Mitophagy and the Brain


Abstract
:1. Introduction
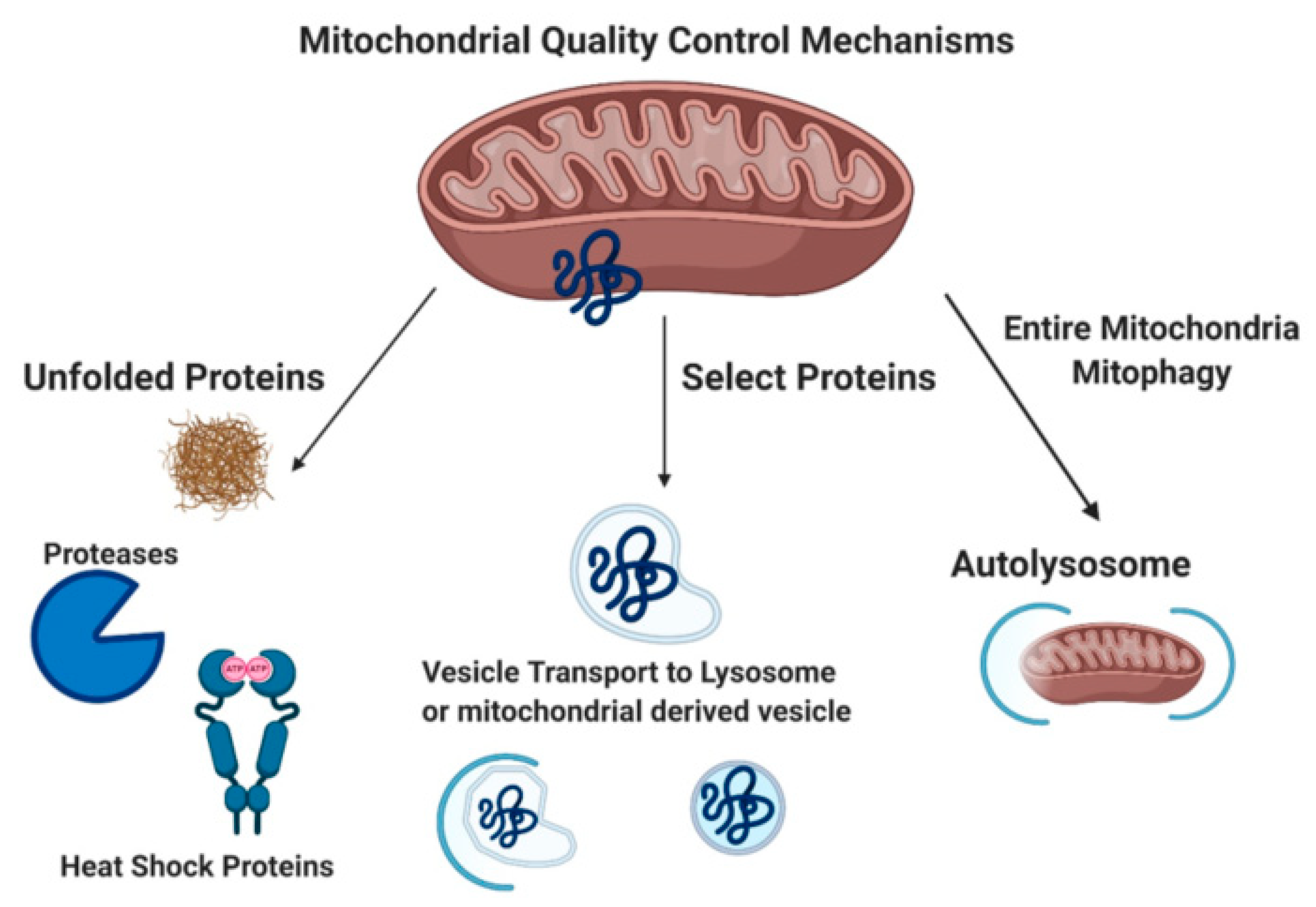
2. Mitophagy: Autophagy for Mitochondria
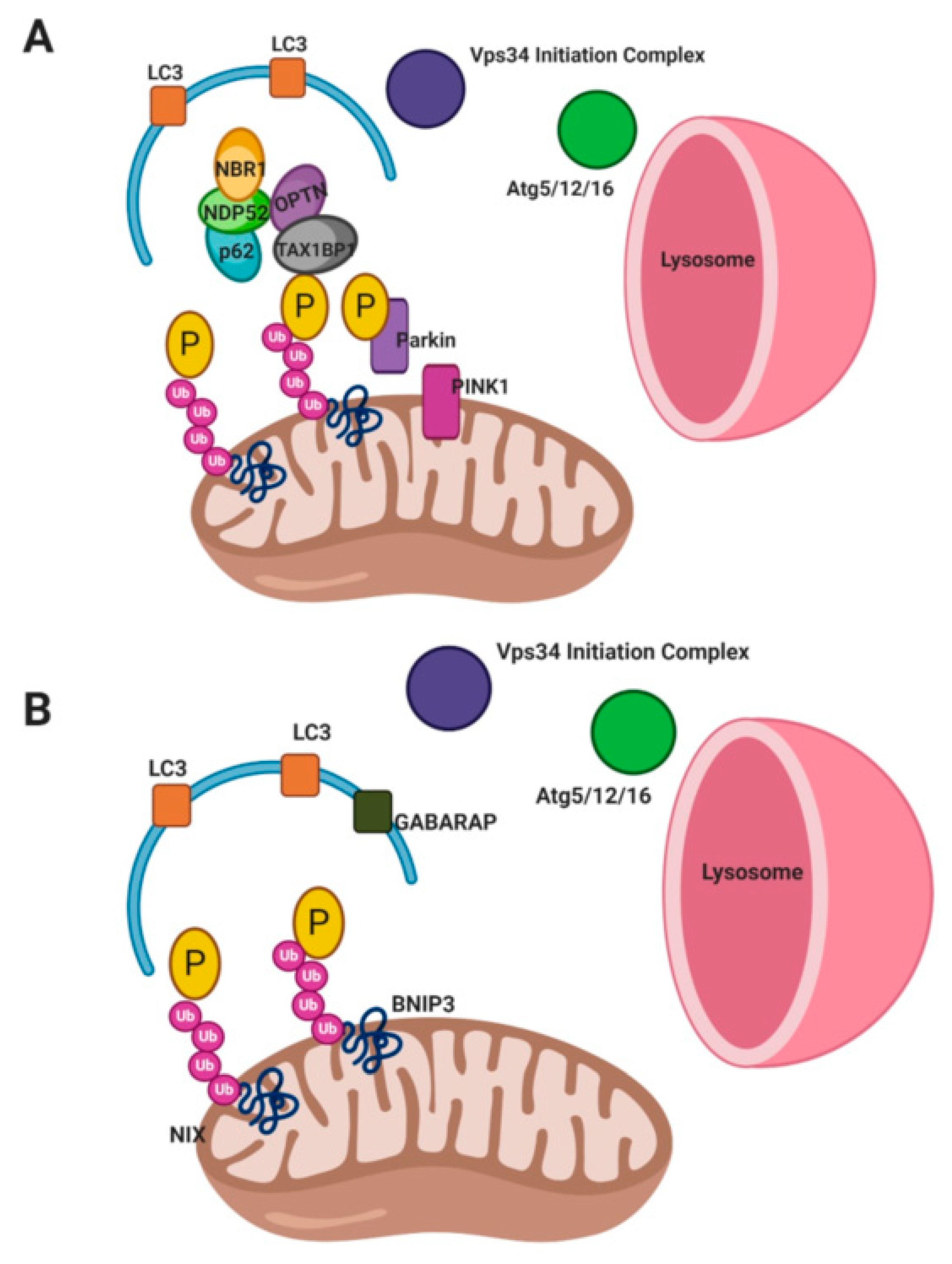
2.1. Non-Receptor Mediated Mitophagy
2.2. Receptor Mediated Mitophagy
2.3. Mitoptosis
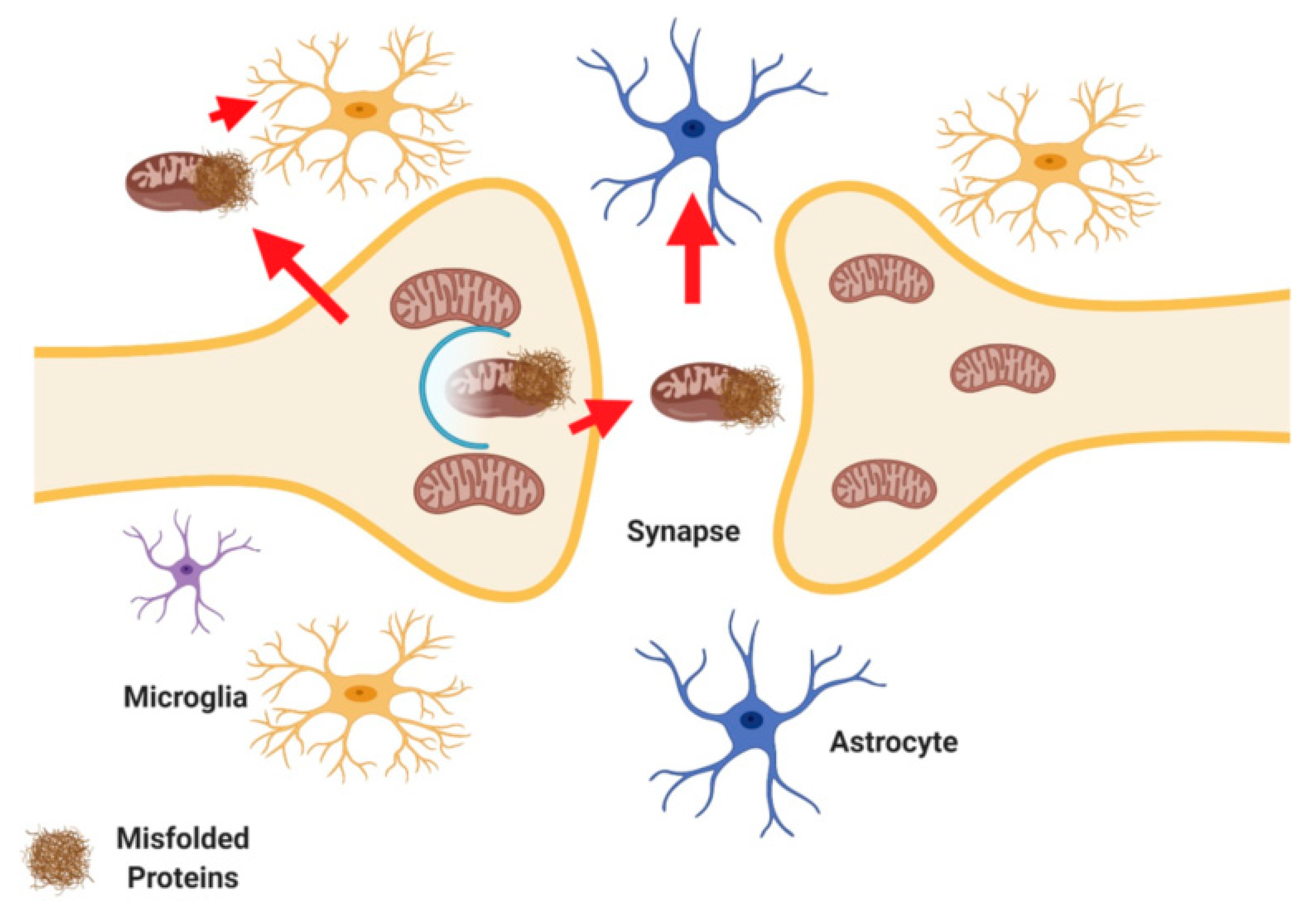
2.4. Transcellular Mitophagy
3. Mitophagy in Aging and Neurodegeneration
3.1. Aging
3.2. Alzheimer’s Disease
3.3. Parkinson’s Disease
3.4. Multiple Sclerosis
3.5. Amyotrophic Lateral Sclerosis
4. Modulating Mitophagy in Neurodegeneration
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Wilkins, H.M.; Swerdlow, R.H. Relationships between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Koppel, S.; Weidling, I.; Hayley, C.; Ji, Y.; Wilkins, H.M. Mitochondria, Cybrids, Aging, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 259–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.W.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Mitophagy and mitoptosis in disease processes. Methods Mol. Biol. 2010, 648, 93–106. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Molina-Porcel, L.; Corrada, M.M.; Raible, K.; Lee, E.B.; Lee, V.M.; Kawas, C.H.; Trojanowski, J.Q. Perforant path synaptic loss correlates with cognitive impairment and Alzheimer’s Disease in the oldest-old. Brain 2014, 137, 2578–2587. [Google Scholar] [CrossRef]
- Kashyap, G.; Bapat, D.; Das, D.; Gowaikar, R.; Amritkar, R.E.; Rangarajan, G.; Ravindranath, V.; Ambika, G. Synapse loss and progress of Alzheimer’s Disease—A network model. Sci. Rep. 2019, 9, 6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Martino Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.; Kato, T.; Di Prospero, N.A.; Ness, S.; Beal, M.F.; Krams, M.; Chen, G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Jeong, Y.Y.; Sheshadri, P.; Su, X.; Cai, Q. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 2020, e201949801. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Marsh-Armstrong, N. Discovery and implications of transcellular mitophagy. Autophagy 2014, 10, 2383–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Liu, L.; Zhu, Y.; Chen, Q. Molecular signaling toward mitophagy and its physiological significance. Exp. Cell Res. 2013, 319, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhang, M.; Kenny, S.J.; Liu, D.; Maeda, M.; Saito, K.; Mathur, A.; Xu, K.; Schekman, R. Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep. 2017, 18, 1586–1603. [Google Scholar] [CrossRef]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 complex—At the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Tassa, A.; Roux, M.P.; Attaix, D.; Bechet, D.M. Class III phosphoinositide 3-kinase-Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem. J. 2003, 376, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Haynes, C.M. Metabolism and the UPR(mt). Mol. Cell 2016, 61, 677–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Voos, W.; Rottgers, K. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim. Biophys. Acta 2002, 1592, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.T.; Naylor, D.J.; Hoj, P.B.; Clark, M.S.; Hoogenraad, N.J. The role of molecular chaperones in mitochondrial protein import and folding. Int. Rev. Cytol. 1997, 174, 127–193. [Google Scholar] [CrossRef]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 2011, 22, 291–300. [Google Scholar] [CrossRef]
- Cadete, V.J.; Deschenes, S.; Cuillerier, A.; Brisebois, F.; Sugiura, A.; Vincent, A.; Turnbull, D.; Picard, M.; McBride, H.M.; Burelle, Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 2016, 594, 5343–5362. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakovic, A.; Shurkewitsch, K.; Seibler, P.; Grunewald, A.; Zanon, A.; Hagenah, J.; Krainc, D.; Klein, C. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 2013, 288, 2223–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Schapira, A.H. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy 2011, 7, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [Green Version]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Fung, G.; Deng, H.; Zhang, J.; Fiesel, F.C.; Springer, W.; Li, X.; Luo, H. NBR1 is dispensable for PARK2-mediated mitophagy regardless of the presence or absence of SQSTM1. Cell Death Dis. 2015, 6, e1943. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Minowa-Nozawa, A.; Nozawa, T.; Okamoto-Furuta, K.; Kohda, H.; Nakagawa, I. Rab35 GTPase recruits NDP52 to autophagy targets. EMBO J. 2017, 36, 2790–2807. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dai, C.; Fan, Y.; Guo, B.; Ren, K.; Sun, T.; Wang, W. From autophagy to mitophagy: The roles of P62 in neurodegenerative diseases. J. Bioenerg. Biomembr. 2017, 49, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Whang, M.I.; Tavares, R.M.; Benjamin, D.I.; Kattah, M.G.; Advincula, R.; Nomura, D.K.; Debnath, J.; Malynn, B.A.; Ma, A. The Ubiquitin Binding Protein TAX1BP1 Mediates Autophagasome Induction and the Metabolic Transition of Activated T Cells. Immunity 2017, 46, 405–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ney, P.A. NIX induces mitochondrial autophagy in reticulocytes. Autophagy 2008, 4, 354–356. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-Terminus Inhibits Mitochondrial Damage and Cell Death without Blocking Autophagy. PLoS ONE 2015, 10, e0129667. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.V.; Suzuki, H.; Marinkovic, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Sprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef]
