Mechanism of Action of Atypical Antipsychotic Drugs in Mood Disorders



Abstract
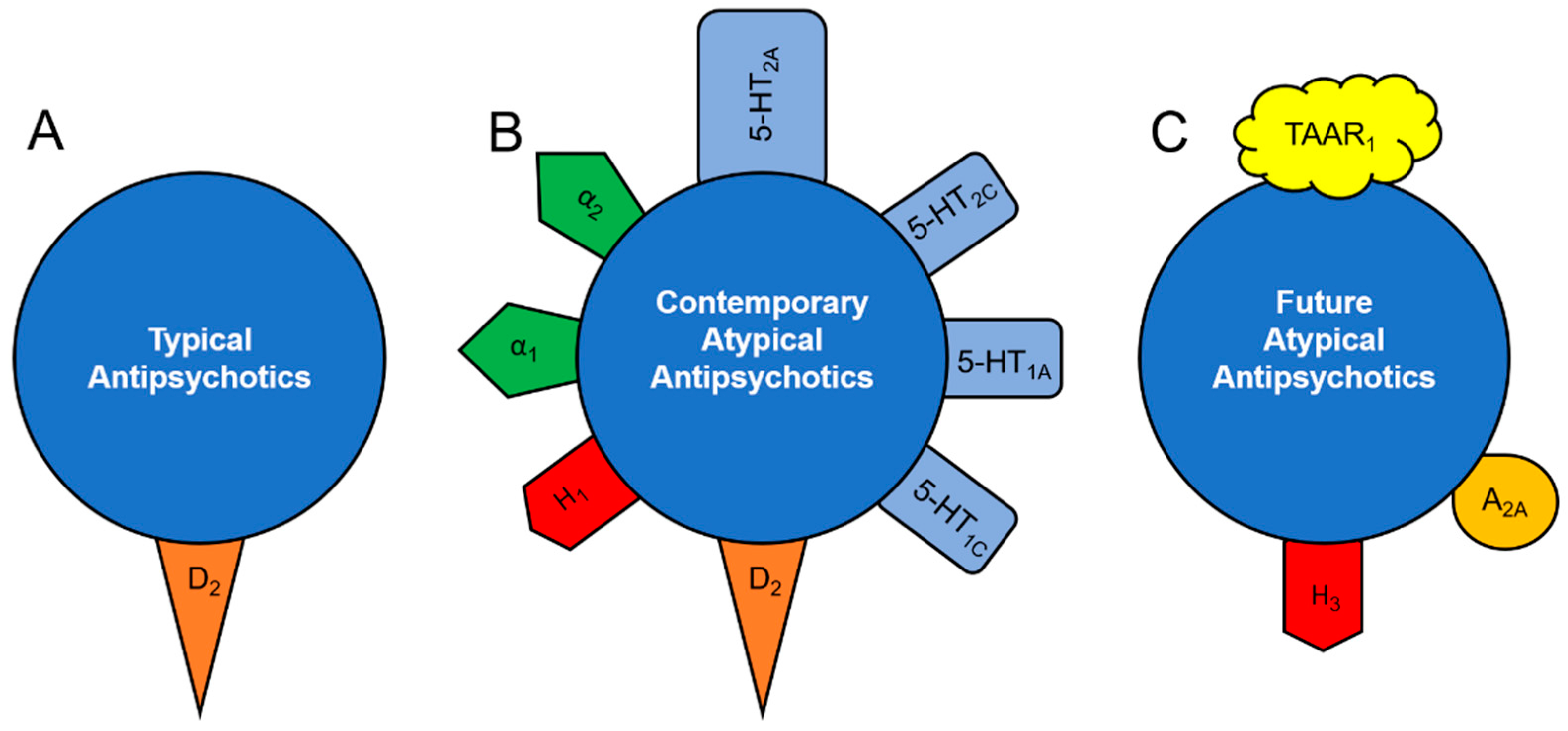
:1. Introduction
2. Mechanism of Action of Atypical Antipsychotics in Mood Disorders
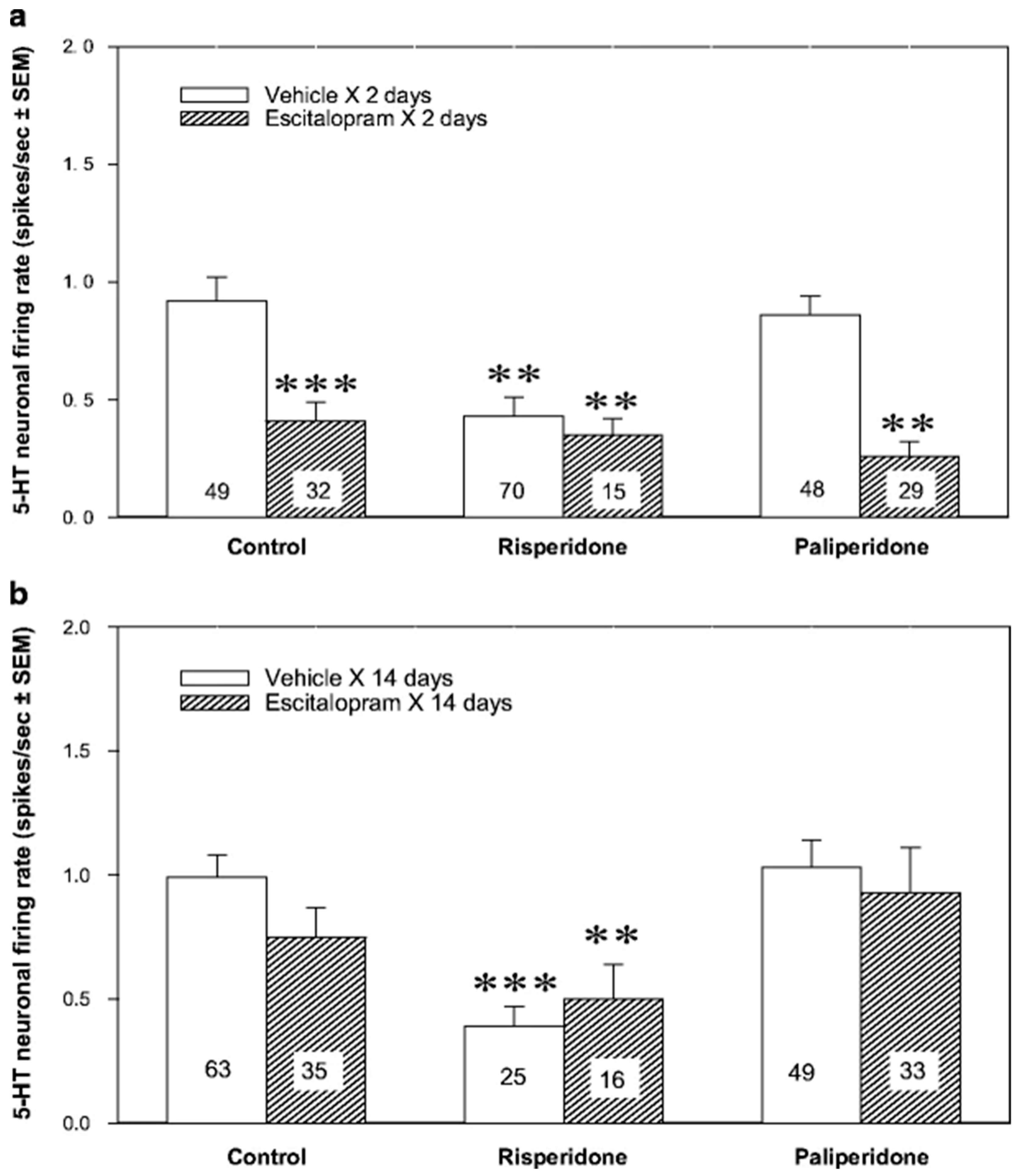
2.1. Serotonergic Mechanisms
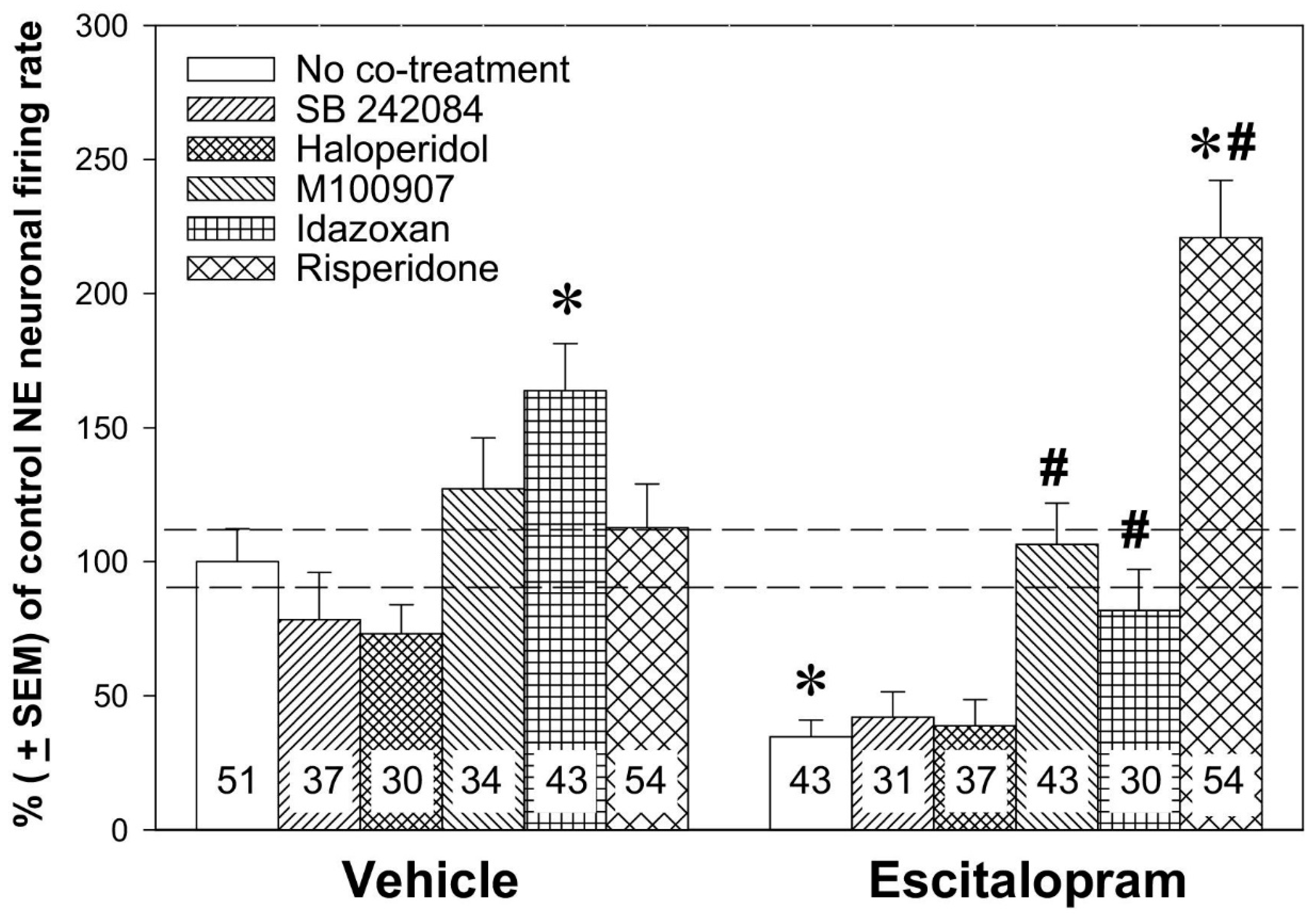
2.2. Noradrenergic Mechanisms
2.3. Dopaminergic Mechanisms
2.4. Histaminergic Mechanisms
2.5. Purinergic Mechanisms
2.6. Trace Aminergic Mechanisms
3. Summary
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT | 5-Hydroxytryptamine (serotonin) |
| CNS | Central nervous system |
| EPM | Elevated plus maze test |
| FST | Forced swim test |
| GABA | γ-Aminobutyric acid |
| LC | Locus coeruleus |
| NAcc | Nucleus accumbens |
| PFC | Prefrontal cortex |
| SNRIs | Selective serotonin reuptake inhibitor |
| SSRIs | Dual serotonin and norepinephrine reuptake inhibitor |
| TAAR | Trace amine-associated receptor |
| VTA | Ventral tegmental area |
References
- Shen, W.W. A history of antipsychotic drug development. Compr. Psychiatry 1999, 40, 407–414. [Google Scholar] [CrossRef]
- Leucht, S.; Corves, C.; Arbter, D.; Engel, R.R.; Li, C.; Davis, J.M. Second-generation versus first-generation antipsychotic drugs for schizophrenia: A meta-analysis. Lancet 2009, 373, 31–41. [Google Scholar] [CrossRef]
- Leucht, S.; Cipriani, A.; Spineli, L.; Mavridis, D.; Orey, D.; Richter, F.; Samara, M.; Barbui, C.; Engel, R.R.; Geddes, J.R.; et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis. Lancet 2013, 382, 951–962. [Google Scholar] [CrossRef]
- Aparasu, R.R.; Jano, E.; Johnson, M.L.; Chen, H. Hospitalization risk associated with typical and atypical antipsychotic use in community-dwelling elderly patients. Am. J. Geriatr. Pharmacother. 2008, 6, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Mauri, M.C.; Moliterno, D.; Rossattini, M.; Colasanti, A. Depression in schizophrenia: Comparison of first- and second-generation antipsychotic drugs. Schizophr. Res. 2008, 99, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Blier, P. Rational site-directed pharmacotherapy for major depressive disorder. Int. J. Neuropsychopharmacol. 2013, 17, 997–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blier, P.; Blondeau, C. Neurobiological bases and clinical aspects of the use of aripiprazole in treatment-resistant major depressive disorder. J. Affect. Disord. 2011, 128 (Suppl. 1), S3–S10. [Google Scholar] [CrossRef]
- Blier, P. Atypical antipsychotics for mood and anxiety disorders: Safe and effective adjuncts? J. Psychiatry Neurosci. 2005, 30, 232–2333. [Google Scholar]
- Berk, M.; Dodd, S. Efficacy of atypical antipsychotics in bipolar disorder. Drugs 2005, 65, 257–269. [Google Scholar] [CrossRef]
- Gao, K.; Gajwani, P.; Elhaj, O.; Calabrese, J.R. Typical and Atypical Antipsychotics in Bipolar Depression. J. Clin. Psychiatry 2005, 66, 1376–1385. [Google Scholar] [CrossRef]
- Cipriani, A.; Barbui, C.; Salanti, G.; Rendell, J.; Brown, R.; Stockton, S.; Purgato, M.; Spineli, L.M.; Goodwin, G.M.; Geddes, J.R. Comparative efficacy and acceptability of antimanic drugs in acute mania: A multiple-treatments meta-analysis. Lancet 2011, 378, 1306–1315. [Google Scholar] [CrossRef]
