Full-Length Transcriptome Assembly of Italian Ryegrass Root Integrated with RNA-Seq to Identify Genes in Response to Plant Cadmium Stress

Abstract
:1. Introduction
2. Results
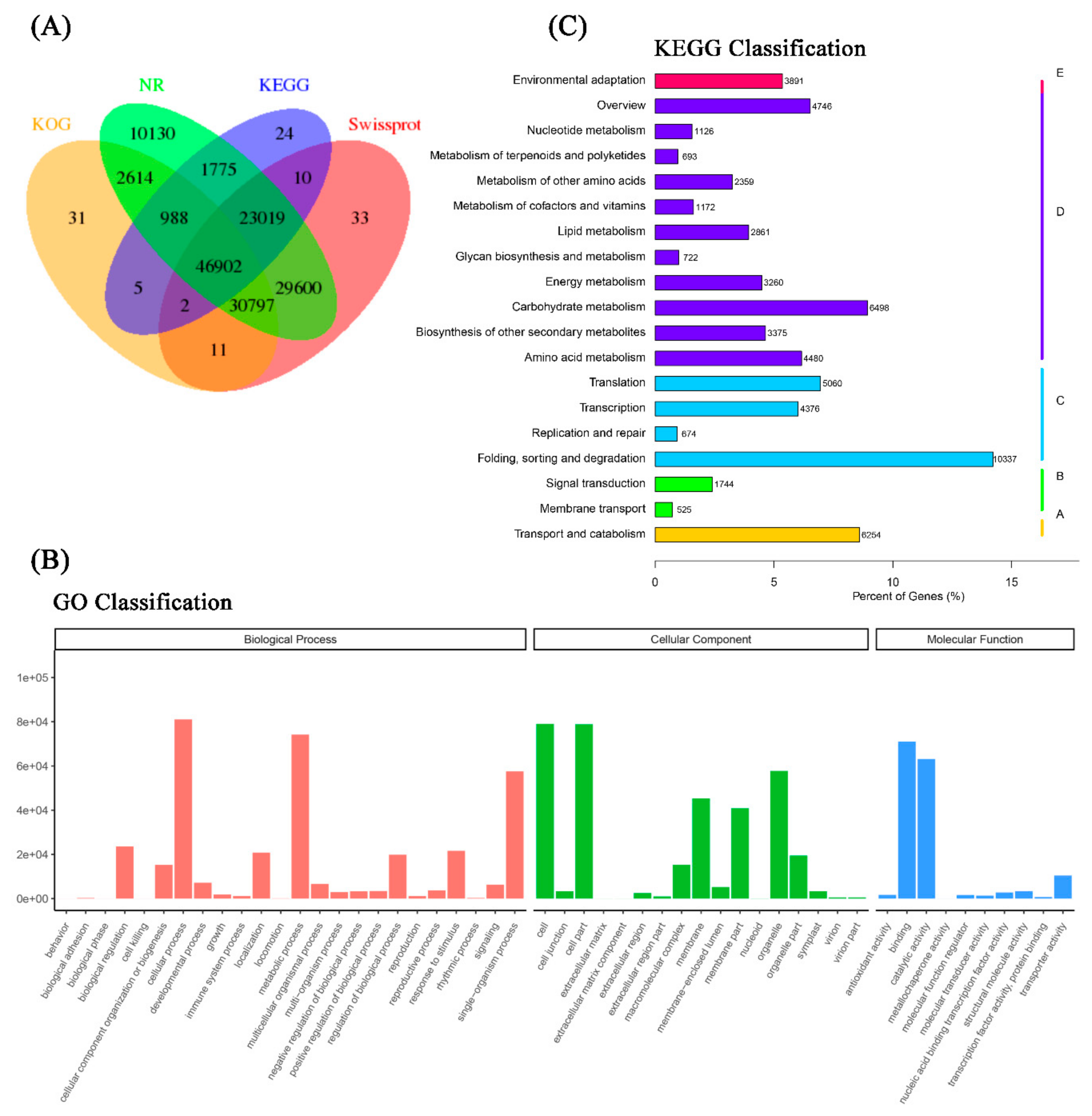
2.1. Assembly of the Full-Length Reference Transcriptomic Database of Italian Ryegrass
2.2. Annotation of the Full Length Reference Transcriptome
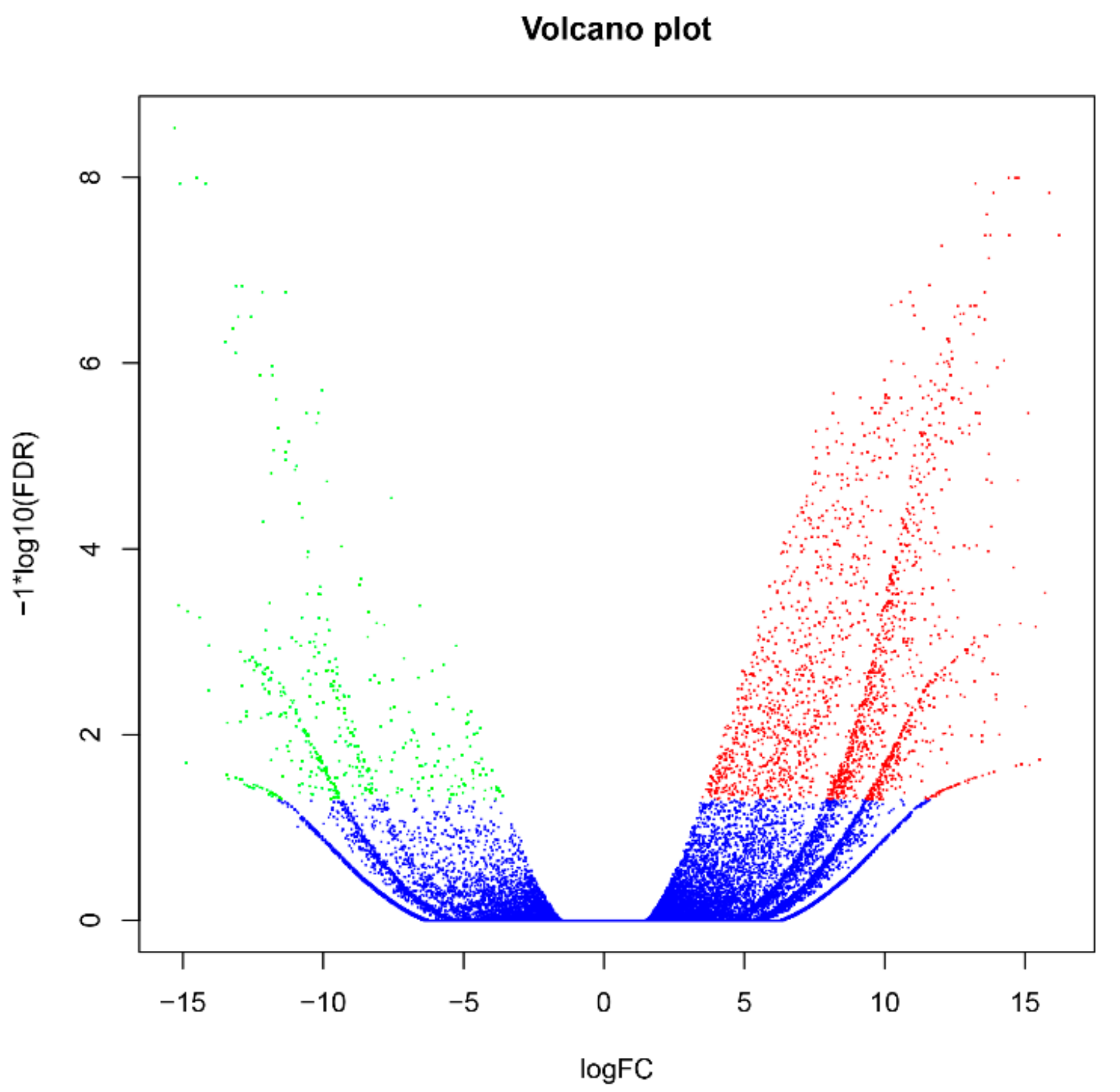
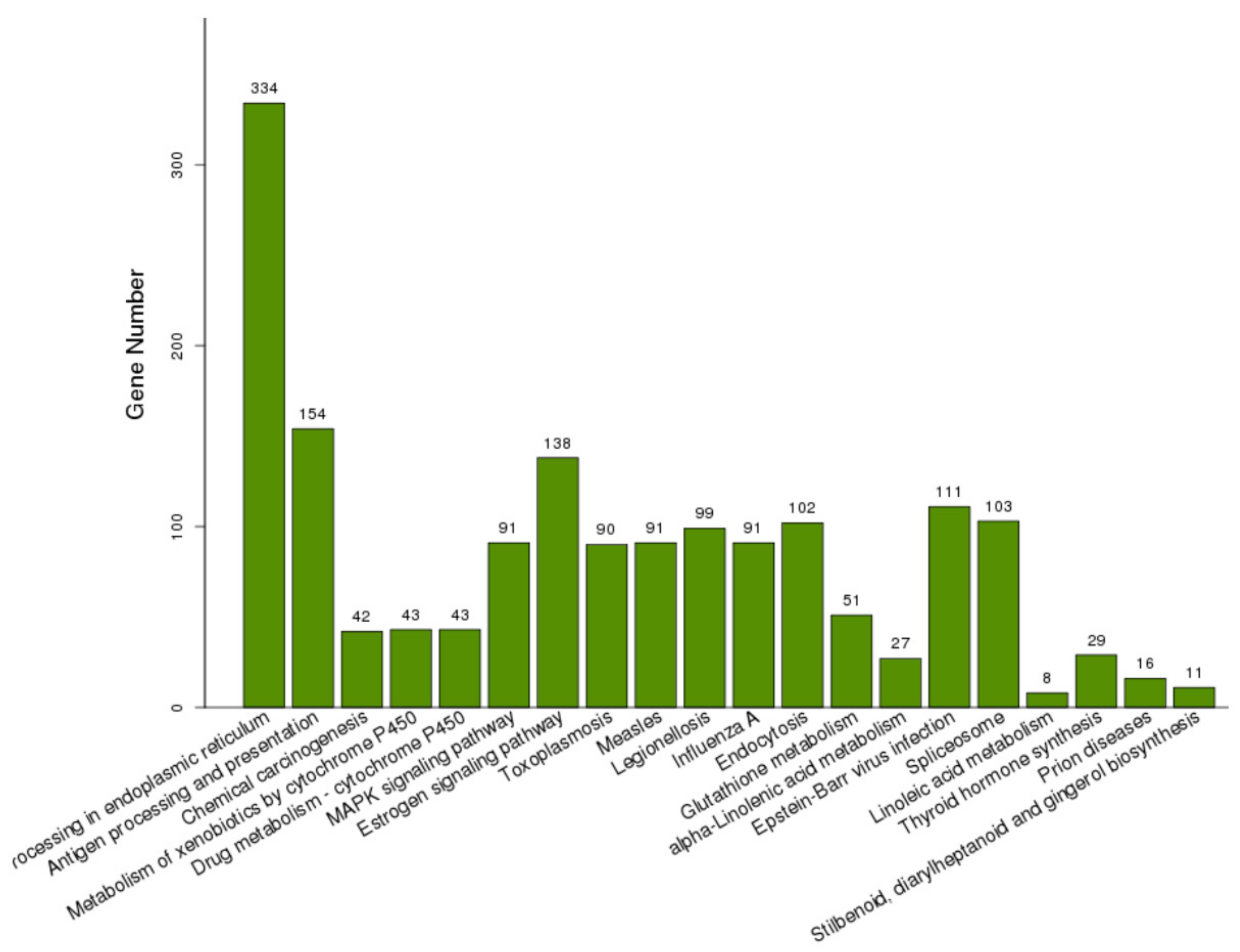
