Biased Ligands Differentially Shape the Conformation of the Extracellular Loop Region in 5-HT2B Receptors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Extended Binding Modes of LSD and Ergotamine
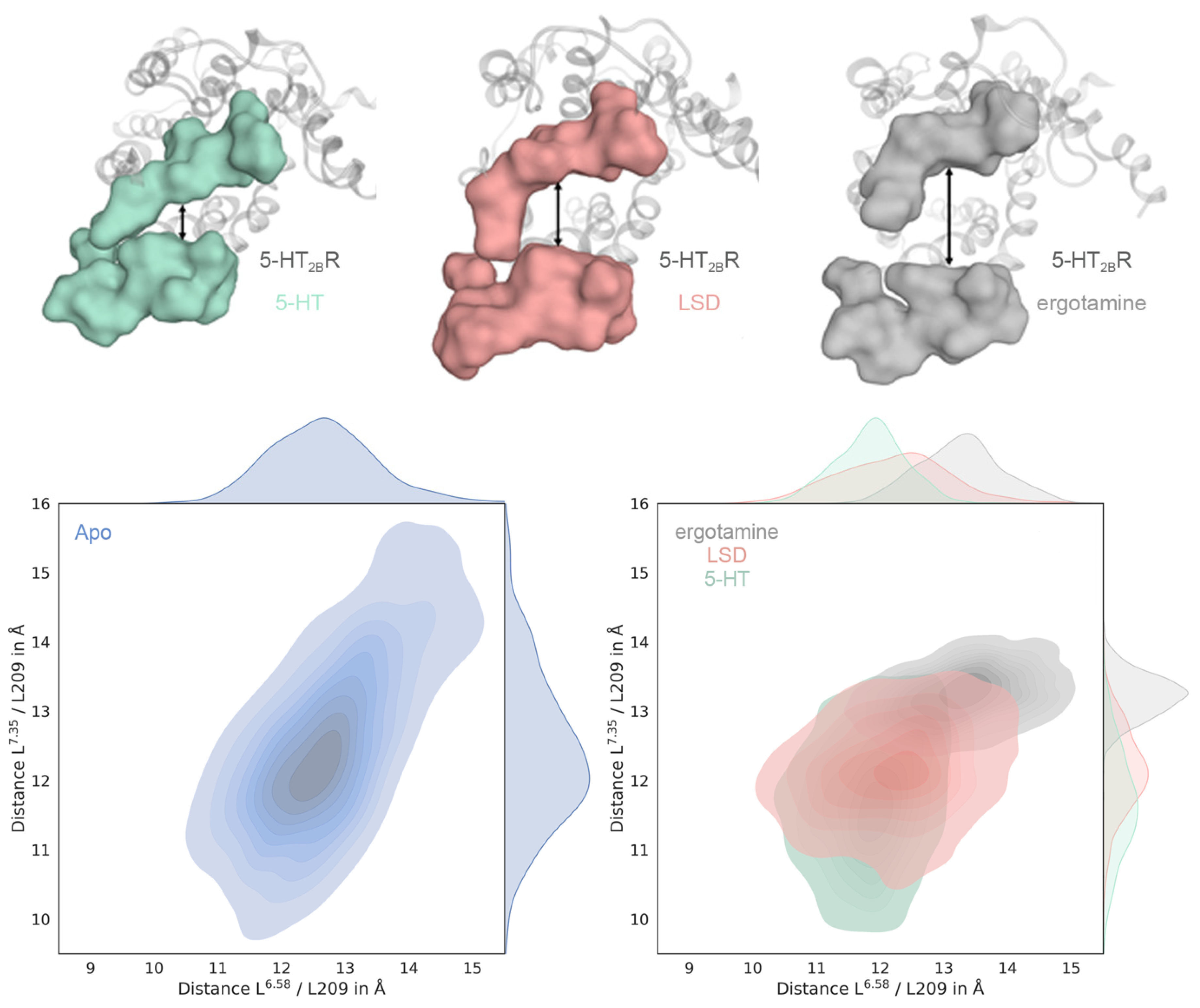
2.2. Ligand-Dependent Shape of the Extracellular Loop Region Is Linked to Biased Properties
2.3. Gq-Biased Ligand LY266097 Shows Stronger Closure of the Extracellular Binding Pocket
2.4. Docking Results Strongly Depend on the Used Receptor Conformation
2.5. Subtype-Specific Differences in Ergotamine Binding
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GPCR | G protein-coupled receptor |
| TM | transmembrane domain |
| ECL | extracellular loop |
| LSD | lysergic acid diethylamide |
| MD | molecular dynamics |
| 5-HT | serotonin |
| PDB | protein data bank |
| MIA | 7-(4-(4-(1-Methyl-1H-indol-4-yl)piperazin-1-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one |
References
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in gpcr drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.; Annibale, P.; Konrad, C.; Hannawacker, A.; Anton, S.E.; Maiellaro, I.; Zabel, U.; Sivaramakrishnan, S.; Falcke, M.; Lohse, M.J. Optical mapping of camp signaling at the nanometer scale. Cell 2020, 182, 1519–1530.e1517. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.A.; Legros, C. The five dimensions of receptor pharmacology exemplified by melatonin receptors: An opinion. Pharmacol. Res. Perspect. 2020, 8, e00556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutkind, J.S.; Kostenis, E. Arrestins as rheostats of gpcr signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Marti-Solano, M.; Crilly, S.E.; Malinverni, D.; Munk, C.; Harris, M.; Pearce, A.; Quon, T.; Mackenzie, A.E.; Wang, X.; Peng, J.; et al. Combinatorial expression of gpcr isoforms affects signalling and drug responses. Nature 2020, 587, 650–656. [Google Scholar] [CrossRef]
- Sommer, M.E.; Selent, J.; Carlsson, J.; De Graaf, C.; Gloriam, D.E.; Keseru, G.M.; Kosloff, M.; Mordalski, S.; Rizk, A.; Rosenkilde, M.M.; et al. The european research network on signal transduction (ernest): Toward a multidimensional holistic understanding of g protein-coupled receptor signaling. Acs Pharmacol. Transl. Sci. 2020, 3, 361–370. [Google Scholar] [CrossRef]
- Sykes, D.A.; Stoddart, L.A.; Kilpatrick, L.E.; Hill, S.J. Binding kinetics of ligands acting at gpcrs. Mol. Cell. Endocrinol. 2019, 485, 9–19. [Google Scholar] [CrossRef]
- Kenakin, T. Biased receptor signaling in drug discovery. Pharmacol. Rev. 2019, 71, 267. [Google Scholar] [CrossRef] [Green Version]