- Dhillon, S. Aripiprazole: A review of its use in the management of mania in adults with bipolar I disorder. Drugs 2012, 72, 133–162. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Taylor, M.J.; Geddes, J. Aripiprazole alone or in combination for acute mania. Cochrane Database Syst. Rev. 2013, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chwieduk, C.M.; Scott, L.J. Asenapine: A review of its use in the management of mania in adults with bipolar I disorder. CNS Drugs 2011, 25, 251–267. [Google Scholar] [CrossRef]
- Vieta, E.; Valenti, M. Pharmacological Management of Bipolar Depression: Acute Treatment, Maintenance, and Prophylaxis. CNS Drugs 2013, 27, 515–529. [Google Scholar] [CrossRef]
- Escudero, M.A.G.; Gutiérrez-Rojas, L.; Lahera, G. Second Generation Antipsychotics Monotherapy as Maintenance Treatment for Bipolar Disorder: A Systematic Review of Long-Term Studies. Psychiatr. Q. 2020, 91, 1047–1060. [Google Scholar] [CrossRef]
- Hershenberg, R.; Gros, D.F.; Brawman-Mintzer, O. Role of Atypical Antipsychotics in the Treatment of Generalized Anxiety Disorder. CNS Drugs 2014, 28, 519–533. [Google Scholar] [CrossRef]
- Zhou, X.; Keitner, G.I.; Qin, B.; Ravindran, A.V.; Bauer, M.; Del Giovane, C.; Zhao, J.; Liu, Y.; Fang, Y.; Zhang, Y.; et al. Atypical Antipsychotic Augmentation for Treatment-Resistant Depression: A Systematic Review and Network Meta-Analysis. Int. J. Neuropsychopharmacol. 2015, 18, pyv060. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Ravindran, A.V.; Qin, B.; Del Giovane, C.; Li, Q.; Bauer, M.; Liu, Y.; Fang, Y.; da Silva, T.; Zhang, Y.; et al. Comparative efficacy, acceptability, and tolerability of augmentation agents in treatment-resistant depression: Systematic review and network meta-analysis. J. Clin. Psychiatry 2015, 76, e487–e498. [Google Scholar] [CrossRef]
- Ceskova, E.; Silhan, P. Novel treatment options in depression and psychosis. Neuropsychiatr. Dis. Treat. 2018, 14, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Lenze, E.J.; Mulsant, B.H.; Blumberger, D.M.; Karp, J.F.; Newcomer, J.W.; Anderson, S.J.; Dew, M.A.; Butters, M.A.; Stack, J.A.; Begley, A.E.; et al. Efficacy, safety, and tolerability of augmentation pharmacotherapy with aripiprazole for treatment-resistant depression in late life: A randomised, double-blind, placebo-controlled trial. Lancet 2015, 386, 2404–2412. [Google Scholar] [CrossRef] [Green Version]
- Markovic, M.; Gallipani, A.; Patel, K.H.; Maroney, M. Brexpiprazole: A New Treatment Option for Schizophrenia and Major Depressive Disorder. Ann. Pharmacother. 2016, 51, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.E.; Thase, M.E. Risperidone: Review of its therapeutic utility in depression. Psychopharmacol. Bull. 2001, 35, 109–129. [Google Scholar] [PubMed]
- Cohen, H.; Zohar, J.; Kaplan, Z.; Arnt, J. Adjunctive treatment with brexpiprazole and escitalopram reduces behavioral stress responses and increase hypothalamic NPY immunoreactivity in a rat model of PTSD-like symptoms. Eur. Neuropsychopharmacol. 2018, 28, 63–74. [Google Scholar] [CrossRef]
- Kim, S.-K.; Fristrup, P.; Abrol, R.; Iii, W.A.G. Structure-Based Prediction of Subtype Selectivity of Histamine H3 Receptor Selective Antagonists in Clinical Trials. J. Chem. Inf. Model. 2011, 51, 3262–3274. [Google Scholar] [CrossRef] [Green Version]