- Ma, K.; Zhang, Z.; Chang, R.; Cheng, H.; Mu, C.; Zhao, T.; Chen, L.; Zhang, C.; Luo, Q.; Lin, J.; et al. Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ. 2020, 27, 1036–1051. [Google Scholar] [CrossRef]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Liu, L.; Cheng, Q.; Li, Y.; Wu, H.; Zhang, W.; Wang, Y.; Sehgal, S.A.; Siraj, S.; Wang, X.; et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017, 18, 495–509. [Google Scholar] [CrossRef]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Desaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef]
- Li, X.X.; Tsoi, B.; Li, Y.F.; Kurihara, H.; He, R.R. Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Kinnally, K.W.; Tedeschi, H. Voltage activation of heart inner mitochondrial membrane channels. J. Bioenerg. Biomembr. 1992, 24, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Lyamzaev, K.G.; Nepryakhina, O.K.; Saprunova, V.B.; Bakeeva, L.E.; Pletjushkina, O.Y.; Chernyak, B.V.; Skulachev, V.P. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): Formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta 2008, 1777, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485. [Google Scholar] [CrossRef]
- Skulachev, V.P. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol. Aspects Med. 1999, 20, 139–184. [Google Scholar] [CrossRef]
- Jangamreddy, J.R.; Los, M.J. Mitoptosis, a novel mitochondrial death mechanism leading predominantly to activation of autophagy. Hepat. Mon. 2012, 12, e6159. [Google Scholar] [CrossRef] [Green Version]
- Tinari, A.; Garofalo, T.; Sorice, M.; Esposti, M.D.; Malorni, W. Mitoptosis: Different pathways for mitochondrial execution. Autophagy 2007, 3, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Programmed death phenomena: From organelle to organism. Ann. N. Y. Acad. Sci. 2002, 959, 214–237. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Corrigendum: Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 539, 123. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Hass, D.T.; Barnstable, C.J. Mitochondrial Uncoupling Protein 2 Knock-out Promotes Mitophagy to Decrease Retinal Ganglion Cell Death in a Mouse Model of Glaucoma. J. Neurosci. 2019, 39, 3582–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Hu, J.; Yan, Q.; Zhu, J.; Zhu, Z.; Chen, Y.; Sun, J.; Zhang, R. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 2016, 13, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.B.; Park, K.S.; Lee, H.K. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef] [Green Version]
- Konari, N.; Nagaishi, K.; Kikuchi, S.; Fujimiya, M. Mitochondria transfer from mesenchymal stem cells structurally and functionally repairs renal proximal tubular epithelial cells in diabetic nephropathy in vivo. Sci. Rep. 2019, 9, 5184. [Google Scholar] [CrossRef] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; De Benedictis, V.; Ruggiero, F.M.; Paradies, G. Decline in cytochrome c oxidase activity in rat-brain mitochondria with aging. Role of peroxidized cardiolipin and beneficial effect of melatonin. J. Bioenerg. Biomembr. 2013, 45, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.M.; Paradies, G. Mitochondrial dysfunction in rat brain with aging Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 2008, 53, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Leslie, S.W.; Chandler, L.J.; Barr, E.M.; Farrar, R.P. Reduced calcium uptake by rat brain mitochondria and synaptosomes in response to aging. Brain Res. 1985, 329, 177–183. [Google Scholar] [CrossRef]
- Stauch, K.L.; Purnell, P.R.; Villeneuve, L.M.; Fox, H.S. Proteomic analysis and functional characterization of mouse brain mitochondria during aging reveal alterations in energy metabolism. Proteomics 2015, 15, 1574–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoni-Freddari, C.; Fattoretti, P.; Casoli, T.; Spagna, C.; Meier-Ruge, W.; Ulrich, J. Morphological plasticity of synaptic mitochondria during aging. Brain Res. 1993, 628, 193–200. [Google Scholar] [CrossRef]
- Bertoni-Freddari, C.; Balietti, M.; Giorgetti, B.; Grossi, Y.; Casoli, T.; Di Stefano, G.; Perretta, G.; Fattoretti, P. Selective decline of the metabolic competence of oversized synaptic mitochondria in the old monkey cerebellum. Rejuvenation Res. 2008, 11, 387–391. [Google Scholar] [CrossRef]