2.3. Identification and Functional Profiles of Differentially Expressed Genes
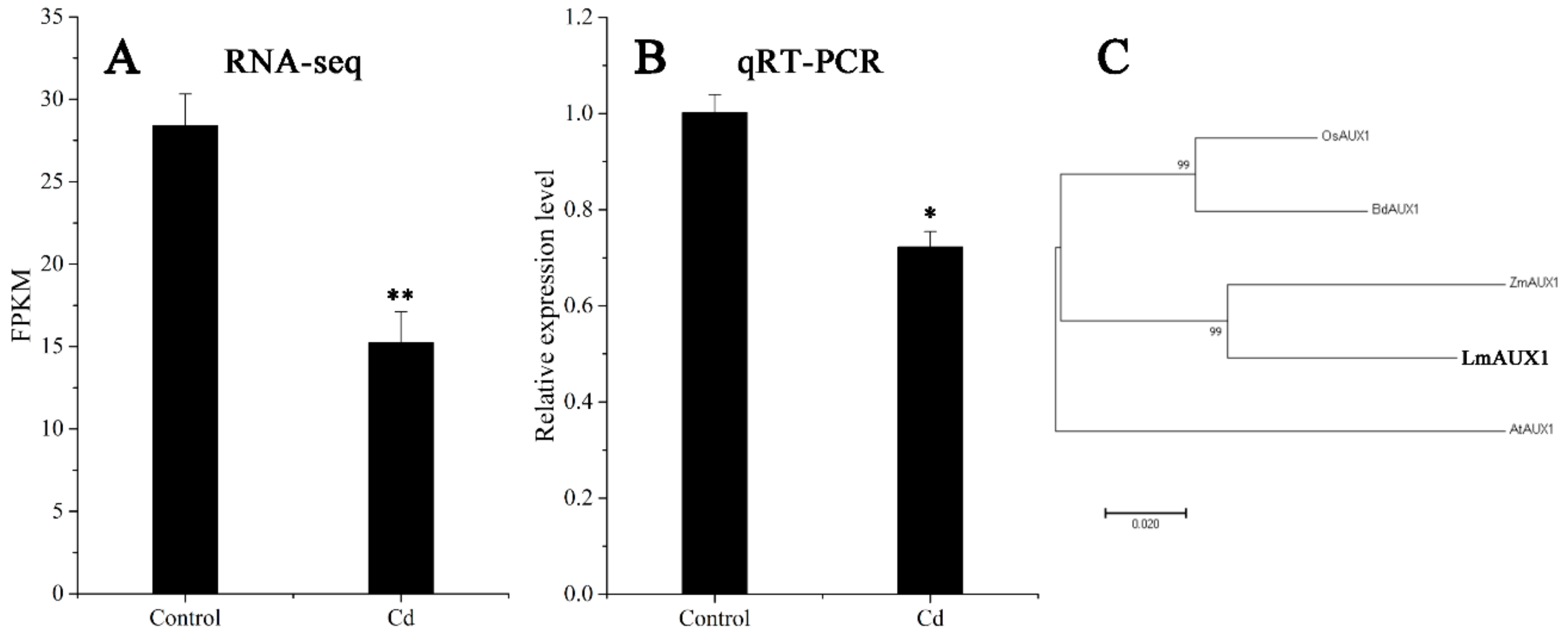
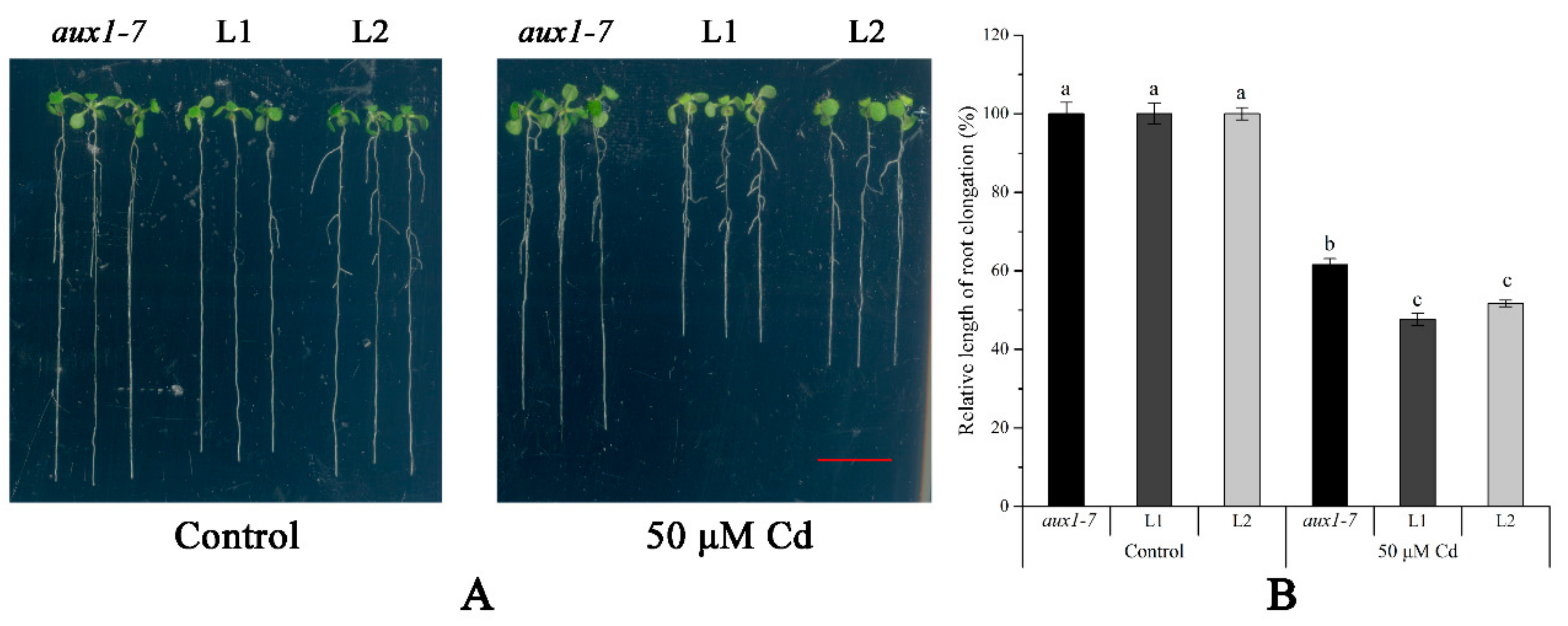
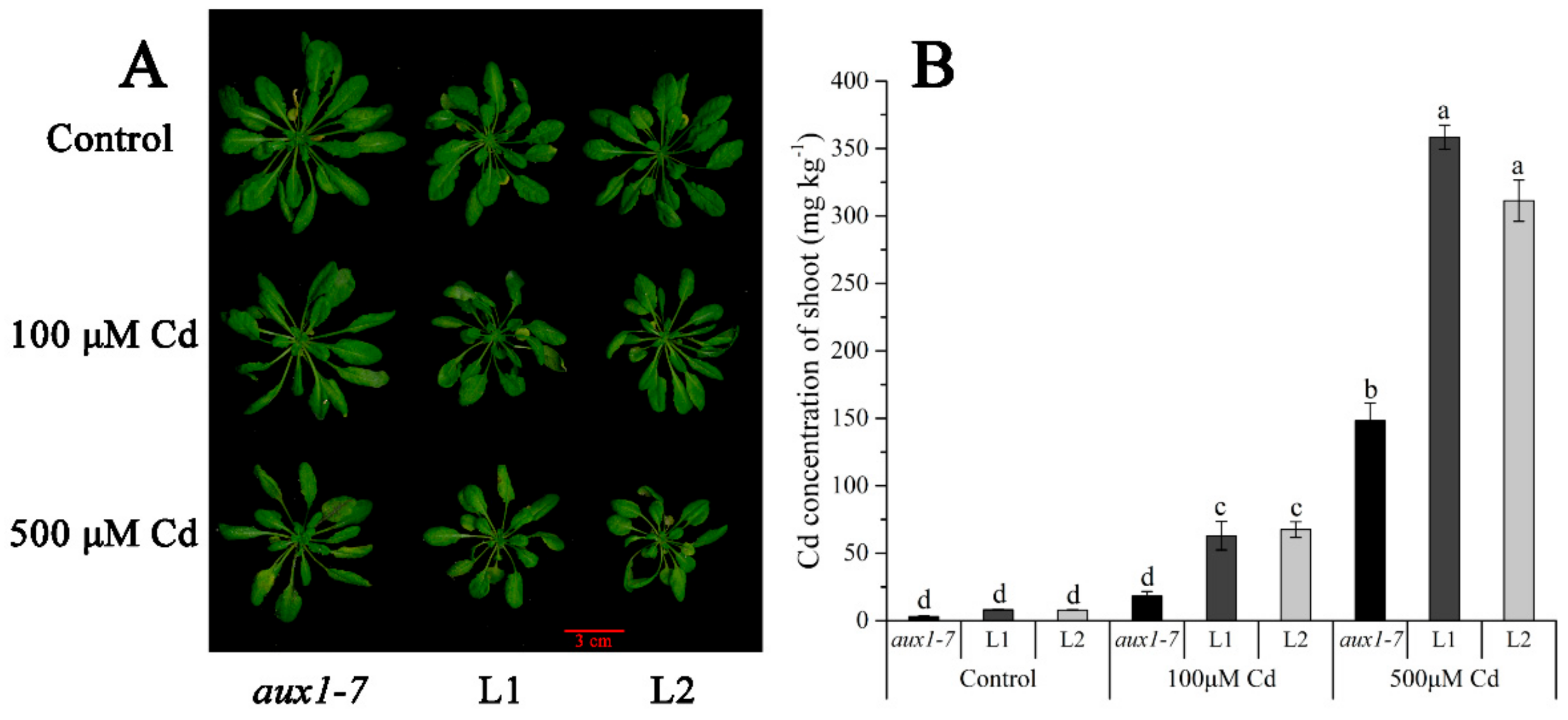
2.4. Functional Validation of LmAUX1 in Response to Cd
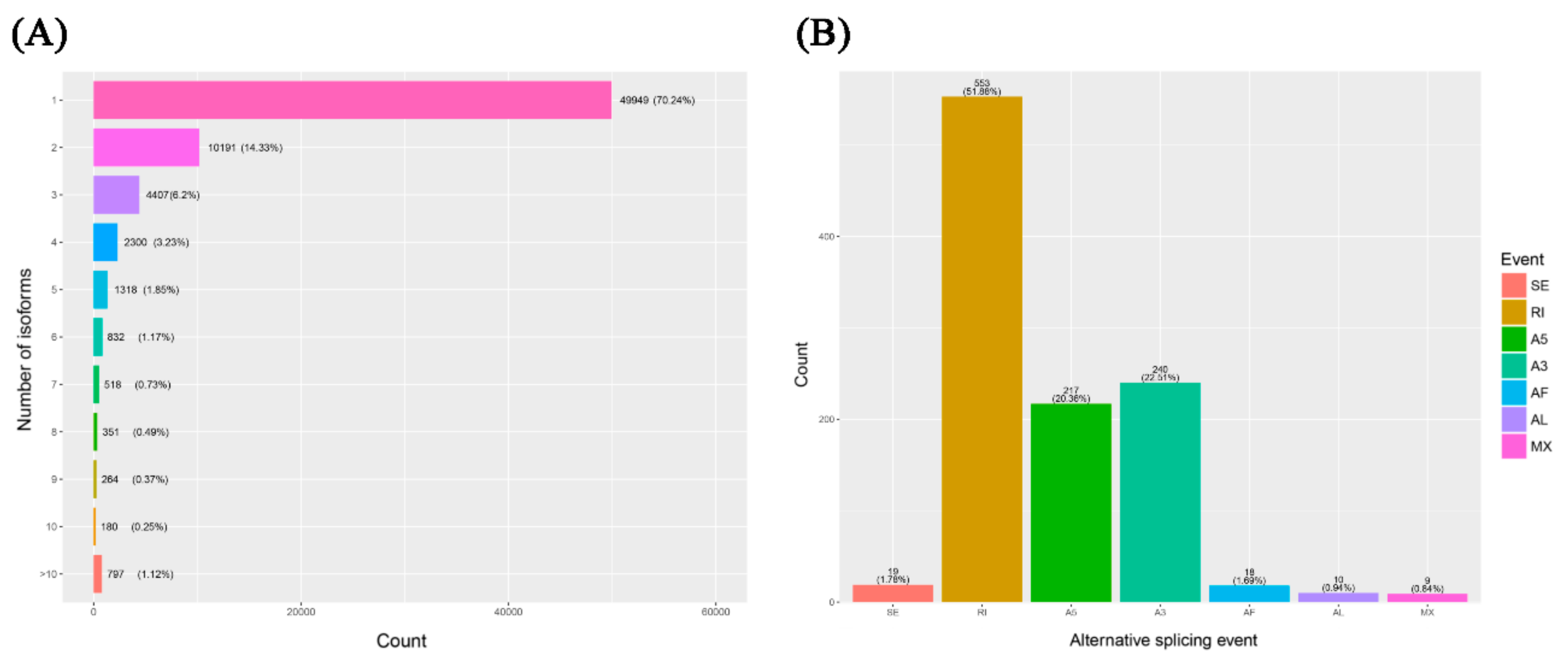
2.5. Alternative Splicing Identification
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Growth Conditions, and Treatment
