Inhibition of BRD4 Reduces Neutrophil Activation and Adhesion to the Vascular Endothelium Following Ischemia Reperfusion Injury

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
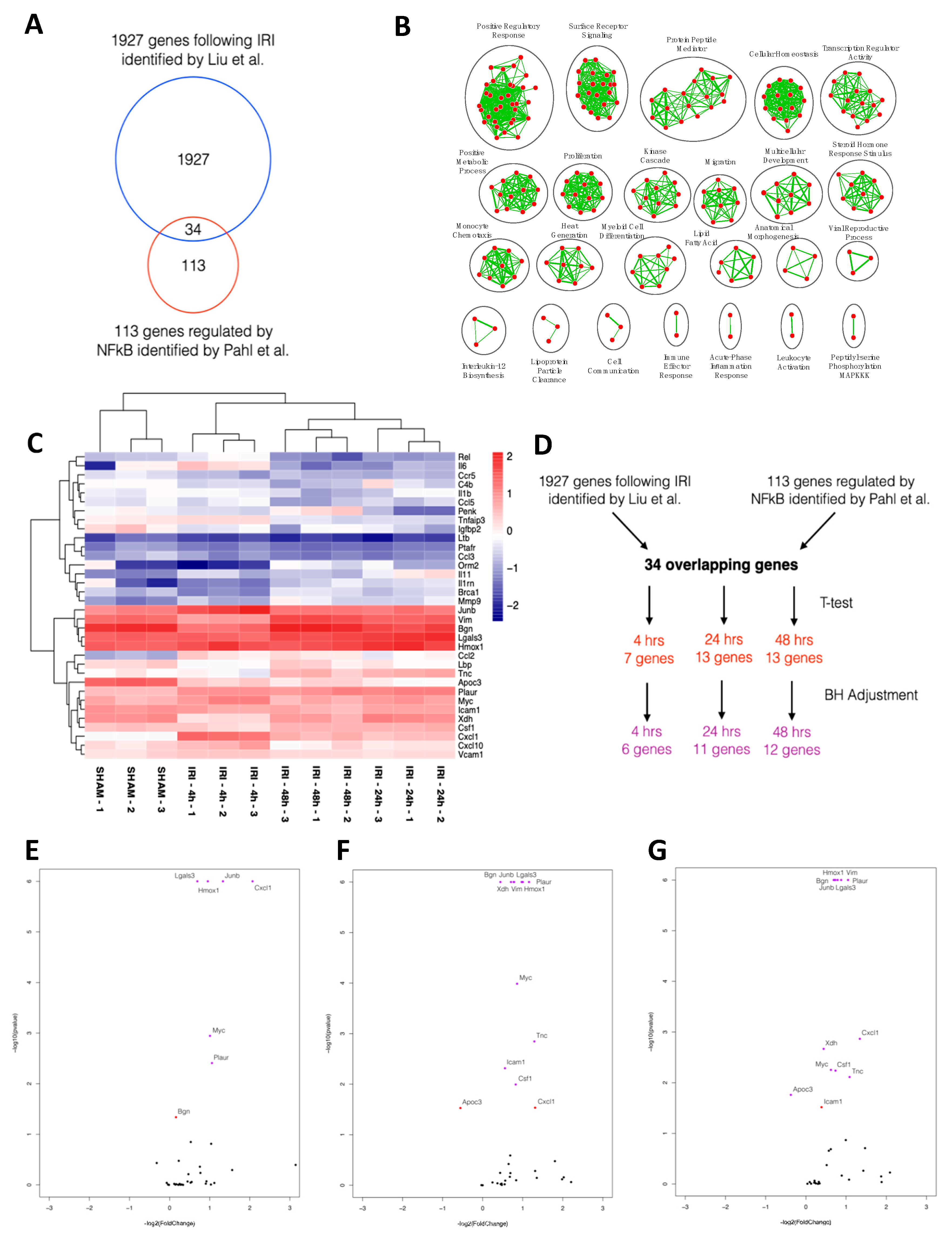
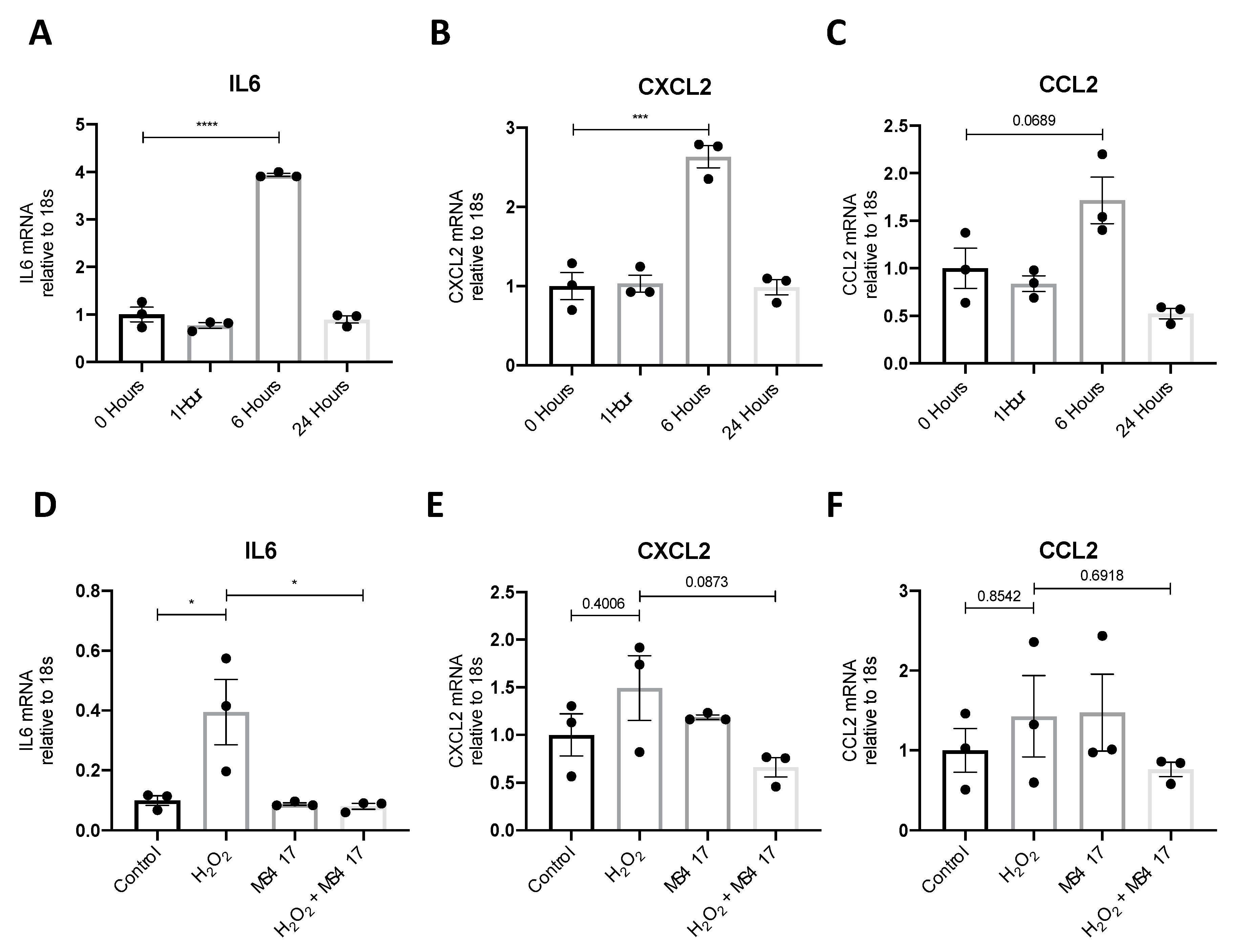
2.1. NFκB-mediated Gene Expression Characterizes Early IRI
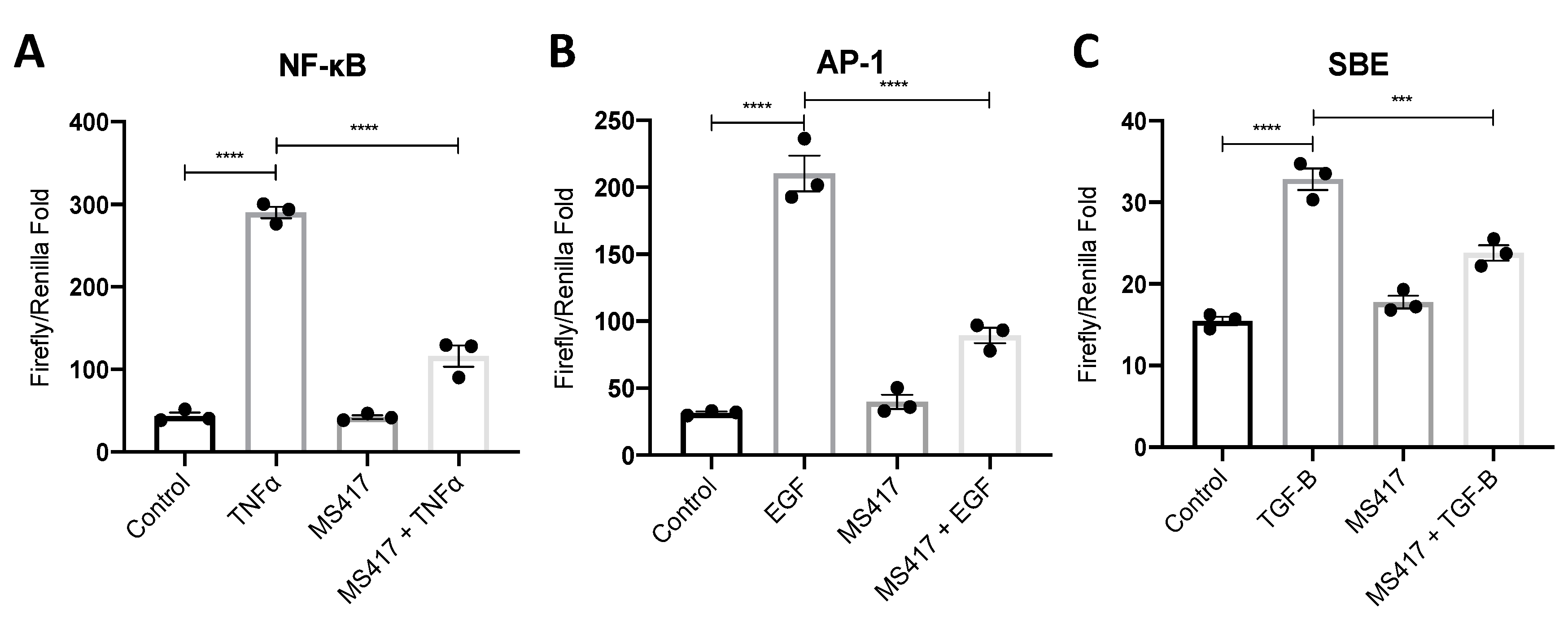
2.2. Inhibiting NFκB-Mediated Gene Expression, In Vitro
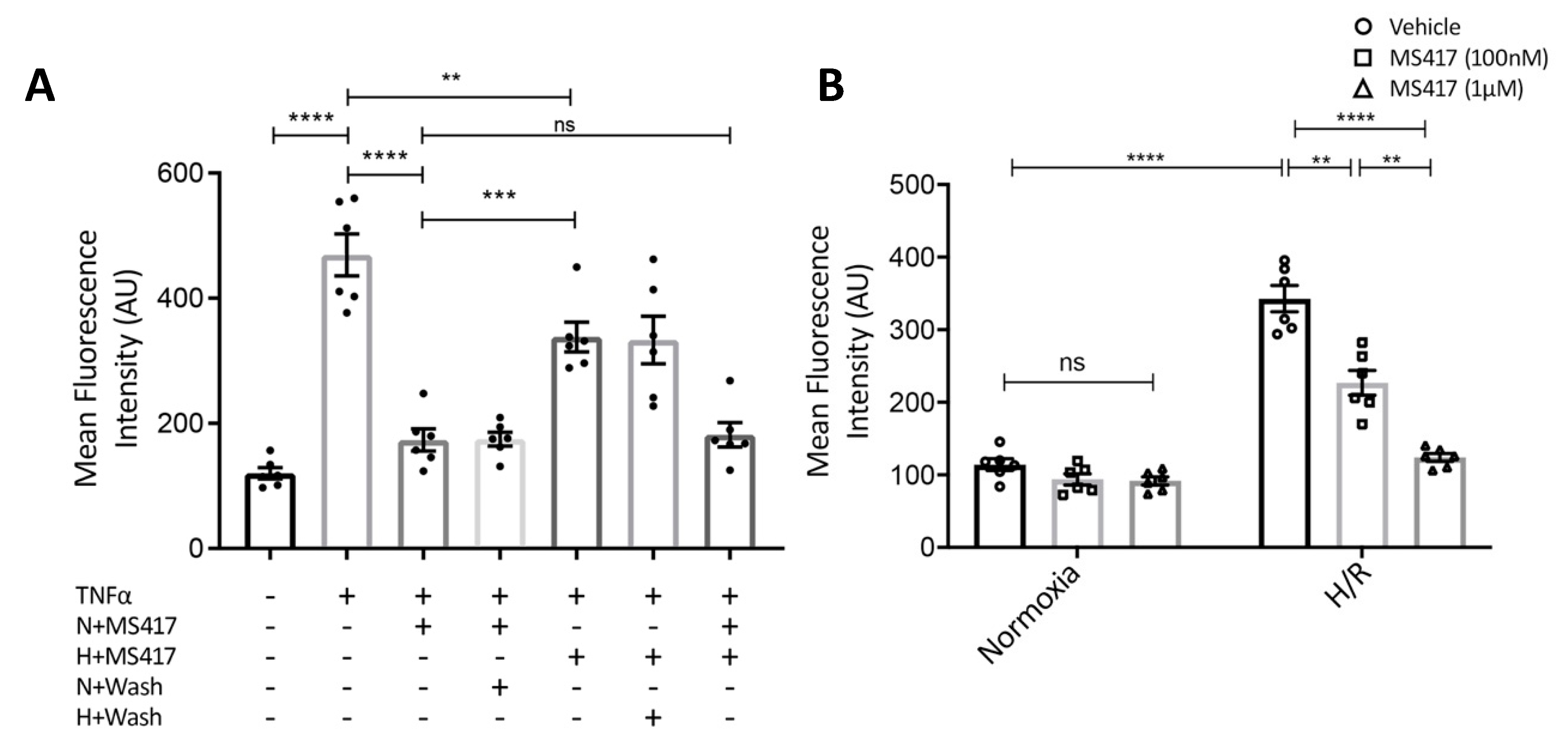
2.3. BRD4 Inhibition Blocks Neutrophil Adhesion to the Activated Endothelial Cells In Vitro
2.4. BRD4 Inhibition Attenuates IL6 Gene Expression Following H2O2-induced Oxidative Stress
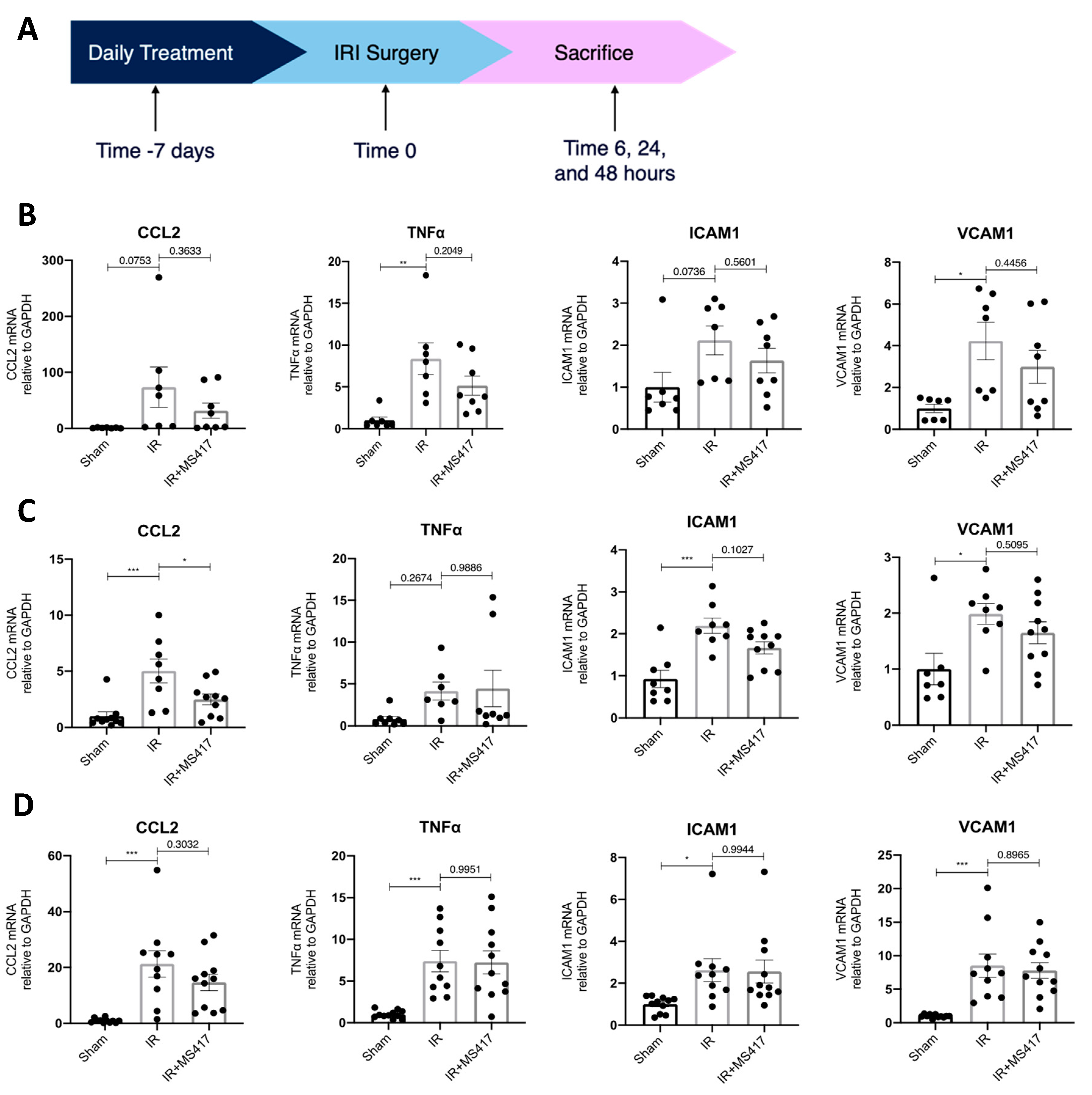
2.5. BRD4 Inhibition Attenuates In Vivo CCL2 Gene Expression Following IRI
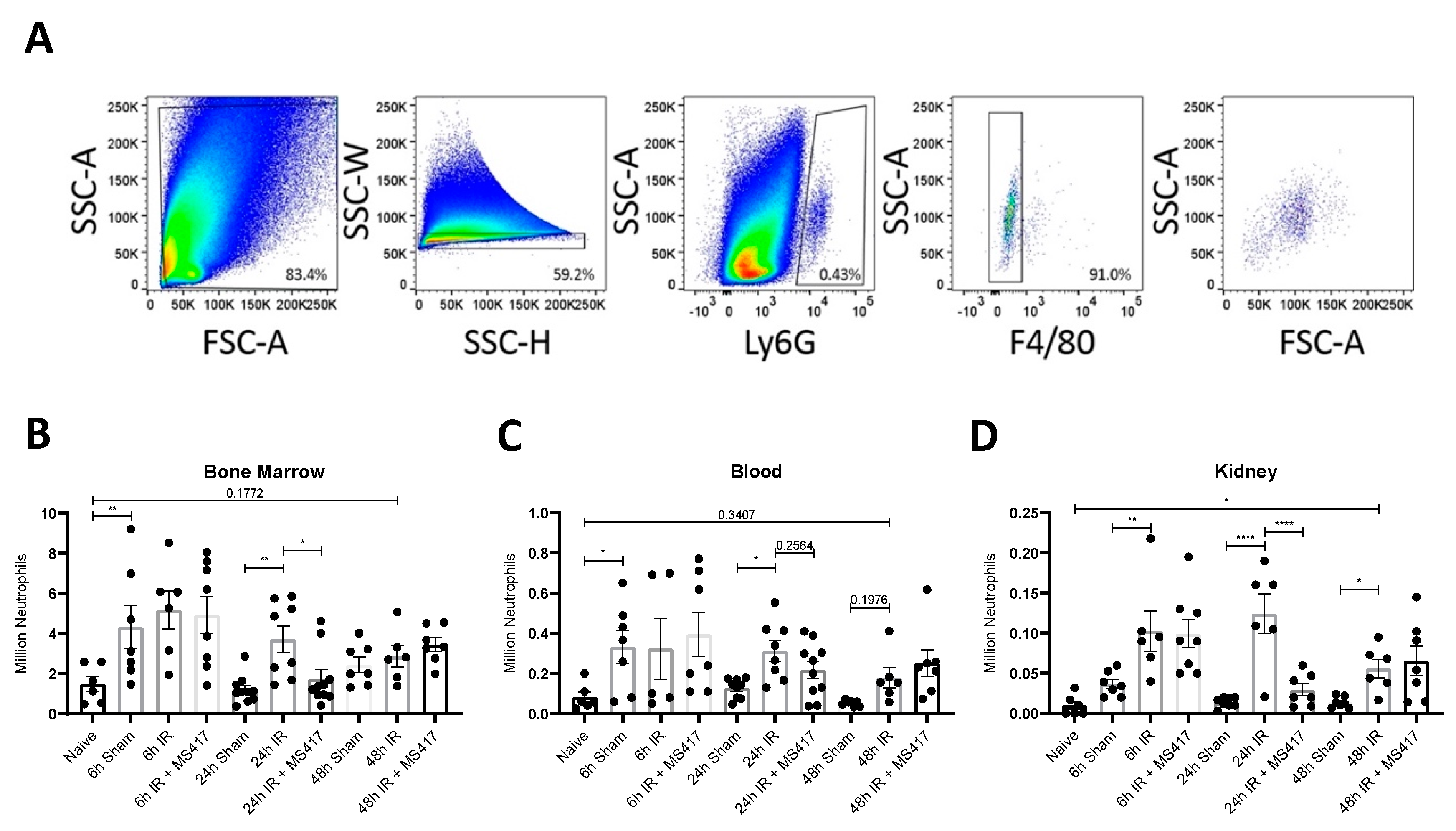
2.6. BRD4 Inhibition Reduces Absolute Neutrophil Counts Following IRI
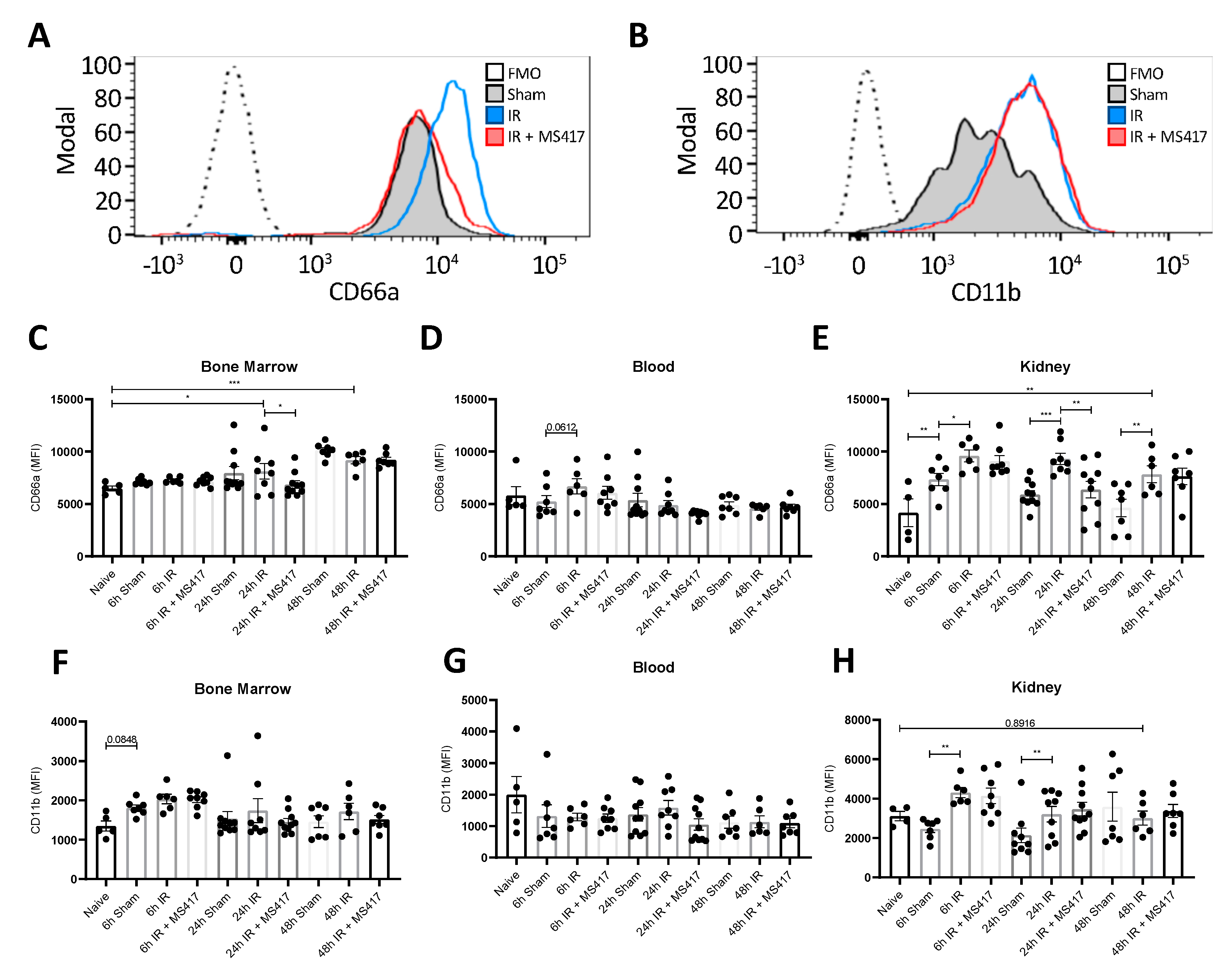
2.7. BRD4 Inhibition Reduces Neutrophil Up-regulation of CD66a Following IRI
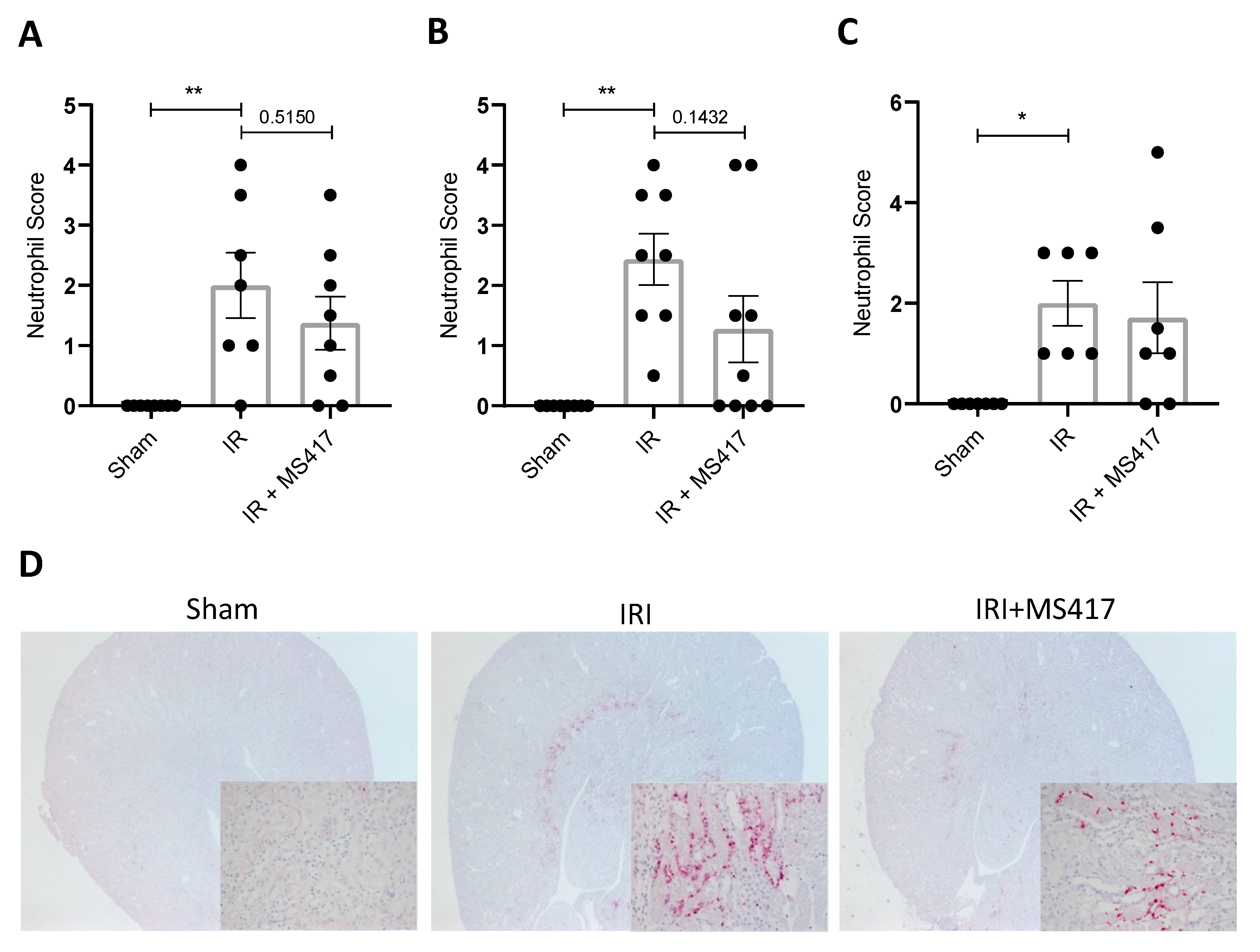
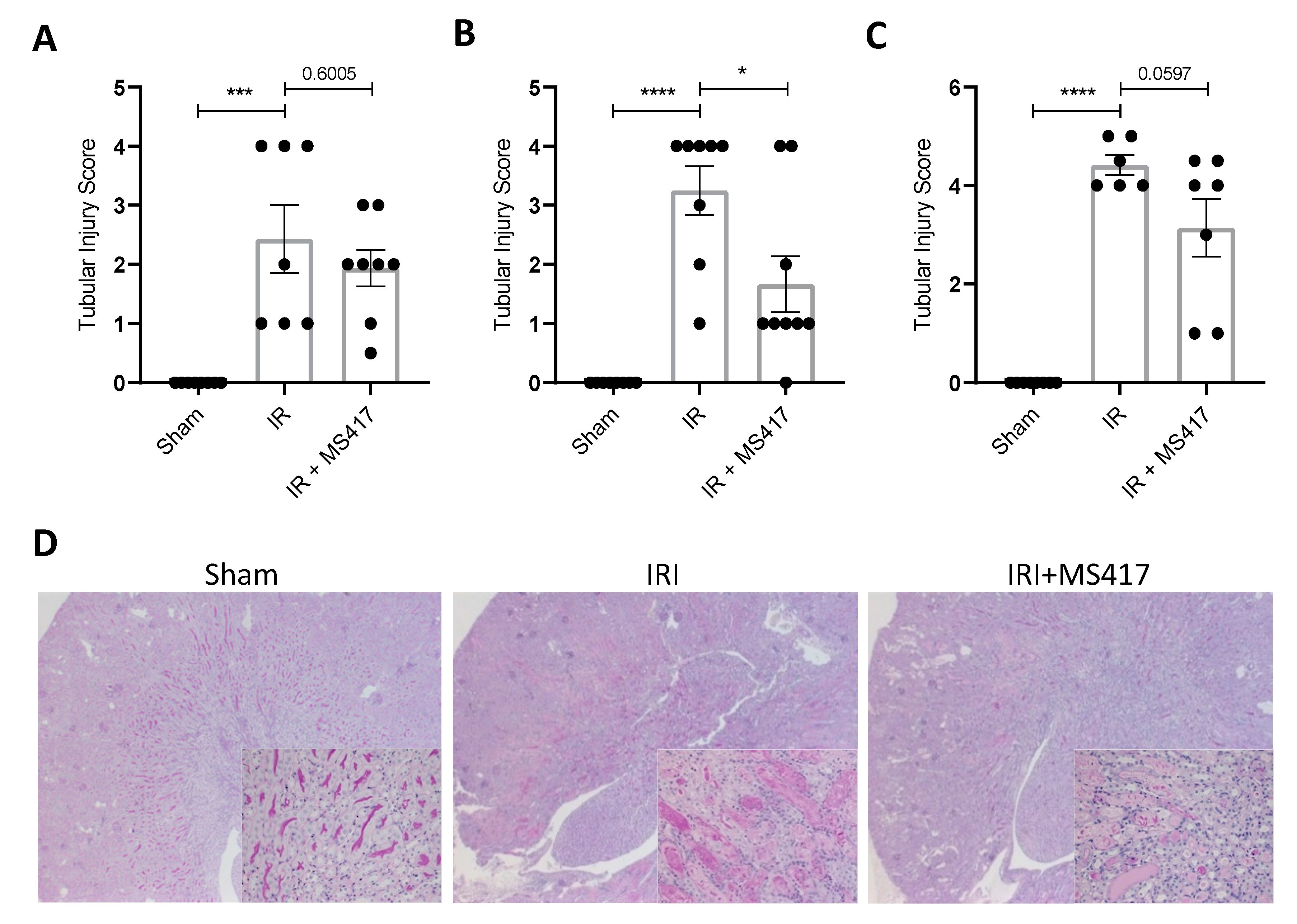
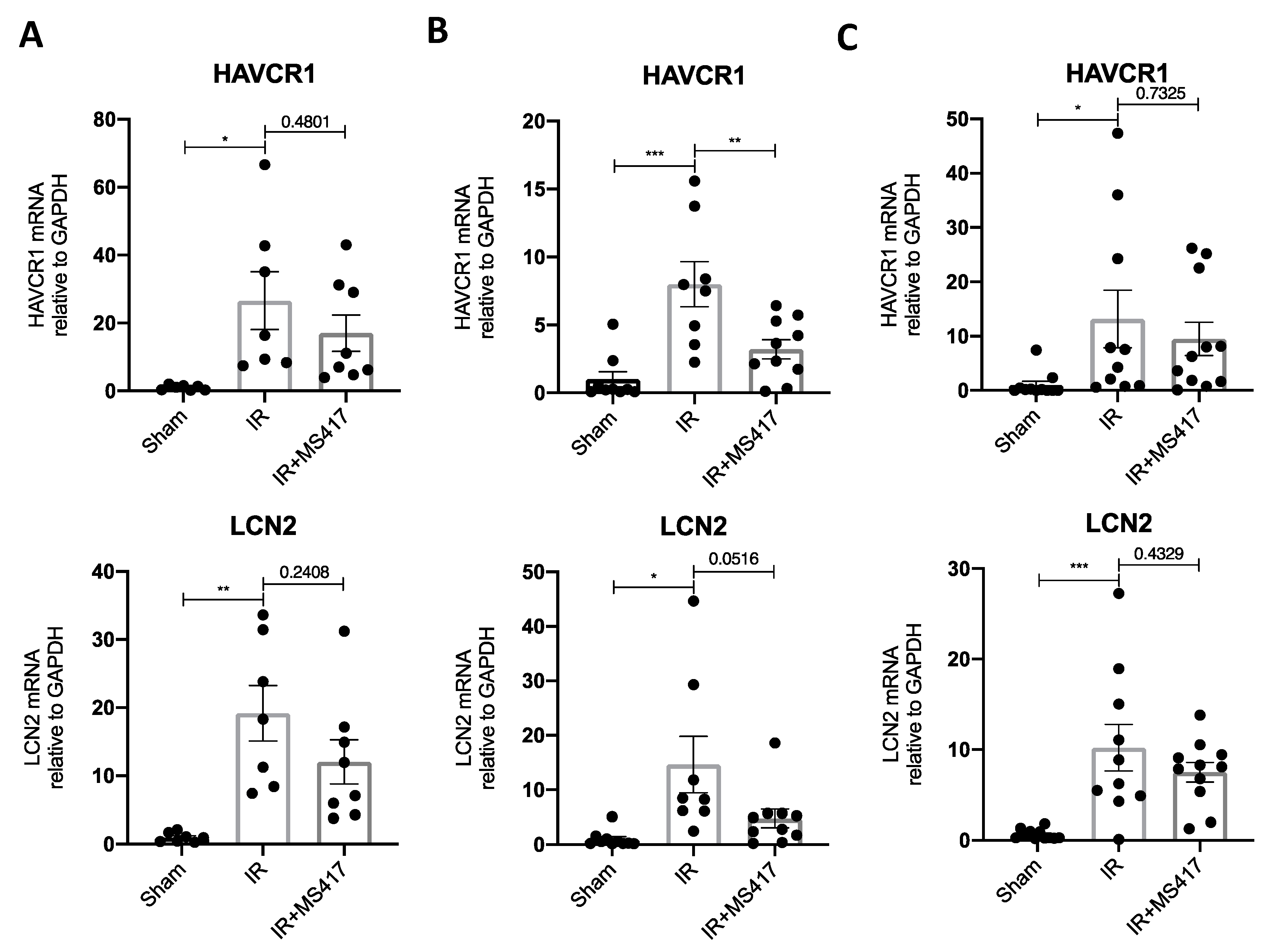
2.8. BRD4 Inhibition Attenuates Tubular Injury Following IRI
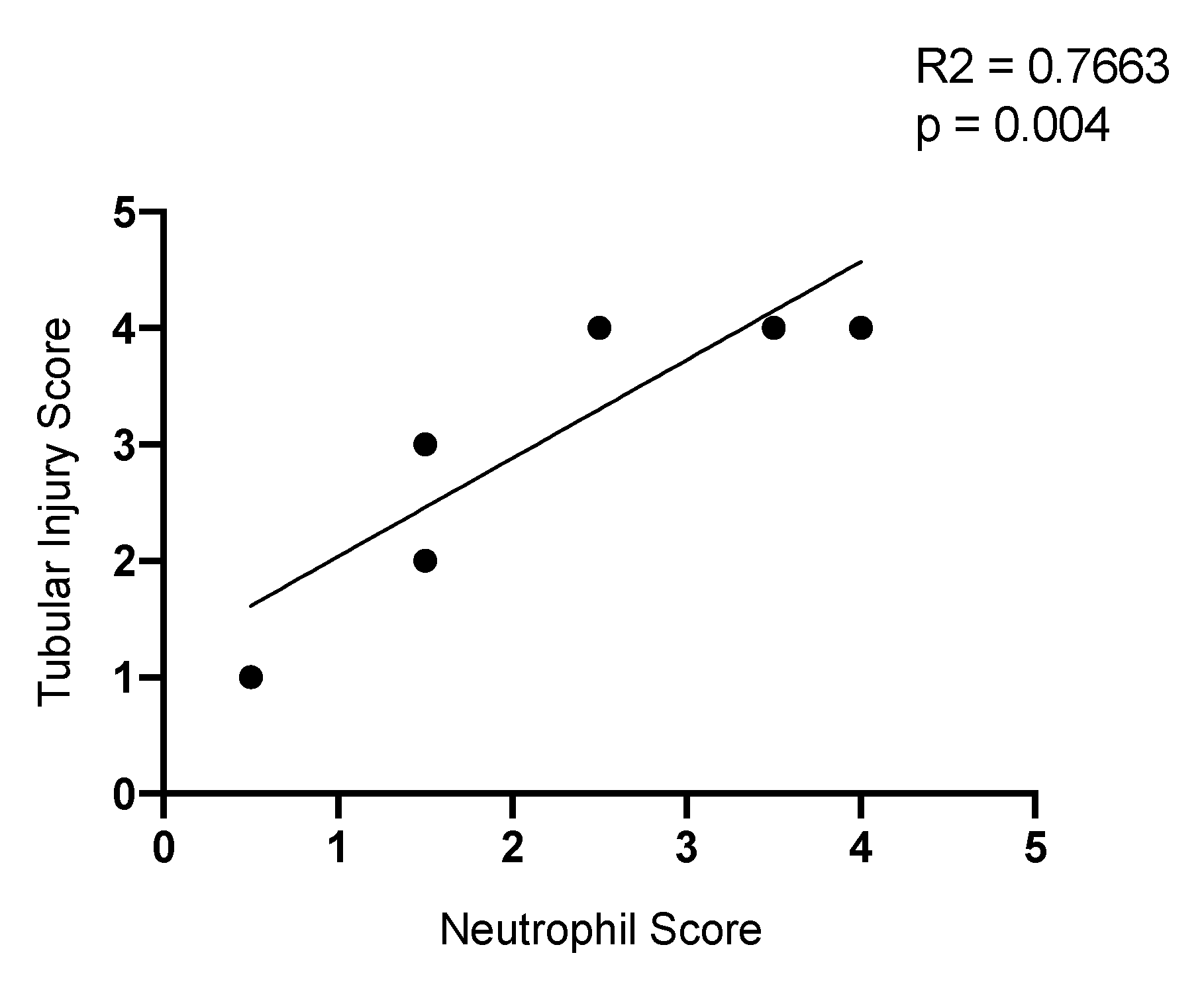
2.9. Neutrophil Infiltration Correlates with Tubule Injury
3. Discussion
4. Materials and Methods
4.1. In Silico Analysis
4.2. Cell Culture
4.3. Luciferase Activity Assay
4.4. H2O2 Time Course
4.5. Isolation of Primary Human Neutrophils
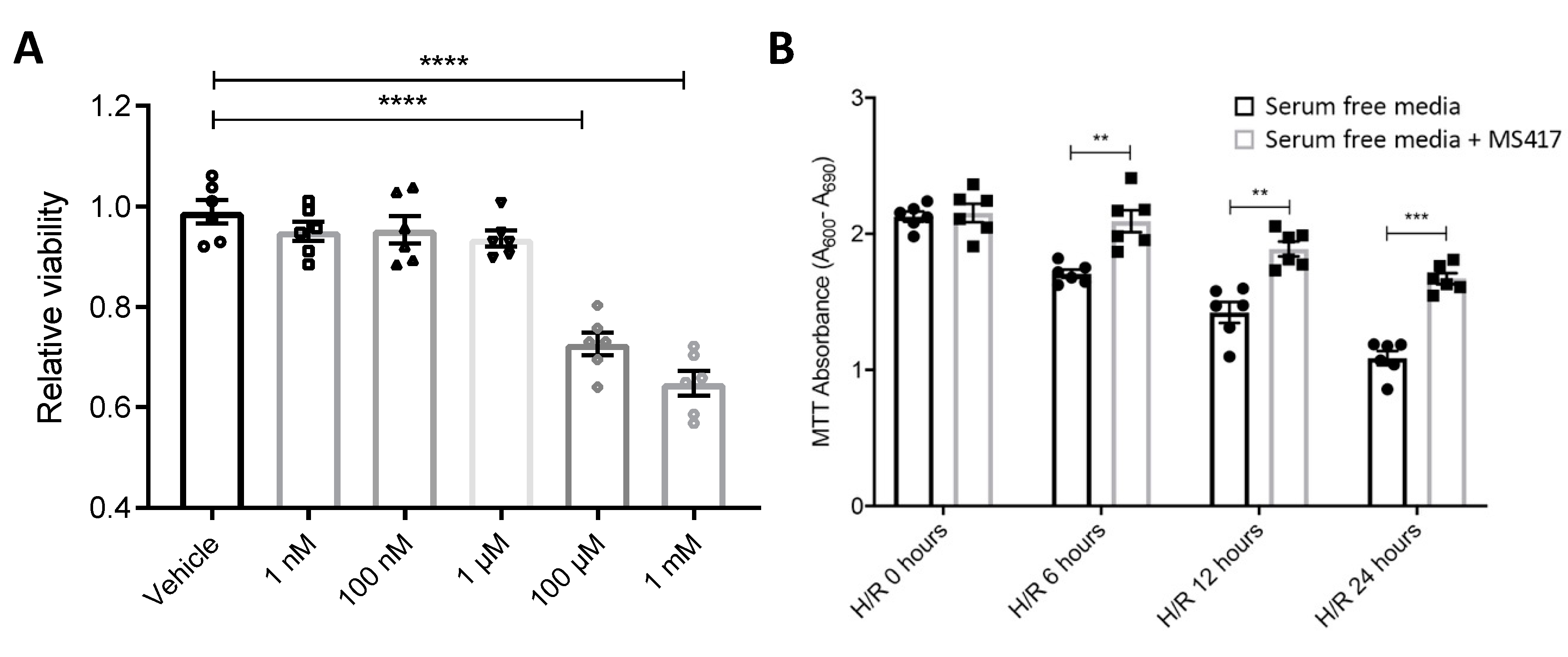
4.6. Hypoxia/re-oxygenation of Endothelial Cells
4.7. Neutrophil-endothelial Adhesion Assay
4.8. Cell Viability Assay
4.9. Quantitative PCR
4.10. Mouse Renal IRI
4.11. Immunohistochemistry
4.12. Flow Cytometry
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKI | Acute kidney injury |
| CKD | Chronic kidney disease |
| ESRD | End-stage renal disease |
| IRI | Ischemia reperfusion injury |
| ROS | Reactive oxygen species |
| NFκB | Nuclear factor-κB |
| IκB | Inhibitor of NFκB |
| IκBβ | IκB kinase β |
| BRD4 | Bromodomain-containing protein 4 |
| BET | Bromodomain and extra-terminal |
| TNF-α | Tumor necrosis factor α |
| HK-2 | Human-kidney 2 |
| AP-1 | Activator protein 1 |
| TGF-β | Transforming growth factor β |
| HUVEC | Human umbilical vein endothelial cells |
| PTEC | Primary human renal epithelial cells |
References
- Kate, R.J.; Perez, R.M.; Mazumdar, D.; Pasupathy, K.S.; Nilakantan, V. Prediction and detection models for acute kidney injury in hospitalized older adults. Bmc Med. Inform. Decis. Mak. 2016, 16, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lameire, N.H.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.; Kellum, J.A.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef]
- Sung, F.L.; Zhu, T.Y.; Au-Yeung, K.K.W.; Siow, Y.L.; Karmin, O. Enhanced MCP-1 expression during ischemia/reperfusion injury is mediated by oxidative stress and NF-κB. Kidney Int. 2002, 62, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.R.; Rabb, H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 2015, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.-M.; Babensee, J.E.; Simon, S.I.; Lu, H.; Perrard, J.L.; Bullard, D.C.; Dai, X.Y.; Bromley, S.K.; Dustin, M.L.; Entman, M.L. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 1999, 163, 5029–5038. [Google Scholar] [PubMed]