- Berlin, M.; Boyce, C.W. Recent advances in the development of histamine H3antagonists. Expert Opin. Ther. Patents 2007, 17, 675–687. [Google Scholar] [CrossRef]
- Yanai, K.; Tashiro, M. The physiological and pathophysiological roles of neuronal histamine: An insight from human positron emission tomography studies. Pharmacol. Ther. 2007, 113, 1–15. [Google Scholar] [CrossRef]
- Domenici, M.R.; Ferrante, A.; Martire, A.; Chiodi, V.; Pepponi, R.; Tebano, M.T.; Popoli, P. Adenosine A2A receptor as potential therapeutic target in neuropsychiatric disorders. Pharmacol. Res. 2019, 147, 104338. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signalling and Neurological Diseases: An Update. CNS Neurol. Disord. Drug. Targets 2017, 16, 257–265. [Google Scholar] [CrossRef]
- Chen, J.-F. Adenosine Receptor Control of Cognition in Normal and Disease. Int. Rev. Neurobiol. 2014, 119, 257–307. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.-L.; Pouzet, B.; Mory, R.; Bradaia, A.; Buchy, D.; Metzler, V.; Chaboz, S.; Zbinden, K.G.; Galley, G.; et al. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol. Psychiatry 2012, 18, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.; Carvalho, A.F.; Puri, B.K.; Maes, M.; Bortolasci, C.C.; Morris, G.; Berk, M. Trace Amine-Associated Receptor 1 (TAAR1): A new drug target for psychiatry? Neurosci. Biobehav. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.D.; Canales, J.J.; Zucchi, R.; Espinoza, S.; Sukhanov, I.; Gainetdinov, R.R. Trace amine-associated receptor 1: A multimodal therapeutic target for neuropsychiatric diseases. Expert Opin. Ther. Targets 2018, 22, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Schotte, A.; Janssen, P.F.M.; Gommeren, W.; Luyten, W.H.M.L.; Van Gompel, P.; Lesage, A.S.; De Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology 1996, 124, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, H.A. Atypical antipsychotic-induced metabolic side effects: Insights from receptor-binding profiles. Mol. Psychiatry 2008, 13, 27–35. [Google Scholar] [CrossRef]
- Li, P.; Snyder, G.L.; Vanover, K.E. Dopamine Targeting Drugs for the Treatment of Schizophrenia: Past, Present and Future. Curr. Top. Med. Chem. 2016, 16, 3385–3403. [Google Scholar] [CrossRef] [Green Version]
- Stark, A.D.; Jordan, S.; Allers, K.A.; Bertekap, R.L.; Chen, R.; Kannan, T.M.; Molski, T.F.; Yocca, F.D.; Sharp, T.; Kikuchi, T.; et al. Interaction of the novel antipsychotic aripiprazole with 5-HT1A and 5-HT2A receptors: Functional receptor-binding and in vivo electrophysiological studies. Psychopharmacology 2006, 190, 373–382. [Google Scholar] [CrossRef]
- Dremencov, E.; El Mansari, M.; Blier, P. Distinct electrophysiological effects of paliperidone and risperidone on the firing activity of rat serotonin and norepinephrine neurons. Psychopharmacology 2007, 194, 63–72. [Google Scholar] [CrossRef]
- Chernoloz, O.; El Mansari, M.; Blier, P. Electrophysiological studies in the rat brain on the basis for aripiprazole augmentation of antidepressants in major depressive disorder. Psychopharmacology 2009, 206, 335–344. [Google Scholar] [CrossRef]