- Tanhauser, S.M.; Laipis, P.J. Multiple deletions are detectable in mitochondrial DNA of aging mice. J. Biol. Chem. 1995, 270, 24769–24775. [Google Scholar] [CrossRef] [Green Version]
- Dzitoyeva, S.; Chen, H.; Manev, H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol. Aging 2012, 33, 2881–2891. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.; Barja, G. 8-oxo-deoxyguanosine levels in heart and brain mitochondrial and nuclear DNA of two mammals and three birds in relation to their different rates of aging. Aging 1999, 11, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA in aging and disease. Sci. Am. 1997, 277, 40–47. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Al-Amin, M.; Zhao, C.; An, F.; Wang, Y.; Huang, Q.; Teng, H.; Song, H. Catechinic acid, a natural polyphenol compound, extends the lifespan of Caenorhabditis elegans via mitophagy pathways. Food Funct. 2020, 11, 5621–5634. [Google Scholar] [CrossRef]
- Fang, E.F.; Waltz, T.B.; Kassahun, H.; Lu, Q.; Kerr, J.S.; Morevati, M.; Fivenson, E.M.; Wollman, B.N.; Marosi, K.; Wilson, M.A.; et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci. Rep. 2017, 7, 46208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Felix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Diagnostic criteria for neuropathologic assessment of Alzheimer’s Disease. Neurobiol. Aging 1997, 18, S85–S88. [Google Scholar] [CrossRef]
- Powers, J.M. Diagnostic criteria for the neuropathologic assessment of Alzheimer’s Disease. Neurobiol. Aging 1997, 18, S53–S54. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Barrio, J.R.; Kepe, V. Cerebral amyloid PET imaging in Alzheimer’s Disease. Acta Neuropathol. 2013, 126, 643–657. [Google Scholar] [CrossRef] [Green Version]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s Disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s Disease. Clin. Nucl. Med. 2014, 39, e413–e422. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; McHugh, P.F. FDG- and amyloid-PET in Alzheimer’s Disease: Is the whole greater than the sum of the parts? Q. J. Nucl. Med. Mol. Imaging 2011, 55, 250–264. [Google Scholar]
- Suppiah, S.; Didier, M.A.; Vinjamuri, S. The Who, When, Why, and How of PET Amyloid Imaging in Management of Alzheimer’s Disease-Review of Literature and Interesting Images. Diagnostics 2019, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Herholz, K.; Salmon, E.; Perani, D.; Baron, J.C.; Holthoff, V.; Frolich, L.; Schonknecht, P.; Ito, K.; Mielke, R.; Kalbe, E.; et al. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage 2002, 17, 302–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valla, J.; Berndt, J.D.; Gonzalez-Lima, F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: Superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci. 2001, 21, 4923–4930. [Google Scholar] [CrossRef] [PubMed]
- Messa, C.; Perani, D.; Lucignani, G.; Zenorini, A.; Zito, F.; Rizzo, G.; Grassi, F.; Del Sole, A.; Franceschi, M.; Gilardi, M.C.; et al. High-resolution technetium-99m-HMPAO SPECT in patients with probable Alzheimer’s Disease: Comparison with fluorine-18-FDG PET. J. Nucl. Med. 1994, 35, 210–216. [Google Scholar] [PubMed]
- Morris, J.K.; Honea, R.A.; Vidoni, E.D.; Swerdlow, R.H.; Burns, J.M. Is Alzheimer’s Disease a systemic disease? Biochim. Biophys. Acta 2014, 1842, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s Disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Fukui, H.; Diaz, F.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2007, 104, 14163–14168. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.D., Jr.; Parks, J.K. Cytochrome c oxidase in Alzheimer’s Disease brain: Purification and characterization. Neurology 1995, 45, 482–486. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Proenca, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s Disease platelets. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Khan, S.M.; Cassarino, D.S.; Abramova, N.N.; Keeney, P.M.; Borland, M.K.; Trimmer, P.A.; Krebs, C.T.; Bennett, J.C.; Parks, J.K.; Swerdlow, R.H.; et al. Alzheimer’s Disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann. Neurol. 2000, 48, 148–155. [Google Scholar] [CrossRef]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s Disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J. Brain energy metabolizing enzymes in Alzheimer’s Disease: Alpha-ketoglutarate dehydrogenase complex and cytochrome oxidase. Ann. N. Y. Acad. Sci. 1997, 826, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s Disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef]
- Parker, W.D., Jr. Cytochrome oxidase deficiency in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1991, 640, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s Disease. Neurology 1990, 40, 1302–1303. [Google Scholar] [CrossRef] [PubMed]