- Hidalgo, A.; Peired, A.J.; Wild, M.K.; Vestweber, D.; Frenette, P.S. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity 2007, 26, 477–489. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, T.H.C.; Marques, P.E.; Proost, P.; Teixeira, M.M.M. Neutrophils: A cornerstone of liver ischemia and reperfusion injury. Lab. Investig. 2018, 98, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Duilio, C.; Ambrosio, G.; Kuppusamy, P.; DiPaula, A.; Becker, L.C.; Zweier, J.L. Neutrophils are primary source of O2radicals during reperfusion after prolonged myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2649–H2657. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.R.; Ko, G.J.; Wasowska, B.A.; Rabb, H. The interaction between ischemia–reperfusion and immune responses in the kidney. J. Mol. Med. 2009, 87, 859–864. [Google Scholar] [CrossRef]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in renal diseases: New and old players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkB kinase complex (IKK) contains two kinase subunits, IKKa and IKKb, necessary for IkB phosphorylation and NF-kB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Liu, R.; Zhong, Y.; Plotnikov, A.N.; Zhang, W.; Zeng, L.; Rusinova, E.; Gerona-Nevarro, G.; Moshkina, N.; Joshua, J. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J. Biol. Chem. 2012, 287, 28840–28851. [Google Scholar] [CrossRef]
- Liu, R.; Zhong, Y.; Li, X.; Chen, H.; Jim, B.; Zhou, M.-M.; Chuang, P.Y.; He, J.C. Role of transcription factor acetylation in diabetic kidney disease. Diabetes 2014, 63, 2440–2453. [Google Scholar] [CrossRef] [Green Version]
- Zwacka, R.M.; Zhou, W.; Zhang, Y.; Darby, C.J.; Dudus, L.; Halldorson, J.; Oberley, L.; Engelhardt, J.F. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-κB activation. Nat. Med. 1998, 4, 698–704. [Google Scholar] [CrossRef]
- Liu, J.; Kumar, S.; Dolzhenko, E.; Alvarado, G.F.; Guo, J.; Lu, C.; Chen, Y.; Li, M.; Dessing, M.C.; Parvez, R.K. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Schütze, S.; Wiegmann, K.; Machleidt, T.; Krönke, M. TNF-induced activation of NF-κB. Immunobiology 1995, 193, 193–203. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Bernard, D.B.; Donohoe, J.F.; Levinsky, N.G. Ischemic damage and repair in the rat proximal tubule: Differences among the S1, S2, and S3 segments. Kidney Int. 1978, 14, 31–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcuratolo, M.; Fertey, J.; Schneider, M.; Schuetz, J.; Leiprecht, N.; Hudjetz, B.; Brodbeck, S.; Corall, S.; Dreer, M.; Schwab, R.M. Papillomavirus-associated tumor formation critically depends on c-Fos expression induced by viral protein E2 and bromodomain protein Brd4. PLoS Pathog. 2016, 12, e1005366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Liu, L.; Yu, Y.; Zhang, R.; Li, Y.; Cao, W.; Xiao, Y.; Fang, G.; Li, Z.; Wang, X. Inhibition of BRD4 attenuates transverse aortic constriction-and TGF-β-induced endothelial-mesenchymal transition and cardiac fibrosis. J. Mol. Cell. Cardiol. 2019, 127, 83–96. [Google Scholar] [CrossRef]
- Zhou, B.; Mu, J.; Gong, Y.; Lu, C.; Zhao, Y.; He, T.; Qin, Z. Brd4 inhibition attenuates unilateral ureteral obstruction-induced fibrosis by blocking TGF-β-mediated Nox4 expression. Redox Biol. 2017, 11, 390–402. [Google Scholar] [CrossRef]
- Mehta, J.L.; Yang, B.C.; Strates, B.S.; Mehta, P. Role of TGF-β1 in platelet-mediated cardioprotection during ischemia-reperfusion in isolated rat hearts. Growth Factors 1999, 16, 179–190. [Google Scholar] [CrossRef]
- Fan, H.; Sun, B.; Gu, Q.; Lafond-Walker, A.; Cao, S.; Becker, L.C. Oxygen radicals trigger activation of NF-κB and AP-1 and upregulation of ICAM-1 in reperfused canine heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1778–H1786. [Google Scholar] [CrossRef] [Green Version]