- Oosterhof, C.A.; El Mansari, M.; Bundgaard, C.; Blier, P. Brexpiprazole Alters Monoaminergic Systems following Repeated Administration: An in Vivo Electrophysiological Study. Int. J. Neuropsychopharmacol. 2016, 19, pyv111. [Google Scholar] [CrossRef] [Green Version]
- Oosterhof, C.A.; El Mansari, M.; Blier, P. Asenapine alters the activity of monoaminergic systems following its subacute and long-term administration: An in vivo electrophysiological characterization. Eur. Neuropsychopharmacol. 2015, 25, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, K.; Górska, A.; Noworyta-Sokołowska, K.; Wojtas, A.; Rogóż, Z.; Gołembiowska, K. The effect of chronic co-treatment with risperidone and novel antidepressant drugs on the dopamine and serotonin levels in the rats frontal cortex. Pharmacol. Rep. 2018, 70, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Hereta, M.; Kamińska, K.; Białoń, M.; Wąsik, A.; Lorenc-Koci, E.; Rogóż, Z. Effect of combined treatment with aripiprazole and antidepressants on the MK-801-induced deficits in recognition memory in novel recognition test and on the release of monoamines in the rat frontal cortex. Behav. Brain Res. 2020, 393, 112769. [Google Scholar] [CrossRef]
- Kaminska, K.; Rogóż, Z. The antidepressant- and anxiolytic-like effects following co-treatment with escitalopram and risperidone in rats. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2016, 67, 471–480. [Google Scholar]
- El Mansari, M.; Sánchez, C.; Chouvet, G.; Renaud, B.; Haddjeri, N. Effects of acute and long-term administration of escitalopram and citalopram on serotonin neurotransmission: An in vivo electrophysiological study in rat brain. Neuropsychopharmacology 2005, 30, 1269–1277. [Google Scholar] [CrossRef]
- Mørk, A.; Kreilgaard, M.; Sánchez, C. The R-enantiomer of citalopram counteracts escitalopram-induced increase in extracellular 5-HT in the frontal cortex of freely moving rats. Neuropharmacology 2003, 45, 167–173. [Google Scholar] [CrossRef]
- Kasamo, K.; Blier, P.; De Montigny, C. Blockade of the serotonin and norepinephrine uptake processes by duloxetine: In vitro and in vivo studies in the rat brain. J. Pharmacol. Exp. Ther. 1996, 277, 278–286. [Google Scholar] [PubMed]
- Mongeau, R.; Weiss, M.; De Montigny, C.; Blier, P. Effect of acute, short- and long-term milnacipran administration on rat locus coeruleus noradrenergic and dorsal raphe serotonergic neurons. Neuropharmacology 1998, 37, 905–918. [Google Scholar] [CrossRef]
- Dong, J.; De Montigny, C.; Blier, P. Assessment of the serotonin reuptake blocking property of YM992: Electrophysiological studies in the rat hippocampus and dorsal raphe. Synapse 1999, 34, 277–289. [Google Scholar] [CrossRef]
- Jean-Claude, P.; Béïque, J.-C.; Blier, P.; De Montigny, C.; Debonnel, G. Potentiation by (-)Pindolol of the Activation of Postsynaptic 5-HT1A Receptors Induced by Venlafaxine. Neuropsychopharmacology 2000, 23, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Guiard, B.P.; Chenu, F.; El Mansari, M.; Blier, P. Characterization of the electrophysiological properties of triple reuptake inhibitors on monoaminergic neurons. Int. J. Neuropsychopharmacol. 