4.2. RNA Isolation, RNA-seq, and PacBio Full-Length ISO-SeqLibrary Preparation and Sequencing
4.3. Assembly of Reference Transcriptomic Database
4.4. Annotation of Gene Function
4.5. Quantification of Gene Expression Levels
4.6. Identification and Function Assessment of DEGs
4.7. Validation of DEGs with qRT-PCR
4.8. Vector Construction and Ectopic Overexpression of LmAUX1 in Arabidopsis Thaliana
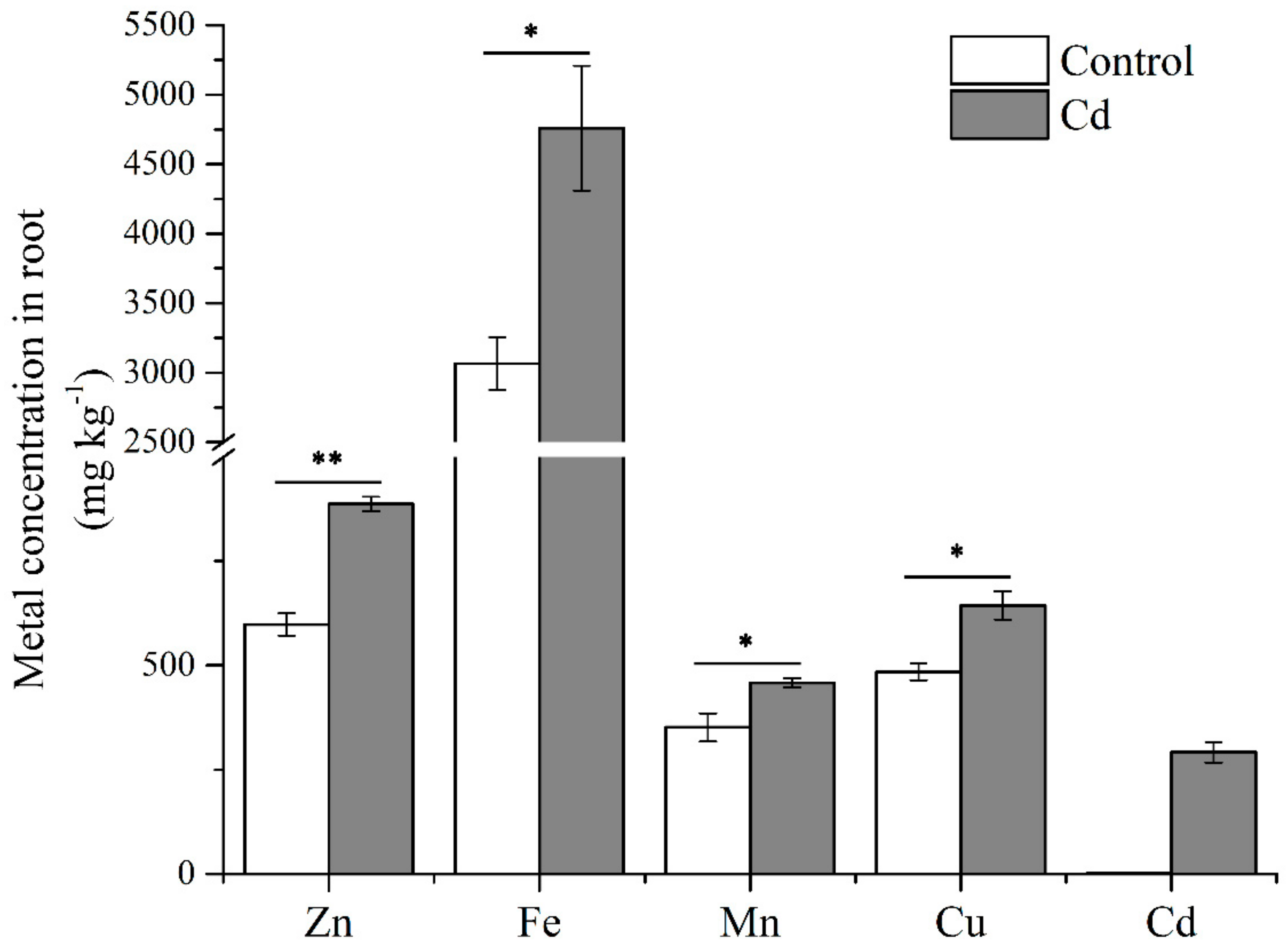
