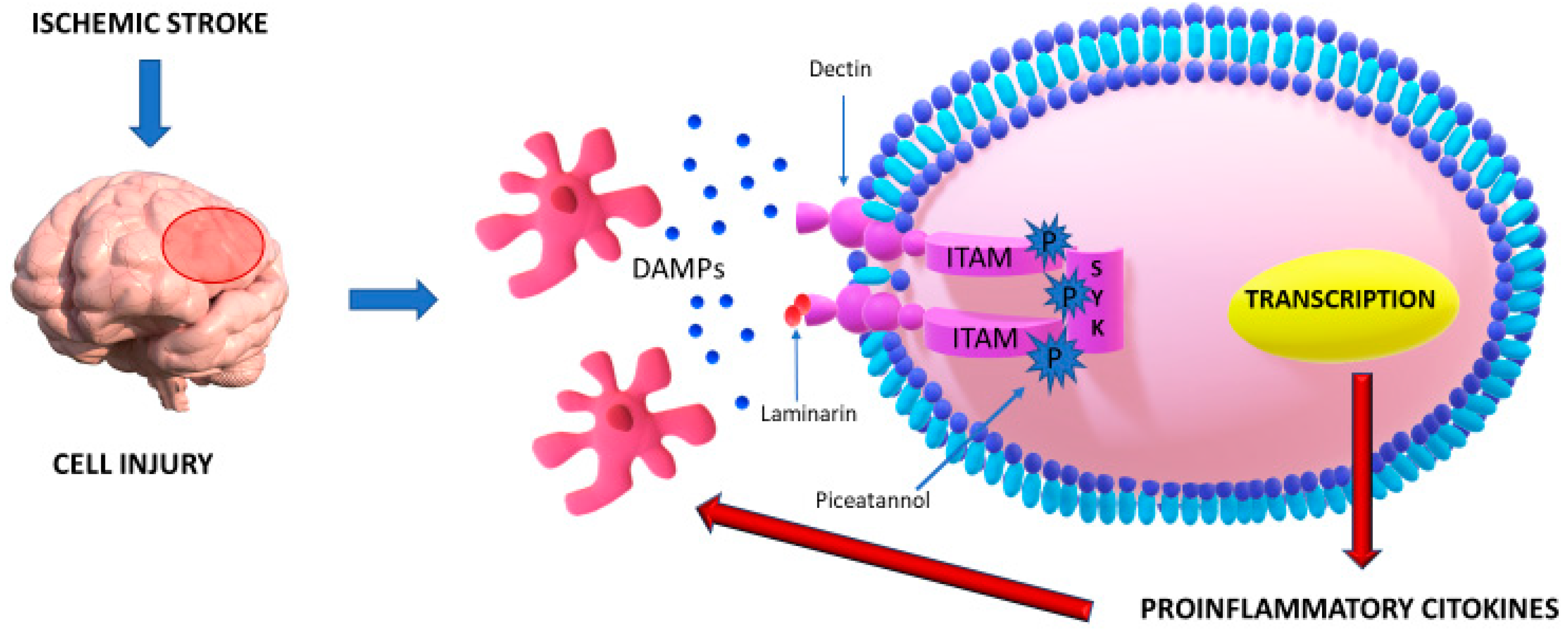
| The inflammatory pathway mediated by Dectin-1/Syk has a pivotal role in neuroinflammation after a stroke. | The crosstalk between Dectin-1 and DAMPs determines phosphorylation of ITAM and, subsequently, of Syk, a kinase which mediates a cascade of neuroinflammation and the release of several cytokines. | [15] |
| The heterodimer CXCL4-CCL5 plays a crucial part in the cerebral injury. | Avoiding the formation of CXCL4-CCL5 heterodimer, MKEY, a cyclic peptide synthesized on the base of CCL5 in mice, restricts the ischemic cerebral lesion and ameliorate neurologic deficits in mice. | [16,17] |
| The microglial IRF5-IRF4 regulatory axis impacts stroke outcomes. | Studies suggested the existence of an IRF5-IRF4regulatory axis in which IRF5 signaling mediates microglial pro-inflammatory responses and IRF4 signaling enhances microglial anti-inflammatory activation. | [18,19] |
| CD200 could play an important role in therapeutic strategies for the treatment of ischemic stroke by way of the inhibition of detrimental leukocyte activation and improvement of stroke-induced lymphopenia. | The loss of interplay between CD200-CD200R will induce microglial proliferation and activation that may exacerbate the process of neuroinflammation and aggravate the prognosis after stroke. | [20] |
| The glial glutamate transporter (GLT-1), an astrocytes’ transporter, performs neuroprotective effects in the initial phases of ischemia. | It determines the formation of a glial scar which has a neuroprotective role because it safeguards salubrious tissues from damages attributable to inflammation. | [21,22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuttolomondo, A.; Puleo, M.G.; Velardo, M.C.; Corpora, F.; Daidone, M.; Pinto, A. Molecular Biology of Atherosclerotic Ischemic Strokes. Int. J. Mol. Sci. 2020, 21, 9372. https://doi.org/10.3390/ijms21249372
Tuttolomondo A, Puleo MG, Velardo MC, Corpora F, Daidone M, Pinto A. Molecular Biology of Atherosclerotic Ischemic Strokes. International Journal of Molecular Sciences. 2020; 21(24):9372. https://doi.org/10.3390/ijms21249372
Chicago/Turabian StyleTuttolomondo, Antonino, Maria Grazia Puleo, Maria Chiara Velardo, Francesca Corpora, Mario Daidone, and Antonio Pinto. 2020. "Molecular Biology of Atherosclerotic Ischemic Strokes" International Journal of Molecular Sciences 21, no. 24: 9372. https://doi.org/10.3390/ijms21249372
APA StyleTuttolomondo, A., Puleo, M. G., Velardo, M. C., Corpora, F., Daidone, M., & Pinto, A. (2020). Molecular Biology of Atherosclerotic Ischemic Strokes. International Journal of Molecular Sciences, 21(24), 9372. https://doi.org/10.3390/ijms21249372