2011, 14, 211–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiard, B.P.; El Mansari, M.; Murphy, D.L.; Blier, P. Altered response to the selective serotonin reuptake inhibitor escitalopram in mice heterozygous for the serotonin transporter: An electrophysiological and neurochemical study. Int. J. Neuropsychopharmacol. 2012, 15, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Mansari, M.; Crnic, A.; Oosterhof, C.A.; Blier, P. Long-term administration of the antidepressant vilazodone modulates rat brain monoaminergic systems. Neuropharmacology 2015, 99, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Blier, P.; Ward, N.M. Is there a role for 5-HT1A agonists in the treatment of depression? Biol. Psychiatry 2003, 53, 193–203. [Google Scholar] [CrossRef]
- Blier, P. Pharmacology of rapid-onset antidepressant treatment strategies. J. Clin. Psychiatry 2001, 62 (Suppl. 1), 12–17. [Google Scholar]
- Blier, P.; Piñeyro, G.; el Mansari, M.; Bergeron, R.; de Montigny, C. Role of somatodendritic 5-HT autoreceptors in modulating 5-HT neurotransmission. Ann. N. Y. Acad. Sci. 1998, 861, 204–216. [Google Scholar] [CrossRef]
- Mongeau, R.; Blier, P.; de Montigny, C. The serotonergic and noradrenergic systems of the hippocampus: Their interactions and the effects of antidepressant treatments. Brain Res. Rev. 1997, 23, 145–195. [Google Scholar] [CrossRef] [Green Version]
- Chernoloz, O.; El Mansari, M.; Blier, P. Effects of Sustained Administration of Quetiapine Alone and in Combination with a Serotonin Reuptake Inhibitor on Norepinephrine and Serotonin Transmission. Neuropsychopharmacol. 2012, 37, 1717–1728. [Google Scholar] [CrossRef] [Green Version]
- Stahl, S.M. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2008. [Google Scholar]
- American Psychiatric Association; DSM-5 Task Force. Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Dremencov, E.; El Mansari, M.; Blier, P. Noradrenergic Augmentation of Escitalopram Response by Risperidone: Electrophysiologic Studies in the Rat Brain. Biol. Psychiatry 2007, 61, 671–678. [Google Scholar] [CrossRef]
- Szabo, S.T.; De Montigny, C.; Blier, P. Modulation of noradrenergic neuronal firing by selective serotonin reuptake blockers. Br. J. Pharmacol. 1999, 126, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Guiard, B.P.; El Mansari, M.; Merali, Z.; Blier, P. Functional interactions between dopamine, serotonin and norepinephrine neurons: An in-vivo electrophysiological study in rats with monoaminergic lesions. Int. J. Neuropsychopharmacol. 2008, 11, 625–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, S.T.; Blier, P. Effects of Serotonin (5-Hydroxytryptamine, 5-HT) Reuptake Inhibition Plus 5-HT2A Receptor Antagonism on the Firing Activity of Norepinephrine Neurons. J. Pharmacol. Exp. Ther. 2002, 302, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, S.T.; Blier, P. Response of the Norepinephrine System to Antidepressant Drugs. CNS Spectrums 2001, 6, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Dawe, G.S.; Huff, K.D.; Vandergriff, J.L.; Sharp, T.; Fromm, M.; Rasmussen, K. Olanzapine activates the rat locus coeruleus: In vivo electrophysiology and c-Fos immunoreactivity. Biol. Psychiatry 2001, 50, 510–520. [Google Scholar] [CrossRef]
- Nasif, F.J.; Cuadra, G.R.; A Ramirez, O. Effects of chronic risperidone on central noradrenergic transmission. Eur. J. Pharmacol. 2000, 394, 67–73. [Google Scholar] [CrossRef]