- Ilter, M.; Mansoor, S.; Sensoy, O. Utilization of biased g protein-coupled receptor signaling towards development of safer and personalized therapeutics. Molecules 2019, 24, 2052. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, H.; Fukuchi, M.; Habata, Y. Potential utility of biased gpcr signaling for treatment of psychiatric disorders. Int. J. Mol. Sci. 2019, 20, 3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, M.C.; Charlton, S.J. Biased agonism in drug discovery-is it too soon to choose a path? Mol. Pharmacol. 2018, 93, 259–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermudez, M.; Nguyen, T.N.; Omieczynski, C.; Wolber, G. Strategies for the discovery of biased gpcr ligands. Drug Discov. Today 2019, 24, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.E.; Kelly, E. A biased view of µ opioid receptors? Mol. Pharmacol. 2019, 96, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Kobilka, B.K. The molecular basis of g protein-coupled receptor activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- DeVree, B.T.; Mahoney, J.P.; Velez-Ruiz, G.A.; Rasmussen, S.G.F.; Kuszak, A.J.; Edwald, E.; Fung, J.J.; Manglik, A.; Masureel, M.; Du, Y.; et al. Allosteric coupling from g protein to the agonist-binding pocket in gpcrs. Nature 2016, 535, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Glukhova, A.; Draper-Joyce, C.J.; Sunahara, R.K.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Rules of engagement: Gpcrs and g proteins. ACS Pharmacol. Transl. Sci. 2018, 1, 73–83. [Google Scholar] [CrossRef]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of gpcr signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Bermudez, M.; Bock, A. Does divergent binding pocket closure drive ligand bias for class a gpcrs? Trends Pharmacol. Sci. 2019. [Google Scholar] [CrossRef]
- Bermudez, M.; Bock, A.; Krebs, F.; Holzgrabe, U.; Mohr, K.; Lohse, M.J.; Wolber, G. Ligand-specific restriction of extracellular conformational dynamics constrains signaling of the m-2 muscarinic receptor. ACS Chem. Biol. 2017, 12, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Holze, J.; Bermudez, M.; Pfeil, E.M.; Kauk, M.; Bödefeld, T.; Irmen, M.; Matera, C.; Dallanoce, C.; De Amici, M.; Holzgrabe, U.; et al. Ligand-specific allosteric coupling controls g-protein-coupled receptor signaling. ACS Pharmacol. Transl. Sci. 2020, 3, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.H.; Siu, F.Y.; et al. Structural features for functional selectivity at serotonin receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, D.; Wang, S.; McCorvy, J.D.; Betz, R.M.; Venkatakrishnan, A.J.; Levit, A.; Lansu, K.; Schools, Z.L.; Che, T.; Nichols, D.E.; et al. Crystal structure of an lsd-bound human serotonin receptor. Cell 2017, 168, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Omieczynski, C.; Nguyen, T.N.; Sribar, D.; Deng, L.; Stepanov, D.; Schaller, D.; Wolber, G.; Bermudez, M. Biasdb: A comprehensive database for biased gpcr ligands. bioRxiv 2019, 742643. [Google Scholar]
- Kim, K.; Che, T.; Panova, O.; DiBerto, J.F.; Lyu, J.; Krumm, B.E.; Wacker, D.; Robertson, M.J.; Seven, A.B.; Nichols, D.E.; et al. Structure of a hallucinogen-activated gq-coupled 5-ht2a serotonin receptor. Cell 2020, 182, 1574–1588.e1519. [Google Scholar] [CrossRef]