- Alberts, M.J.; Ioannou, P.; Deucher, R.; Gilbert, J.; Lee, J.; Middleton, L.; Roses, A.D. Isolation of a cytochrome oxidase gene overexpressed in Alzheimer’s Disease brain. Mol. Cell Neurosci. 1992, 3, 461–470. [Google Scholar] [CrossRef]
- Curti, D.; Rognoni, F.; Gasparini, L.; Cattaneo, A.; Paolillo, M.; Racchi, M.; Zani, L.; Bianchetti, A.; Trabucchi, M.; Bergamaschi, S.; et al. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer’s Disease (AD) patients. Neurosci. Lett. 1997, 236, 13–16. [Google Scholar] [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; Lezi, E.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.P.; Swerdlow, R.H.; Miller, S.W.; Davis, R.E.; Parks, J.K.; Parker, W.D.; Tuttle, J.B. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s Disease. J. Neurosci. 1997, 17, 4612–4622. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s Disease cybrids enhances Abeta toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J. Neurosci. Res. 2007, 85, 3416–3428. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Keeney, P.M.; Borland, M.K.; Simon, F.A.; Almeida, J.; Swerdlow, R.H.; Parks, J.P.; Parker, W.D., Jr.; Bennett, J.P., Jr. Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer’s Disease worsen with passage in culture. Neurobiol. Dis. 2004, 15, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Baloyannis, S.J. Mitochondria in Alzheimer’s Disease: An Electron Microscopy Study. Alzheimer Dis. Treat. 2019. [Google Scholar] [CrossRef] [Green Version]
- Berti, V.; Mosconi, L.; Glodzik, L.; Li, Y.; Murray, J.; De Santi, S.; Pupi, A.; Tsui, W.; De Leon, M.J. Structural brain changes in normal individuals with a maternal history of Alzheimer’s. Neurobiol. Aging 2011, 32, 2325-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s Disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; Mistur, R.; Switalski, R.; Brys, M.; Glodzik, L.; Rich, K.; Pirraglia, E.; Tsui, W.; De Santi, S.; de Leon, M.J. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology 2009, 72, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; Rinne, J.O.; Tsui, W.H.; Murray, J.; Li, Y.; Glodzik, L.; McHugh, P.; Williams, S.; Cummings, M.; Pirraglia, E.; et al. Amyloid and metabolic positron emission tomography imaging of cognitively normal adults with Alzheimer’s parents. Neurobiol. Aging 2013, 34, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Hui, D.; Chalise, P.; Sharma, P.; Wang, X.; Andrews, S.J.; Pa, J.; Mahnken, J.D.; Morris, J.; Wilkins, H.M.; et al. Exploratory analysis of mtDNA haplogroups in two Alzheimer’s longitudinal cohorts. Alzheimers Dement. 2020, 16, 1164–1172. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Patterson, C.; McFall, G.P.; Gross, A.; Michaelis, E.K.; Goate, A.; Swerdlow, R.H.; Pa, J. Mitochondrial haplogroups & a nuclear encoded mitochondrial polygenic risk score interact to influence dementia risk. Neurobiol. Aging 2020, 87, 138-e7. [Google Scholar]
- Carrieri, G.; Bonafe, M.; De Luca, M.; Rose, G.; Varcasia, O.; Bruni, A.; Maletta, R.; Nacmias, B.; Sorbi, S.; Corsonello, F.; et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s Disease. Hum. Genet. 2001, 108, 194–198. [Google Scholar] [CrossRef]
- Coto, E.; Gomez, J.; Alonso, B.; Corao, A.I.; Diaz, M.; Menendez, M.; Martinez, C.; Calatayud, M.T.; Moris, G.; Alvarez, V. Late-onset Alzheimer’s Disease is associated with mitochondrial DNA 7028C/haplogroup H and D310 poly-C tract heteroplasmy. Neurogenetics 2011, 12, 345–346. [Google Scholar] [CrossRef]
- Fesahat, F.; Houshmand, M.; Panahi, M.S.; Gharagozli, K.; Mirzajani, F. Do haplogroups H and U act to increase the penetrance of Alzheimer’s Disease? Cell Mol. Neurobiol. 2007, 27, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, S. Mitochondrial haplogroups associated with Japanese Alzheimer’s patients. J. Bioenerg. Biomembr. 2009, 41, 407–410. [Google Scholar] [CrossRef] [PubMed]
- van der Walt, J.M.; Dementieva, Y.A.; Martin, E.R.; Scott, W.K.; Nicodemus, K.K.; Kroner, C.C.; Welsh-Bohmer, K.A.; Saunders, A.M.; Roses, A.D.; Small, G.W.; et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci. Lett. 2004, 365, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Y.; Mok, K.Y.; Kwok, T.C.Y.; Mok, V.C.T.; Guo, Q.; Ip, F.C.; Chen, Y.; Mullapudi, N.; Alzheimer’s Disease Neuroimaging, I.; et al. Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat. Commun. 2019, 10, 3310. [Google Scholar] [CrossRef] [Green Version]