- Blier, P.; Szabo, S.T. Potential mechanisms of action of atypical antipsychotic medications in treatment-resistant depression and anxiety. J. Clin. Psychiatry 2005, 66 (Suppl. 8), 30–40. [Google Scholar]
- Tremblay, P.; Blier, P. Catecholaminergic Strategies for the Treatment of Major Depression. Curr. Drug Targets 2006, 7, 149–158. [Google Scholar] [CrossRef]
- Dremencov, E.; El Mansari, M.; Blier, P. Effects of sustained serotonin reuptake inhibition on the firing of dopamine neurons in the rat ventral tegmental area. J. Psychiatry Neurosci. 2009, 34, 223–229. [Google Scholar]
- El Mansari, M.; Guiard, B.P.; Chernoloz, O.; Ghanbari, R.; Katz, N.; Blier, P. Relevance of Norepinephrine-Dopamine Interactions in the Treatment of Major Depressive Disorder. CNS Neurosci. Ther. 2010, 16, e1–e17. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, E.O. The role of histamine on cognition. Behav. Brain Res. 2009, 199, 183–189. [Google Scholar] [CrossRef]
- Dere, E.; Zlomuzica, A.; Silva, M.D.S.; Ruocco, L.; Sadile, A.; Huston, J. Neuronal histamine and the interplay of memory, reinforcement and emotions. Behav. Brain Res. 2010, 215, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Panula, P.; Nuutinen, S. The histaminergic network in the brain: Basic organization and role in disease. Nat. Rev. Neurosci. 2013, 14, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Flik, G.; Cremers, T.I.H.F.; Folgering, J.H.; Dremencov, E.; Westerink, B.H.C. The role of cortical and hypothalamic histamine-3 receptors in the modulation of central histamine neurotransmission: An in vivo electrophysiology and microdialysis study. Eur. J. Neurosci. 2011, 34, 1747–1755. [Google Scholar] [CrossRef]
- Flik, G.; Folgering, J.H.A.; Cremers, T.I.H.F.; Westerink, B.H.C.; Dremencov, E. Interaction Between Brain Histamine and Serotonin, Norepinephrine, and Dopamine Systems: In Vivo Microdialysis and Electrophysiology Study. J. Mol. Neurosci. 2015, 56, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, C.; Morales, L.; Cano, M.V.; Sancho, I.; Alguacil, L.F. Effects of histamine H 3 receptor ligands in experimental models of anxiety and depression. Psychopharmacology 1999, 142, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Bertaina-Anglade, V.; Drieu-La-Rochelle, C.; Mocaër, E.; Seguin, L. Memory facilitating effects of agomelatine in the novel object recognition memory paradigm in the rat. Pharmacol. Biochem. Behav. 2011, 98, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, F.; Yamauchi, M.; Oyama, M.; Okuma, K.; Onozawa, K.; Nagayama, T.; Shinei, R.; Ishikawa, M.; Sato, Y.; Kakui, N. Anxiolytic-like profiles of histamine H3 receptor agonists in animal models of anxiety: A comparative study with antidepressants and benzodiazepine anxiolytic. Psychopharmacology 2009, 205, 177–187. [Google Scholar] [CrossRef]
- Arias-Montaño, J.A.; Floran, B.; Garcia, M.; Aceves, J.; Young, J.M. Histamine H(3) receptor-mediated inhibition of depolarization-induced, dopamine D(1) receptor-dependent release of [(3)H]-gamma-aminobutryic acid from rat striatal slices. Br. J. Pharmacol. 2001, 133, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, H.; Knigge, U.; Kjaer, A.; Warberg, J. Interactions of histaminergic and serotonergic neurons in the hypothalamic regulation of prolactin and ACTH secretion. Neuroendocrinology 1996, 64, 329–336. [Google Scholar] [CrossRef]