- McCorvy, J.D.; Wacker, D.; Wang, S.; Agegnehu, B.; Liu, J.; Lansu, K.; Tribo, A.R.; Olsen, R.H.J.; Che, T.; Jin, J.; et al. Structural determinants of 5-ht2b receptor activation and biased agonism. Nat. Struct. Mol. Biol. 2018, 25, 787–796. [Google Scholar] [CrossRef]
- Yin, J.; Chen, K.-Y.M.; Clark, M.J.; Hijazi, M.; Kumari, P.; Bai, X.-c.; Sunahara, R.K.; Barth, P.; Rosenbaum, D.M. Structure of a d2 dopamine receptor–g-protein complex in a lipid membrane. Nature 2020, 584, 125–129. [Google Scholar] [CrossRef]
- Crews, C. Analysis of ergot alkaloids. Toxins 2015, 7, 2024–2050. [Google Scholar] [CrossRef] [Green Version]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3d pharmacophore modeling. Wires Comput. Mol. Sci. 2020, e1468. [Google Scholar] [CrossRef] [Green Version]
- Vass, M.; Podlewska, S.; de Esch, I.J.P.; Bojarski, A.J.; Leurs, R.; Kooistra, A.J.; de Graaf, C. Aminergic gpcr–ligand interactions: A chemical and structural map of receptor mutation data. J. Med. Chem. 2019, 62, 3784–3839. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Penner, U.; Dorow, R.; Pertz, H.H.; Jähnichen, S.; Horowski, R.; Latté, K.P.; Palla, D.; Schurad, B. Lisuride, a dopamine receptor agonist with 5-ht2b receptor antagonist properties: Absence of cardiac valvulopathy adverse drug reaction reports supports the concept of a crucial role for 5-ht2b receptor agonism in cardiac valvular fibrosis. Clin. Neuropharmacol. 2006, 29, 80–86. [Google Scholar] [CrossRef] [PubMed]
- McCorvy, J.D.; Butler, K.V.; Kelly, B.; Rechsteiner, K.; Karpiak, J.; Betz, R.M.; Kormos, B.L.; Shoichet, B.K.; Dror, R.O.; Jin, J.; et al. Structure-inspired design of beta-arrestin-biased ligands for aminergic gpcrs. Nat. Chem. Biol. 2018, 14, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, M.; Rakers, C.; Wolber, G. Structural characteristics of the allosteric binding site represent a key to subtype selective modulators of muscarinic acetylcholine receptors. Mol. Inform. 2015, 34, 526–530. [Google Scholar] [CrossRef]
- Rataj, K.; Kelemen, Á.A.; Brea, J.; Loza, M.I.; Bojarski, A.J.; Keserű, G.M. Fingerprint-based machine learning approach to identify potent and selective 5-ht2br ligands. Molecules 2018, 23, 1137. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Espigares, I.; Torrens-Fontanals, M.; Tiemann, J.K.S.; Aranda-García, D.; Ramírez-Anguita, J.M.; Stepniewski, T.M.; Worp, N.; Varela-Rial, A.; Morales-Pastor, A.; Medel-Lacruz, B.; et al. Gpcrmd uncovers the dynamics of the 3d-gpcrome. Nat. Methods 2020, 17, 777–787. [Google Scholar] [CrossRef]
- Torrens-Fontanals, M.; Stepniewski, T.M.; Aranda-García, D.; Morales-Pastor, A.; Medel-Lacruz, B.; Selent, J. How do molecular dynamics data complement static structural data of gpcrs. Int. J. Mol. Sci. 2020, 21, 5933. [Google Scholar] [CrossRef]
- Martí-Solano, M.; Iglesias, A.; de Fabritiis, G.; Sanz, F.; Brea, J.; Loza, M.I.; Pastor, M.; Selent, J. Detection of new biased agonists for the serotonin 5-ht2b receptor: Modeling and experimental validation. Mol. Pharmacol. 2015, 87, 740. [Google Scholar] [CrossRef] [Green Version]