- Lakatos, A.; Derbeneva, O.; Younes, D.; Keator, D.; Bakken, T.; Lvova, M.; Brandon, M.; Guffanti, G.; Reglodi, D.; Saykin, A.; et al. Association between mitochondrial DNA variations and Alzheimer’s Disease in the ADNI cohort. Neurobiol. Aging 2010, 31, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhao, X.L.; Xu, L.L.; Wang, C.F.; Wei, L.F.; Liu, Z.; Yang, H.; Wang, P.; Xie, Z.H.; Bi, J.Z. Mitophagy in APPsw/PS1dE9 transgenic mice and APPsw stably expressing in HEK293 cells. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4595–4602. [Google Scholar]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s Disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Kurosawa, M.; Matsumoto, G.; Sumikura, H.; Hatsuta, H.; Murayama, S.; Sakurai, T.; Shimogori, T.; Hattori, N.; Nukina, N. Serine 403-phosphorylated p62/SQSTM1 immunoreactivity in inclusions of neurodegenerative diseases. Neurosci. Res. 2016, 103, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Martins, R.N. PRKAG2 Gene Expression Is Elevated and its Protein Levels Are Associated with Increased Amyloid-beta Accumulation in the Alzheimer’s Disease Brain. J. Alzheimers Dis. 2020, 74, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonskaya, I.; Shekoyan, A.R.; Hebron, M.L.; Desforges, N.; Algarzae, N.K.; Moussa, C.E. Diminished parkin solubility and co-localization with intraneuronal amyloid-beta are associated with autophagic defects in Alzheimer’s Disease. J. Alzheimers Dis. 2013, 33, 231–247. [Google Scholar] [CrossRef]
- Yan, X.; Wang, B.; Hu, Y.; Wang, S.; Zhang, X. Abnormal Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 138. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, C.E. Parkinson’s disease. BMJ 2007, 335, 441–445. [Google Scholar] [CrossRef]
- Mann, V.M.; Cooper, J.M.; Krige, D.; Daniel, S.E.; Schapira, A.H.; Marsden, C.D. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson’s disease. Brain 1992, 115 Pt 2, 333–342. [Google Scholar] [CrossRef]
- Gatt, A.P.; Duncan, O.F.; Attems, J.; Francis, P.T.; Ballard, C.G.; Bateman, J.M. Dementia in Parkinson’s disease is associated with enhanced mitochondrial complex I deficiency. Mov. Disord. 2016, 31, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.I.; Spitz, E.; Leehey, M.; Hoffer, B.J.; Boyson, S.J. Platelet mitochondrial respiratory chain function in Parkinson’s disease. Mov. Disord. 1997, 12, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Krige, D.; Carroll, M.T.; Cooper, J.M.; Marsden, C.D.; Schapira, A.H. Platelet mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann. Neurol. 1992, 32, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Esteves, A.R.; Swerdlow, R.H.; Cardoso, S.M. A cybrid cell model for the assessment of the link between mitochondrial deficits and sporadic Parkinson’s disease. Methods Mol. Biol. 2015, 1265, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, A.R.; Domingues, A.F.; Ferreira, I.L.; Januario, C.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 2008, 8, 219–228. [Google Scholar] [CrossRef]
- Esteves, A.R.; Lu, J.; Rodova, M.; Onyango, I.; Lezi, E.; Dubinsky, R.; Lyons, K.E.; Pahwa, R.; Burns, J.M.; Cardoso, S.M.; et al. Mitochondrial respiration and respiration-associated proteins in cell lines created through Parkinson’s subject mitochondrial transfer. J. Neurochem. 2010, 113, 674–682. [Google Scholar] [CrossRef]
- Dolle, C.; Flones, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Gu, G.; Reyes, P.E.; Golden, G.T.; Woltjer, R.L.; Hulette, C.; Montine, T.J.; Zhang, J. Mitochondrial DNA deletions/rearrangements in parkinson disease and related neurodegenerative disorders. J. Neuropathol. Exp. Neurol. 2002, 61, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, S.; Condroyer, C.; Klebe, S.; Honore, A.; Tison, F.; Brefel-Courbon, C.; Durr, A.; Brice, A.; French Parkinson’s Disease Genetics Study, G. Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology 2012, 78, 1449–1450. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, K.; Song, W.; Chen, X.; Cao, B.; Huang, R.; Zhao, B.; Guo, X.; Burgunder, J.; Li, J.; et al. VPS35 Asp620Asn and EIF4G1 Arg1205His mutations are rare in Parkinson disease from southwest China. Neurobiol. Aging 2013, 34, 1709-e7. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, K.S.; Sindhu, K.M.; Mohanakumar, K.P. Acute intranigral infusion of rotenone in rats causes progressive biochemical lesions in the striatum similar to Parkinson’s disease. Brain Res. 2005, 1049, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [Green Version]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Zeng, X.S.; Geng, W.S.; Jia, J.J. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro 2018, 10, 1759091418777438. [Google Scholar] [CrossRef]