- Knigge, U.; Søe-Jensen, P.; Jørgensen, H.; Kjær, A.; Møller, M.; Warberg, J.; Kjær, A. Stress-induced release of anterior pituitary hormones: Effect of H3 receptor-mediated inhibition of histaminergic activity or posterior hypothalamic lesion. Neuroendocrinology 1999, 69, 44–53. [Google Scholar] [CrossRef]
- Andersen, M.B.; Fuxe, K.; Werge, T.; Gerlach, J. The adenosine A2A receptor agonist CGS 21680 exhibits antipsychotic-like activity in Cebus apella monkeys. Behav. Pharmacol. 2002, 13, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Popoli, P.; Rimondini, R.; Reggio, R.; Kehr, J.; Fuxe, K. Adenosine A2A and group I metabotropic glutamate receptors synergistically modulate the binding characteristics of dopamine D2 receptors in the rat striatum. Neuropharmacology 1999, 38, 129–140. [Google Scholar] [CrossRef]
- Rimondini, R.; Fuxe, K.; Ferré, S. Multiple intramembrane receptor–receptor interactions in the regulation of striatal dopamine D2 receptors. NeuroReport 1999, 10, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Cabiale, Z.; Hurd, Y.; Guidolin, D.; Finnman, U.B.; Zoli, M.; Agnati, L.F.; Vanderhaeghen, J.J.; Fuxe, K.; Ferré, S. Adenosine A2A agonist CGS 21680 decreases the affinity of dopamine D2 receptors for dopamine in human striatum. NeuroReport 2001, 12, 1831–1834. [Google Scholar] [CrossRef]
- Fuxe, K.; Ferré, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A2A and Dopamine D2 Heteromeric Receptor Complexes and Their Function. J. Mol. Neurosci. 2005, 26, 209–220. [Google Scholar] [CrossRef]
- Dremencov, E.; Lacinova, L.; Flik, G.; Folgering, J.H.; Cremers, T.I.; Westerink, B.H. Purinergic regulation of brain catecholamine neurotransmission: In vivo electrophysiology and microdialysis study in rats. Gen. Physiol. Biophys. 2016, 36, 431–441. [Google Scholar] [CrossRef] [Green Version]
- El Yacoubi, M.; Ledent, C.; Parmentier, M.; Bertorelli, R.; Ongini, E.; Costentin, J.; Vaugeois, J.-M. Adenosine A2A receptor antagonists are potential antidepressants: Evidence based on pharmacology and A2A receptor knockout mice. Br. J. Pharmacol. 2001, 134, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Kaster, M.P.; Rosa, A.O.; Rosso, M.M.; Goulart, E.C.; Dos Santos, A.R.S.; Rodrigues, A.L.S. Adenosine administration produces an antidepressant-like effect in mice: Evidence for the involvement of A1 and A2A receptors. Neurosci. Lett. 2004, 355, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Kaster, M.P.; Dos Santos, A.R.S.; Rodrigues, A.L.S. Involvement of 5-HT1A receptors in the antidepressant-like effect of adenosine in the mouse forced swimming test. Brain Res. Bull. 2005, 67, 53–61. [Google Scholar] [CrossRef]
- Lindemann, L.; Hoener, M.C. A renaissance in trace amines inspired by a novel GPCR family. Trends Pharmacol. Sci. 2005, 26, 274–281. [Google Scholar] [CrossRef]