- Labute, P. Protonate3d: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins Struct. Funct. Bioinform. 2009, 75, 187–205. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking11edited by f. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3d pharmacophores. J. Comput. Aided Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. Ligandscout: 3-d pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of mmff94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. Opm database and ppm web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- McKinney, W. Data structures for statistical computing in python. SciPy 2010, 445, 51–56. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2d graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.; Botvinnik, O.; Gelbart, M.; Ostblom, J.; Hobson, P.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Warmenhoven, J.; et al. Mwaskom/Seaborn: V0.11.0 (sepetmber 2020); Zenodo: New York City, NY, USA, 2020. [Google Scholar]
- Bock, A.; Bermudez, M.; Krebs, F.; Matera, C.; Chirinda, B.; Sydow, D.; Dallanoce, C.; Holzgrabe, U.; De Amici, M.; Lohse, M.J.; et al. Ligand binding ensembles determine graded agonist efficacies at a g protein-coupled receptor. J. Biol. Chem. 2016, 291, 16375–16389. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Machalz, D.; Yan, Q.; Sorensen, E.J.; Wolber, G.; Bureik, M. Importance of asparagine-381 and arginine-487 for substrate recognition in cyp4z1. Biochem. Pharmacol. 2020, 174, 113850. [Google Scholar] [CrossRef]
- Mortier, J.; Prévost, J.R.C.; Sydow, D.; Teuchert, S.; Omieczynski, C.; Bermudez, M.; Frédérick, R.; Wolber, G. Arginase structure and inhibition: Catalytic site plasticity reveals new modulation possibilities. Sci. Rep. 2017, 7, 13616. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denzinger, K.; Nguyen, T.N.; Noonan, T.; Wolber, G.; Bermudez, M. Biased Ligands Differentially Shape the Conformation of the Extracellular Loop Region in 5-HT2B Receptors. Int. J. Mol. Sci. 2020, 21, 9728. https://doi.org/10.3390/ijms21249728
Denzinger K, Nguyen TN, Noonan T, Wolber G, Bermudez M. Biased Ligands Differentially Shape the Conformation of the Extracellular Loop Region in 5-HT2B Receptors. International Journal of Molecular Sciences. 2020; 21(24):9728. https://doi.org/10.3390/ijms21249728
Chicago/Turabian StyleDenzinger, Katrin, Trung Ngoc Nguyen, Theresa Noonan, Gerhard Wolber, and Marcel Bermudez. 2020. "Biased Ligands Differentially Shape the Conformation of the Extracellular Loop Region in 5-HT2B Receptors" International Journal of Molecular Sciences 21, no. 24: 9728. https://doi.org/10.3390/ijms21249728
APA StyleDenzinger, K., Nguyen, T. N., Noonan, T., Wolber, G., & Bermudez, M. (2020). Biased Ligands Differentially Shape the Conformation of the Extracellular Loop Region in 5-HT2B Receptors. International Journal of Molecular Sciences, 21(24), 9728. https://doi.org/10.3390/ijms21249728