- von Wrangel, C.; Schwabe, K.; John, N.; Krauss, J.K.; Alam, M. The rotenone-induced rat model of Parkinson’s disease: Behavioral and electrophysiological findings. Behav. Brain Res. 2015, 279, 52–61. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Fiesel, F.C.; Truban, D.; Castanedes Casey, M.; Lin, W.L.; Soto, A.I.; Tacik, P.; Rousseau, L.G.; Diehl, N.N.; Heckman, M.G.; et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 2018, 14, 1404–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, C.B.; Jakimovski, D.; Kavak, K.S.; Ramanathan, M.; Benedict, R.H.B.; Zivadinov, R.; Weinstock-Guttman, B. Epidemiology and treatment of multiple sclerosis in elderly populations. Nat. Rev. Neurol. 2019, 15, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, H.; Nogueras, L.; Gil-Sanchez, A.; Hervas, J.V.; Valcheva, P.; Gonzalez-Mingot, C.; Martin-Gari, M.; Canudes, M.; Peralta, S.; Solana, M.J.; et al. Impairment of Mitochondrial Redox Status in Peripheral Lymphocytes of Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 938. [Google Scholar] [CrossRef]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009, 5, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Le, W. Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci. Bull. 2015, 31, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Igci, M.; Baysan, M.; Yigiter, R.; Ulasli, M.; Geyik, S.; Bayraktar, R.; Bozgeyik, I.; Bozgeyik, E.; Bayram, A.; Cakmak, E.A. Gene expression profiles of autophagy-related genes in multiple sclerosis. Gene 2016, 588, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Parks, J.K.; Pattee, G.; Parker, W.D., Jr. Role of mitochondria in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 185–190. [Google Scholar] [CrossRef]
- Beeldman, E.; Raaphorst, J.; Klein Twennaar, M.; de Visser, M.; Schmand, B.A.; de Haan, R.J. The cognitive profile of ALS: A systematic review and meta-analysis update. J. Neurol. Neurosurg. Psychiatry 2016, 87, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Gubbay, S.S.; Kahana, E.; Zilber, N.; Cooper, G.; Pintov, S.; Leibowitz, Y. Amyotrophic lateral sclerosis. A study of its presentation and prognosis. J. Neurol. 1985, 232, 295–300. [Google Scholar] [CrossRef]
- Norris, F.; Shepherd, R.; Denys, E.; Kwei, U.; Mukai, E.; Elias, L.; Holden, D.; Norris, H. Onset, natural history and outcome in idiopathic adult motor neuron disease. J. Neurol. Sci. 1993, 118, 48–55. [Google Scholar] [CrossRef]
- Rothstein, J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 2009, 65 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Deng, H.X.; Bigio, E.H.; Zhai, H.; Fecto, F.; Ajroud, K.; Shi, Y.; Yan, J.; Mishra, M.; Ajroud-Driss, S.; Heller, S.; et al. Differential involvement of optineurin in amyotrophic lateral sclerosis with or without SOD1 mutations. Arch. Neurol. 2011, 68, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Blitterswijk, M.; van Vught, P.W.; van Es, M.A.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 2012, 33, 1016-e1. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, T.; Irie, T.; Kawabata, R.; Yoshida, A.; Maruyama, H.; Kawakami, H. Optineurin with amyotrophic lateral sclerosis-related mutations abrogates inhibition of interferon regulatory factor-3 activation. Neurosci. Lett. 2011, 505, 279–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [Green Version]
- Vielhaber, S.; Kunz, D.; Winkler, K.; Wiedemann, F.R.; Kirches, E.; Feistner, H.; Heinze, H.J.; Elger, C.E.; Schubert, W.; Kunz, W.S. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 2000, 123 Pt 7, 1339–1348. [Google Scholar] [CrossRef] [Green Version]
- Duffy, L.M.; Chapman, A.L.; Shaw, P.J.; Grierson, A.J. Review: The role of mitochondria in the pathogenesis of amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2011, 37, 336–352. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Trimmer, P.A.; Miller, S.W.; Maguire, D.J.; Sheehan, J.P.; Maguire, R.S.; Pattee, G.; Juel, V.C.; et al. Mitochondria in sporadic amyotrophic lateral sclerosis. Exp. Neurol. 1998, 153, 135–142. [Google Scholar] [CrossRef]
- Hart, M.N.; Cancilla, P.A.; Frommes, S.; Hirano, A. Anterior horn cell degeneration and Bunina-type inclusions associated with dementia. Acta Neuropathol. 1977, 38, 225–228. [Google Scholar] [CrossRef]
- Mizuno, Y.; Amari, M.; Takatama, M.; Aizawa, H.; Mihara, B.; Okamoto, K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2006, 249, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.E.; Gonzalez, D.M.; Monachelli, G.M.; Costa, J.J.; Nicola, A.F.; Sica, R.E. Morphological abnormalities in mitochondria of the skin of patients with sporadic amyotrophic lateral sclerosis. Arq. Neuropsiquiatr. 2012, 70, 40–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H. The neurodegenerative mitochondriopathies. J. Alzheimers Dis. 2009, 17, 737–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalan-Garcia, M.; Garrabou, G.; Moren, C.; Guitart-Mampel, M.; Hernando, A.; Diaz-Ramos, A.; Gonzalez-Casacuberta, I.; Juarez, D.L.; Bano, M.; Enrich-Bengoa, J.; et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin. Sci. 2016, 130, 1741–1751. [Google Scholar] [CrossRef] [Green Version]
- Horvath, R.; Fu, K.; Johns, T.; Genge, A.; Karpati, G.; Shoubridge, E.A. Characterization of the mitochondrial DNA abnormalities in the skeletal muscle of patients with inclusion body myositis. J. Neuropathol. Exp. Neurol. 1998, 57, 396–403. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbull, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790. [Google Scholar] [CrossRef]