- Humes, H.D.; Cieslinski, D.A.; Coimbra, T.M.; Messana, J.M.; Galvao, C. Epidermal growth factor enhances renal tubule cell regeneration and repair and accelerates the recovery of renal function in postischemic acute renal failure. J. Clin. Investig. 1989, 84, 1757–1761. [Google Scholar] [CrossRef]
- Wu, W.; Liu, C.; Farrar, C.A.; Ma, L.; Dong, X.; Sacks, S.H.; Li, K.; Zhou, W. Collectin-11 Promotes the Development of Renal Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2017, 29, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Diacovo, T.G.; Roth, S.J.; Buccola, J.M.; Bainton, D.F.; Springer, T.A. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood 1996, 88, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, S.; Yuen, D.A.; Bajwa, A.; Huang, Y.-W.; Sokollik, C.; Huang, L.; Lam, G.Y.; Tole, S.; Liu, G.-Y.; Pan, J. Slit2 prevents neutrophil recruitment and renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2013, 8, 1274–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnahoo, K.K.; Shames, B.D.; Harken, A.H.; Meldrum, D.R. The role of tumor necrosis factor in renal ischemia-reperfusion injury. J. Urol. 1999, 162, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Flecha, B.; Cutrin, J.C.; Boveris, A. Time course and mechanism of oxidative stress and tissue damage in rat liver subjected to in vivo ischemia-reperfusion. J. Clin. Investig. 1993, 91, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, L.; Weng, X.; Chen, H.; Du, Y.; Diao, C.; Chen, Z.; Liu, X. Inhibition of Brd4 alleviates renal ischemia/reperfusion injury-induced apoptosis and endoplasmic reticulum stress by blocking FoxO4-mediated oxidative stress. Redox Biol. 2019, 24, 101195. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; van der Schoot, C.E.; Hoogerwerf, M.; Roos, D. Cross-linking of the carcinoembryonic antigen-like glycoproteins CD66 and CD67 induces neutrophil aggregation. J. Immunol. 1993, 151, 4934–4940. [Google Scholar]
- Stocks, S.C.; Ruchaud-Sparagano, M.h.; Kerr, M.A.; Grunert, F.; Haslett, C.; Dransfield, I. CD66: Role in the regulation of neutrophil effector function. Eur. J. Immunol. 1996, 26, 2924–2932. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Hoogerwerf, M.; Van der Laan, L.J.; Nagel, G.; van der Schoot, C.E.; Grunert, F.; Roos, D. CD66 nonspecific cross-reacting antigens are involved in neutrophil adherence to cytokine-activated endothelial cells. J. Cell Biol. 1992, 118, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Simon, S.I.; Burns, A.R.; Taylor, A.D.; Gopalan, P.K.; Lynam, E.B.; Sklar, L.A.; Smith, C.W. L-selectin (CD62L) cross-linking signals neutrophil adhesive functions via the Mac-1 (CD11b/CD18) beta 2-integrin. J. Immunol. 1995, 155, 1502–1514. [Google Scholar]
- Christmas, S.E.; De La Mata Espinosa, C.T.; Halliday, D.; Buxton, C.A.; Cummerson, J.A.; Johnson, P.M. Levels of expression of complement regulatory proteins CD46, CD55 and CD59 on resting and activated human peripheral blood leucocytes. Immunology 2006, 119, 522–528. [Google Scholar] [CrossRef]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity 2018, 48, 364–379. [Google Scholar] [CrossRef] [Green Version]