- Borowsky, B.; Adham, N.; Jones, K.A.; Raddatz, R.; Artymyshyn, R.; Ogozalek, K.L.; Durkin, M.M.; Lakhlani, P.P.; Bonini, J.A.; Pathirana, S.; et al. Trace amines: Identification of a family of mammalian G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 8966–8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, M.D.; Gainetdinov, R.R.; Hoener, M.C.; Shahid, M. Pharmacology of human trace amine-associated receptors: Therapeutic opportunities and challenges. Pharmacol. Ther. 2017, 180, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Revel, F.G.; Meyer, C.A.; Bradaia, A.; Jeanneau, K.; Calcagno, E.; André, C.B.; Haenggi, M.; Miss, M.T.; Galley, G.; Norcross, R.D.; et al. Brain-Specific Overexpression of Trace Amine-Associated Receptor 1 Alters Monoaminergic Neurotransmission and Decreases Sensitivity to Amphetamine. Neuropsychopharmacology 2012, 37, 2580–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revel, F.G.; Moreau, J.-L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef] [Green Version]
- Grinchii, D.; Hoener, C.; Dremencov, E. P.538 Ligands of trace amine-associated receptor 1 modulate in vivo excitability of rat serotonin and dopamine, but not norepinephrine neurons. Eur. Neuropsychopharmacol. 2019, 29, S379–S380. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug | Neurophysiological Effect | |
|---|---|---|
| On its Own (Monotherapy) | In Combination with SSRIs | |
| Currently Used Atypical Antipsychotic Drugs | ||
| Aripiprazole | 5-HT neurons ↓ (acute) 5-HT neurons ↑ (subchronic, chronic) NE neurons ↑ (subchronic, chronic) | SSRI-induced 5-HT inhibition (subchronic) ↓ SSRI-induced NE inhibition (subchronic, chronic) ↓ SSRI-induced DA inhibition (subchronic, chronic) ↓ |
| Asenapine | 5-HT neurons ↑ (subchronic) NE neurons ↑ (chronic) DA neurons ↑ (chronic) | |
| Brexpiprazole | 5-HT neurons ↑ (subchronic, chronic) NE neurons↑ (subchronic, chronic) | |
| Olanzapine | NE neurons ↑ (acute) | |
| Paliperidone | SSRI-induced 5-HT inhibition (subchronic) ↓ SSRI-induced NE inhibition (subchronic, chronic) ↓ | |
| Risperidone | 5-HT neurons↓ (acute, subchronic, chronic) | SSRI-induced NE inhibition (subchronic, chronic) ↓ |
| Quetiapine | NE neurons ↑ (subchronic, chronic) | SSRI-induced NE inhibition (subchronic, chronic) ↓ |
| Putative Novel Atypical Antipsychotic Drugs | ||
| Thioperamide (H3/4 partial agonist) | DA neurons ↑ (acute) | |
| ZM 241385 (A2A antagonist) | DA neurons ↑ (acute) | |
| RO5263397 (TAAR1 partial agonist) | 5-HT neurons ↑ (acute) DA neurons ↑ (acute) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grinchii, D.; Dremencov, E. Mechanism of Action of Atypical Antipsychotic Drugs in Mood Disorders. Int. J. Mol. Sci. 2020, 21, 9532. https://doi.org/10.3390/ijms21249532
Grinchii D, Dremencov E. Mechanism of Action of Atypical Antipsychotic Drugs in Mood Disorders. International Journal of Molecular Sciences. 2020; 21(24):9532. https://doi.org/10.3390/ijms21249532
Chicago/Turabian StyleGrinchii, Daniil, and Eliyahu Dremencov. 2020. "Mechanism of Action of Atypical Antipsychotic Drugs in Mood Disorders" International Journal of Molecular Sciences 21, no. 24: 9532. https://doi.org/10.3390/ijms21249532
APA StyleGrinchii, D., & Dremencov, E. (2020). Mechanism of Action of Atypical Antipsychotic Drugs in Mood Disorders. International Journal of Molecular Sciences, 21(24), 9532. https://doi.org/10.3390/ijms21249532