- Fujita, K.; Yamauchi, M.; Shibayama, K.; Ando, M.; Honda, M.; Nagata, Y. Decreased cytochrome c oxidase activity but unchanged superoxide dismutase and glutathione peroxidase activities in the spinal cords of patients with amyotrophic lateral sclerosis. J. Neurosci. Res. 1996, 45, 276–281. [Google Scholar] [CrossRef]
- Shrivastava, M.; Subbiah, V. Elevated caspase 3 activity and cytosolic cytochrome c in NT2 cybrids containing amyotrophic lateral sclerosis subject mtDNA. Int. J. Neurosci. 2016, 126, 839–849. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Huntley, M.L.; Gao, J.; Termsarasab, P.; Wang, L.; Zeng, S.; Thammongkolchai, T.; Liu, Y.; Cohen, M.L.; Wang, X. Association between TDP-43 and mitochondria in inclusion body myositis. Lab. Investig. 2019, 99, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Nogalska, A.; D’Agostino, C.; Terracciano, C.; Engel, W.K.; Askanas, V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 2010, 177, 1377–1387. [Google Scholar] [CrossRef]
- Vattemi, G.; Nogalska, A.; King Engel, W.; D’Agostino, C.; Checler, F.; Askanas, V. Amyloid-beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion-body myositis. Acta Neuropathol. 2009, 117, 569–574. [Google Scholar] [CrossRef]
- Watanabe, M.; Dykes-Hoberg, M.; Culotta, V.C.; Price, D.L.; Wong, P.C.; Rothstein, J.D. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol. Dis. 2001, 8, 933–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.M.; Ding, W.X.; Nishimune, H. Impaired Mitophagy Plays a Role in Denervation of Neuromuscular Junctions in ALS Mice. Front. Neurosci. 2017, 11, 473. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffoli, R.; Bartalucci, A.; Frati, A.; Fornai, F. Ultrastructural studies of ALS mitochondria connect altered function and permeability with defects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 341. [Google Scholar] [CrossRef] [Green Version]
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.J.; Zhao, D.; Milner, T.A.; Manfredi, G. Parkin is a disease modifier in the mutant SOD1 mouse model of ALS. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef]
- Almeida, S.; Gascon, E.; Tran, H.; Chou, H.J.; Gendron, T.F.; Degroot, S.; Tapper, A.R.; Sellier, C.; Charlet-Berguerand, N.; Karydas, A.; et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013, 126, 385–399. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef]
- Fang, E.F. Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy 2019, 15, 1112–1114. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 2018, 14, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Staats, K.A.; Hernandez, S.; Schonefeldt, S.; Bento-Abreu, A.; Dooley, J.; Van Damme, P.; Liston, A.; Robberecht, W.; Van Den Bosch, L. Rapamycin increases survival in ALS mice lacking mature lymphocytes. Mol. Neurodegener. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Chen, J. Salidroside Protects Dopaminergic Neurons by Enhancing PINK1/Parkin-Mediated Mitophagy. Oxid. Med. Cell Longev. 2019, 2019, 9341018. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Choi, Y.; Park, J.; Nah, W.; Park, J.; Jung, Y.; Lee, J.; Lee, H.; Park, S.; Hwang, S.; et al. Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological alpha-synuclein. Sci. Adv. 2020, 6, eaba1193. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, J.; Gao, C.; Wu, M.; Du, Q.; Tsoi, B.; Wang, Q.; Yang, D.; Shen, J. Nitration of Drp1 provokes mitophagy activation mediating neuronal injury in experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 2019, 143, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, R.; Jing, X.; Chen, J.; Yang, D.; Shen, J. Acteoside ameliorates experimental autoimmune encephalomyelitis through inhibiting peroxynitrite-mediated mitophagy activation. Free Radic. Biol. Med. 2020, 146, 79–91. [Google Scholar] [CrossRef]
- Kovacs, J.R.; Li, C.; Yang, Q.; Li, G.; Garcia, I.G.; Ju, S.; Roodman, D.G.; Windle, J.J.; Zhang, X.; Lu, B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012, 19, 144–152. [Google Scholar] [CrossRef]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef]
- Irie, J.; Inagaki, E.; Fujita, M.; Nakaya, H.; Mitsuishi, M.; Yamaguchi, S.; Yamashita, K.; Shigaki, S.; Ono, T.; Yukioka, H.; et al. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr. J. 2020, 67, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Tsubota, K. The first human clinical study for NMN has started in Japan. NPJ Aging Mech. Dis. 2016, 2, 16021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.Z.; No, M.H.; Heo, J.W.; Park, D.H.; Kang, J.H.; Kim, J.H.; Seo, D.Y.; Han, J.; Jung, S.J.; Kwak, H.B. Effects of Acute Exercise on Mitochondrial Function, Dynamics, and Mitophagy in Rat Cardiac and Skeletal Muscles. Int. Neurourol. J. 2019, 23, S22–S31. [Google Scholar] [CrossRef]