- Bongoni, A.K.; Lu, B.; Salvaris, E.J.; Roberts, V.; Fang, D.; McRae, J.L.; Fisicaro, N.; Dwyer, K.M.; Cowan, P.J. Overexpression of human CD55 and CD59 or treatment with human CD55 protects against renal ischemia-reperfusion injury in mice. J. Immunol. 2017, 198, 4837–4845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, J.; Ma, Q.; Kelly, C.; Mitsnefes, M.; Mori, K.; Barasch, J.; Devarajan, P. Kidney NGAL is a novel early marker of acute injury following transplantation. Pediatric Nephrol. 2006, 21, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, V.S.; Ozer, J.S.; Dieterle, F.; Collings, F.B.; Ramirez, V.; Troth, S.; Muniappa, N.; Thudium, D.; Gerhold, D.; Holder, D.J. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat. Biotechnol. 2010, 28, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovcanin, B.; Medic, B.; Kocic, G.; Cebovic, T.; Ristic, M.; Prostran, M. Molecular dissection of renal ischemia-reperfusion: Oxidative stress and cellular events. Curr. Med. Chem. 2016, 23, 1965–1980. [Google Scholar] [CrossRef]
- Cao, C.C.; Ding, X.Q.; Ou, Z.L.; Liu, C.F.; Li, P.; Wang, L.; Zhu, C.F. In vivo transfection of NF-κB decoy oligodeoxynucleotides attenuate renal ischemia/reperfusion injury in rats. Kidney Int. 2004, 65, 834–845. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Nickkholgh, A.; Yi, X.; Bruns, H.; Gross, M.L.; Hoffmann, K.; Mohr, E.; Zorn, M.; Büchler, M.W.; Schemmer, P. Melatonin protects kidney grafts from ischemia/reperfusion injury through inhibition of NF-kB and apoptosis after experimental kidney transplantation. J. Pineal Res. 2009, 46, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Markó, L.; Vigolo, E.; Hinze, C.; Park, J.-K.; Roël, G.; Balogh, A.; Choi, M.; Wübken, A.; Cording, J.; Blasig, I.E. Tubular epithelial NF-κB activity regulates ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 2658–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Chen, G.; Zheng, X.; Sun, H.; Zhang, X.; Zhang, Z.-X.; Xiang, Y.; Ichim, T.E.; Garcia, B.; Luke, P. Small interfering RNA targeting RelB protects against renal ischemia-reperfusion injury. Transplantation 2009, 87, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Donnahoo, K.K.; Meldrum, D.R.; Shenkar, R.; Chung, C.-S.; Abraham, E.; Harken, A.H. Early renal ischemia, with or without reperfusion, activates NFκB and increases TNF-α bioactivity in the kidney. J. Urol. 2000, 163, 1328–1332. [Google Scholar] [CrossRef]
- Kim, J.W.; Jin, Y.C.; Kim, Y.M.; Rhie, S.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Ha, Y.L.; Chang, K.C. Daidzein administration in vivo reduces myocardial injury in a rat ischemia/reperfusion model by inhibiting NF-kB activation. Life Sci. 2009, 84, 227–234. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhou, M.-M. Bromodomain: An acetyl-lysine binding domain. Febs Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.-F. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 2014, 33, 2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.; McMahon, S.; Anand, P.; Shah, H.; Thomas, S.; Salunga, H.T.; Huang, Y.; Zhang, R.; Sahadevan, A.; Lemieux, M.E. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017, 9, eaah5084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Zeng, S.; Zou, Y.; Shi, M.; Qiu, Q.; Xiao, Y.; Chen, G.; Yang, X.; Liang, L.; Xu, H. The suppression of bromodomain and extra-terminal domain inhibits vascular inflammation by blocking NF-κB and MAPK activation. Br. J. Pharmacol. 2017, 174, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Liu, J.; Yuan, Y.; Zhang, X.; Dong, Z. Protective effect of the BET protein inhibitor JQ1 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2018, 315, F469–F478. [Google Scholar] [CrossRef] [PubMed]
- DeMars, K.M.; Yang, C.; Candelario-Jalil, E. Neuroprotective effects of targeting BET proteins for degradation with dBET1 in aged mice subjected to ischemic stroke. Neurochem. Int. 2019, 127, 94–102. [Google Scholar] [CrossRef]
- Ranghino, A.; Bruno, S.; Bussolati, B.; Moggio, A.; Dimuccio, V.; Tapparo, M.; Biancone, L.; Gontero, P.; Frea, B.; Camussi, G. The effects of glomerular and tubular renal progenitors and derived extracellular vesicles on recovery from acute kidney injury. Stem Cell Res. Ther. 2017, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Wernig, G.; Chen, S.-Y.; Cui, L.; Van Neste, C.; Tsai, J.M.; Kambham, N.; Vogel, H.; Natkunam, Y.; Gilliland, D.G.; Nolan, G. Unifying mechanism for different fibrotic diseases. Proc. Natl. Acad. Sci. USA 2017, 114, 4757–4762. [Google Scholar] [CrossRef] [Green Version]
- James, M.T.; Bhatt, M.; Pannu, N.; Tonelli, M. Long-term outcomes of acute kidney injury and strategies for improved care. Nat. Rev. Nephrol. 2020, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J.; Williams, W.W.; Colvin, R.B.; Meehan, S.M.; Springer, T.A.; Gutiérrez-Ramos, J.-C.; Bonventre, J.V. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J. Clin. Investig. 1996, 97, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.M.; Schneider, J.E.; Chapman, S.J.; Jefferson, A.; Digby, J.E.; Mankia, K.; Chen, Y.; McAteer, M.A.; Wood, K.J.; Choudhury, R.P. In vivo quantification of VCAM-1 expression in renal ischemia reperfusion injury using non-invasive magnetic resonance molecular imaging. PLoS ONE 2010, 5, e12800. [Google Scholar] [CrossRef] [Green Version]
- Spertini, O.; Luscinskas, F.W.; Kansas, G.S.; Munro, J.M.; Griffin, J.D.; Gimbrone, M.A.; Tedder, T.F. Leukocyte adhesion molecule-1 (LAM-1, L-selectin) interacts with an inducible endothelial cell ligand to support leukocyte adhesion. J. Immunol. 1991, 147, 2565–2573. [Google Scholar]
- Read, M.A.; Neish, A.S.; Luscinskas, F.W.; Palombella, V.J.; Maniatis, T.; Collins, T. The proteasome pathway is required for cytokine-induced endothelial-leukocyte adhesion molecule expression. Immunity 1995, 2, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Anto-Michel, N.; Blankenbach, H.; Wiedemann, A.; Buscher, K.; Hohmann, J.D.; Lim, B.; Bäuml, M.; Marki, A.; Mauler, M. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Dunne, J.L.; Ballantyne, C.M.; Beaudet, A.L.; Ley, K. Control of leukocyte rolling velocity in TNF-α–induced inflammation by LFA-1 and Mac-1. Blood J. Am. Soc. Hematol. 2002, 99, 336–341. [Google Scholar] [CrossRef]
- Dunne, J.L.; Collins, R.G.; Beaudet, A.L.; Ballantyne, C.M.; Ley, K. Mac-1, but not LFA-1, uses intercellular adhesion molecule-1 to mediate slow leukocyte rolling in TNF-α-induced inflammation. J. Immunol. 2003, 171, 6105–6111. [Google Scholar] [CrossRef] [Green Version]
- Furze, R.C.; Rankin, S.M. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008, 125, 281–288. [Google Scholar] [CrossRef]
- Jagels, M.A.; Chambers, J.D.; Arfors, K.E.; Hugli, T.E. C5a-and tumor necrosis factor-alpha-induced leukocytosis occurs independently of beta 2 integrins and L-selectin: Differential effects on neutrophil adhesion molecule expression in vivo. Blood 1995, 85, 2900–2909. [Google Scholar] [CrossRef] [Green Version]