- Kwon, I.; Jang, Y.; Lee, Y. Endurance Exercise-Induced Autophagy/Mitophagy Coincides with a Reinforced Anabolic State and Increased Mitochondrial Turnover in the Cortex of Young Male Mouse Brain. J. Mol. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Drake, J.C.; Yan, Z. Exercise-Induced Mitophagy in Skeletal Muscle and Heart. Exerc. Sport Sci. Rev. 2019, 47, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, T.C.; Marques-Aleixo, I.; Beleza, J.; Oliveira, P.J.; Ascensao, A.; Magalhaes, J. Physical Exercise and Brain Mitochondrial Fitness: The Possible Role Against Alzheimer’s Disease. Brain Pathol. 2016, 26, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Tryon, L.D.; Pauly, M.; Hood, D.A. Role of PGC-1alpha during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015, 308, C710–C719. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.K.; Vidoni, E.D.; Johnson, D.K.; Van Sciver, A.; Mahnken, J.D.; Honea, R.A.; Wilkins, H.M.; Brooks, W.M.; Billinger, S.A.; Swerdlow, R.H.; et al. Aerobic exercise for Alzheimer’s Disease: A randomized controlled pilot trial. PLoS ONE 2017, 12, e0170547. [Google Scholar] [CrossRef]
- Baker, L.D.; Frank, L.L.; Foster-Schubert, K.; Green, P.S.; Wilkinson, C.W.; McTiernan, A.; Cholerton, B.A.; Plymate, S.R.; Fishel, M.A.; Watson, G.S.; et al. Aerobic exercise improves cognition for older adults with glucose intolerance, a risk factor for Alzheimer’s Disease. J. Alzheimers Dis. 2010, 22, 569–579. [Google Scholar] [CrossRef]
- Vidoni, E.D.; Johnson, D.K.; Morris, J.K.; Van Sciver, A.; Greer, C.S.; Billinger, S.A.; Donnelly, J.E.; Burns, J.M. Dose-Response of Aerobic Exercise on Cognition: A Community-Based, Pilot Randomized Controlled Trial. PLoS ONE 2015, 10, e0131647. [Google Scholar] [CrossRef] [Green Version]
- Tomporowski, P.D. Effects of acute bouts of exercise on cognition. Acta Psychol. 2003, 112, 297–324. [Google Scholar] [CrossRef]
- Nichol, K.; Deeny, S.P.; Seif, J.; Camaclang, K.; Cotman, C.W. Exercise improves cognition and hippocampal plasticity in APOE epsilon4 mice. Alzheimers Dement. 2009, 5, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, M.C. Amyotrophic lateral sclerosis and the neuroprotective potential of exercise. J. Physiol. 2009, 587, 3759–3760. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, J.P.; Silvestre, R.; Pinto, A.C.; de Carvalho, M. Exercise and amyotrophic lateral sclerosis. Neurol. Sci. 2012, 33, 9–15. [Google Scholar] [CrossRef] [PubMed]
- David, F.J.; Robichaud, J.A.; Leurgans, S.E.; Poon, C.; Kohrt, W.M.; Goldman, J.G.; Comella, C.L.; Vaillancourt, D.E.; Corcos, D.M. Exercise improves cognition in Parkinson’s disease: The PRET-PD randomized, clinical trial. Mov. Disord. 2015, 30, 1657–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armon, C. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology 2008, 71, 865–866. [Google Scholar] [CrossRef] [PubMed]
- Bello-Haas, V.D.; Florence, J.M.; Kloos, A.D.; Scheirbecker, J.; Lopate, G.; Hayes, S.M.; Pioro, E.P.; Mitsumoto, H. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology 2007, 68, 2003–2007. [Google Scholar] [CrossRef]
- Dalbello-Haas, V.; Florence, J.M.; Krivickas, L.S. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst. Rev. 2008, CD005229. [Google Scholar] [CrossRef] [Green Version]
- Wing, R.R.; Vazquez, J.A.; Ryan, C.M. Cognitive effects of ketogenic weight-reducing diets. Int. J. Obes. Relat. Metab. Disord. 1995, 19, 811–816. [Google Scholar]
- Ari, C.; Murdun, C.; Goldhagen, C.; Koutnik, A.P.; Bharwani, S.R.; Diamond, D.M.; Kindy, M.; D’Agostino, D.P.; Kovacs, Z. Exogenous Ketone Supplements Improved Motor Performance in Preclinical Rodent Models. Nutrients 2020, 12, 2459. [Google Scholar] [CrossRef]
- Hernandez, A.R.; Hernandez, C.M.; Campos, K.; Truckenbrod, L.; Federico, Q.; Moon, B.; McQuail, J.A.; Maurer, A.P.; Bizon, J.L.; Burke, S.N. A Ketogenic Diet Improves Cognition and Has Biochemical Effects in Prefrontal Cortex That Are Dissociable From Hippocampus. Front. Aging Neurosci. 2018, 10, 391. [Google Scholar] [CrossRef]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Z.; Zuo, Z. Chronic intermittent fasting improves cognitive functions and brain structures in mice. PLoS ONE 2013, 8, e66069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swerdlow, N.S.; Wilkins, H.M. Mitophagy and the Brain. Int. J. Mol. Sci. 2020, 21, 9661. https://doi.org/10.3390/ijms21249661
Swerdlow NS, Wilkins HM. Mitophagy and the Brain. International Journal of Molecular Sciences. 2020; 21(24):9661. https://doi.org/10.3390/ijms21249661
Chicago/Turabian StyleSwerdlow, Natalie S., and Heather M. Wilkins. 2020. "Mitophagy and the Brain" International Journal of Molecular Sciences 21, no. 24: 9661. https://doi.org/10.3390/ijms21249661
APA StyleSwerdlow, N. S., & Wilkins, H. M. (2020). Mitophagy and the Brain. International Journal of Molecular Sciences, 21(24), 9661. https://doi.org/10.3390/ijms21249661