- Terashima, T.; English, D.; Hogg, J.C.; vanEeden, S.F. Release of polymorphonuclear leukocytes from the bone marrow by interleukin-8. Blood J. Am. Soc. Hematol. 1998, 92, 1062–1069. [Google Scholar]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaar, A.L.; Sweeney, L.E.; MacDonald, A.J.; Alexis, N.E.; Chen, C.; Tal-Singer, R. SB-656933, a novel CXCR2 selective antagonist, inhibits ex vivo neutrophil activation and ozone-induced airway inflammation in humans. Br. J. Clin. Pharmacol. 2011, 72, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Winkler, J.W.; Colas, R.A.; Arnardottir, H.; Cheng, C.-Y.C.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem. Biol. 2013, 20, 188–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opfermann, P.; Derhaschnig, U.; Felli, A.; Wenisch, J.; Santer, D.; Zuckermann, A.; Dworschak, M.; Jilma, B.; Steinlechner, B. A pilot study on reparixin, a CXCR 1/2 antagonist, to assess safety and efficacy in attenuating ischaemia–reperfusion injury and inflammation after on-pump coronary artery bypass graft surgery. Clin. Exp. Immunol. 2015, 180, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Yang, J.; Zhao, Y.; Ivanciuc, T.; Sun, H.; Garofalo, R.P.; Brasier, A.R. BRD4 couples NF-κB/RelA with airway inflammation and the IRF-RIG-I amplification loop in respiratory syncytial virus infection. J. Virol. 2017, 91, e00007–e00017. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, M.P.; Stahl, G.L.; Longhurst, J.C. In vivo and in vitro assessment of porcine neutrophil activation responses to chemoattractants: Flow cytometric evidence for the selective absence of formyl peptide receptors. J. Leukoc. Biol. 1990, 47, 355–365. [Google Scholar] [CrossRef]
- Lehr, H.-A.; Krombach, F.; Münzing, S.; Bodlaj, R.; Glaubitt, S.I.; Seiffge, D.; Hübner, C.; Von Andrian, U.H.; Messmer, K. In vitro effects of oxidized low density lipoprotein on CD11b/CD18 and L-selectin presentation on neutrophils and monocytes with relevance for the in vivo situation. Am. J. Pathol. 1995, 146, 218–227. [Google Scholar]
- Videm, V.; Strand, E. Changes in neutrophil surface-receptor expression after stimulation with FMLP, endotoxin, interleukin-8 and activated complement compared to degranulation. Scand. J. Immunol. 2004, 59, 25–33. [Google Scholar] [CrossRef]
- Nicholson, G.C.; Tennant, R.C.; Carpenter, D.C.; Sarau, H.M.; Kon, O.M.; Barnes, P.J.; Salmon, M.; Vessey, R.S.; Tal-Singer, R.; Hansel, T.T. A novel flow cytometric assay of human whole blood neutrophil and monocyte CD11b levels: Upregulation by chemokines is related to receptor expression, comparison with neutrophil shape change, and effects of a chemokine receptor (CXCR2) antagonist. Pulm. Pharmacol. Ther. 2007, 20, 52–59. [Google Scholar] [CrossRef]
- Fine, N.; Barzilay, O.; Sun, C.; Wellappuli, N.; Tanwir, F.; Chadwick, J.W.; Oveisi, M.; Tasevski, N.; Prescott, D.; Gargan, M. Primed PMNs in healthy mouse and human circulation are first responders during acute inflammation. Blood Adv. 2019, 3, 1622–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakschevitz, F.S.; Hassanpour, S.; Rubin, A.; Fine, N.; Sun, C.; Glogauer, M. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp. Cell Res. 2016, 342, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Fine, N.; Hassanpour, S.; Borenstein, A.; Sima, C.; Oveisi, M.; Scholey, J.; Cherney, D.; Glogauer, M. Distinct oral neutrophil subsets define health and periodontal disease states. J. Dent. Res. 2016, 95, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Mathias, J.R.; Perrin, B.J.; Liu, T.X.; Kanki, J.; Look, A.T.; Huttenlocher, A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J. Leukoc. Biol. 2006, 80, 1281–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, S.; Leitch, A.E.; Duffin, R.; Haslett, C.; Rossi, A.G. Neutrophil apoptosis: Relevance to the innate immune response and inflammatory disease. J. Innate Immun. 2010, 2, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Woodfin, A.; Voisin, M.-B.; Beyrau, M.; Colom, B.; Caille, D.; Diapouli, F.-M.; Nash, G.B.; Chavakis, T.; Albelda, S.M.; Rainger, G.E. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 2011, 12, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Colom, B.; Bodkin, J.V.; Beyrau, M.; Woodfin, A.; Ody, C.; Rourke, C.; Chavakis, T.; Brohi, K.; Imhof, B.A.; Nourshargh, S. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity 2015, 42, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Muenzner, P.; Naumann, M.; Meyer, T.F.; Gray-Owen, S.D. Pathogenic Neisseria trigger expression of their carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1; previously CD66a) receptor on primary endothelial cells by activating the immediate early response transcription factor, nuclear factor-κB. J. Biol. Chem. 2001, 276, 24331–24340. [Google Scholar]
- Oguiza, A.; Recio, C.; Lazaro, I.; Mallavia, B.; Blanco, J.; Egido, J.; Gomez-Guerrero, C. Peptide-based inhibition of IκB kinase/nuclear factor-κB pathway protects against diabetes-associated nephropathy and atherosclerosis in a mouse model of type 1 diabetes. Diabetologia 2015, 58, 1656–1667. [Google Scholar] [CrossRef] [Green Version]
- Ozkok, A.; Ravichandran, K.; Wang, Q.; Ljubanovic, D.; Edelstein, C.L. NF-κB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol. Lett. 2016, 240, 105–113. [Google Scholar] [CrossRef]
- Yu, X.; Meng, X.; Xu, M.; Zhang, X.; Zhang, Y.; Ding, G.; Huang, S.; Zhang, A.; Jia, Z. Celastrol ameliorates cisplatin nephrotoxicity by inhibiting NF-κB and improving mitochondrial function. EBioMedicine 2018, 36, 266–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadatomo, A.; Inoue, Y.; Ito, H.; Karasawa, T.; Kimura, H.; Watanabe, S.; Mizushina, Y.; Nakamura, J.; Kamata, R.; Kasahara, T. Interaction of neutrophils with macrophages promotes IL-1β maturation and contributes to hepatic ischemia–reperfusion injury. J. Immunol. 2017, 199, 3306–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Sharkey, D.; Cantley, L.G. Tubular GM-CSF promotes late MCP-1/CCR2-mediated fibrosis and inflammation after ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2019, 30, 1825–1840. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.P.; Garnier, J.-M.; Huang, D.C.S.; Burns, C.J. Evaluation of functional groups as acetyl-lysine mimetics for BET bromodomain inhibition. MedChemComm 2014, 5, 1834–1842. [Google Scholar] [CrossRef]
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin nephrotoxicity: A review. Am. J. Med Sci. 2007, 334, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelmayer, W.C.; Waikar, S.S.; Mogun, H.; Solomon, D.H. Nonselective and cyclooxygenase-2-selective NSAIDs and acute kidney injury. Am. J. Med. 2008, 121, 1092–1098. [Google Scholar] [CrossRef]
- Khalili, H.; Bairami, S.; Kargar, M. Antibiotics induced acute kidney injury: Incidence, risk factors, onset time and outcome. Acta Med. Iran. 2013, 51, 871–878. [Google Scholar]
- Kpemissi, M.; Eklu-Gadegbeku, K.; Veerapur, V.P.; Negru, M.; Taulescu, M.; Chandramohan, V.; Hiriyan, J.; Banakar, S.M.; Thimmaiah, N.V.; Suhas, D.S. Nephroprotective activity of Combretum micranthum G. Don in cisplatin induced nephrotoxicity in rats: In-vitro, in-vivo and in-silico experiments. Biomed. Pharmacother. 2019, 116, 108961. [Google Scholar] [CrossRef]
- Ridzuan, N.R.A.; Rashid, N.A.; Othman, F.; Budin, S.B.; Hussan, F.; Teoh, S.L. Protective role of natural products in cisplatin-induced nephrotoxicity. Mini Rev. Med. Chem. 2019, 19, 1134–1143. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, W.; Sun, X.; Wang, Y.; Zhou, M. Kaempferol ameliorates Cisplatin induced nephrotoxicity by modulating oxidative stress, inflammation and apoptosis via ERK and NF-κB pathways. Amb. Express 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Garg, A.X.; Devereaux, P.J.; Hill, A.; Sood, M.; Aggarwal, B.; Dubois, L.; Hiremath, S.; Guzman, R.; Iyer, V.; James, M. Oral curcumin in elective abdominal aortic aneurysm repair: A multicentre randomized controlled trial. CMAJ 2018, 190, E1273–E1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van, J.A.D.; Clotet-Freixas, S.; Zhou, J.; Batruch, I.; Sun, C.; Glogauer, M.; Rampoldi, L.; Elia, Y.; Mahmud, F.H.; Sochett, E. Peptidomic analysis of urine from youths with early type 1 diabetes reveals novel bioactivity of uromodulin peptides in vitro. Mol. Cell. Proteom. 2020, 19, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.R.; Konvalinka, A.; Song, X.; Zhou, X.; John, R.; Pei, Y.; Scholey, J.W. Connectivity mapping of a chronic kidney disease progression signature identified lysine deacetylases as novel therapeutic targets. Kidney Int. 2020, 1, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Konvalinka, A.; Zhou, J.; Dimitromanolakis, A.; Drabovich, A.P.; Fang, F.; Gurley, S.; Coffman, T.; John, R.; Zhang, S.-L.; Diamandis, E.P. Determination of an angiotensin II-regulated proteome in primary human kidney cells by stable isotope labeling of amino acids in cell culture (SILAC). J. Biol. Chem. 2013, 288, 24834–24847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allport, J.R.; Ding, H.T.; Ager, A.; Steeber, D.A.; Tedder, T.F.; Luscinskas, F.W. L-selectin shedding does not regulate human neutrophil attachment, rolling, or transmigration across human vascular endothelium in vitro. J. Immunol. 1997, 158, 4365–4372. [Google Scholar] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reid, S.; Fine, N.; Bhosle, V.K.; Zhou, J.; John, R.; Glogauer, M.; Robinson, L.A.; Scholey, J.W. Inhibition of BRD4 Reduces Neutrophil Activation and Adhesion to the Vascular Endothelium Following Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2020, 21, 9620. https://doi.org/10.3390/ijms21249620
Reid S, Fine N, Bhosle VK, Zhou J, John R, Glogauer M, Robinson LA, Scholey JW. Inhibition of BRD4 Reduces Neutrophil Activation and Adhesion to the Vascular Endothelium Following Ischemia Reperfusion Injury. International Journal of Molecular Sciences. 2020; 21(24):9620. https://doi.org/10.3390/ijms21249620
Chicago/Turabian StyleReid, Shelby, Noah Fine, Vikrant K. Bhosle, Joyce Zhou, Rohan John, Michael Glogauer, Lisa A. Robinson, and James W. Scholey. 2020. "Inhibition of BRD4 Reduces Neutrophil Activation and Adhesion to the Vascular Endothelium Following Ischemia Reperfusion Injury" International Journal of Molecular Sciences 21, no. 24: 9620. https://doi.org/10.3390/ijms21249620
APA StyleReid, S., Fine, N., Bhosle, V. K., Zhou, J., John, R., Glogauer, M., Robinson, L. A., & Scholey, J. W. (2020). Inhibition of BRD4 Reduces Neutrophil Activation and Adhesion to the Vascular Endothelium Following Ischemia Reperfusion Injury. International Journal of Molecular Sciences, 21(24), 9620. https://doi.org/10.3390/ijms21249620