4.9. Determination of Metal Concentrations
4.10. Microscopic Imaging of GUS Staining and Fluorescence of DII-VENUS
4.11. Alternative Splicing Identification
4.12. Accession Numbers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Charmet, G.; Balfourier, F.; Chatard, V. Taxonomic relationships and interspecific hybridization in the genus Lolium (grasses). Genet. Resour. Crop Evolut. 1996, 43, 319–327. [Google Scholar] [CrossRef]
- Aguirre, A.A.; Studer, B.; Frei, U.L.; übberstedt, T. Prospects for hybrid breeding in bioenergy grasses. BioEnergy Res. 2012, 5, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Fè, D.; Ashraf, B.; Byrne, S.; Czaban, A.; Roulund, N.; Lenk, I.; Asp, T. Prospects for introducing genomic selection info forage grass breeding. In EGF at 50: The Future of European Grasslands. In Proceedings of the 25th General Meeting of the European Grassland Federation, Aberystwyth, Wales, 7–11 September 2014; Centre for Agriculture and Biosciences International: Wallingford, UK, 2014; pp. 830–832. Available online: https://www.cabdirect.org/cabdirect/abstract/20143369178 (accessed on 7 September 2014).
- Byrne, S.L.; Nagy, I.; Pfeifer, M.; Armstead, I.; Swain, S.; Studer, B.; Mayer, K.; Campbell, J.D.; Czaban, A.; Hentrup, S.; et al. A synteny-based draft genome sequence of the forage grass Lolium perenne. Plant J. 2015, 84, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Stewart, A.; Cai, H.W. Italian ryegrass. In Genetics, Genomics and Breeding of Forage Crops; Kole, C., Yamada, T., Cai, H.W., Eds.; CRC Press: Boca Raton, FL, USA, 2013; pp. 36–57. [Google Scholar] [CrossRef]
- Yasuda, M.; Takenouchi, Y.; Nitta, Y.; Ishii, Y.; Ohta, K. Italian ryegrass (Lolium multiflorum Lam) as a high-potential bio-ethanol resource. BioEnergy Res. 2015, 8, 1303–1309. [Google Scholar] [CrossRef]
- Zhu, T.; Fu, D.; Yang, F. Effect of saponin on the phytoextraction of Pb, Cd and Zn from soil using Italian ryegrass. Bull. Environ. Contam. Toxicol. 2015, 94, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Daisei, U.; Naoki, Y.; Izumi, K.; Feng, H.C.; Tsuyu, A.; Masahiro, Y.; Ma, J.F. Gene limiting cadmium accumulation in rice. Proc. Natl. Acad. Sci. USA 2010, 107, 16500–16505. [Google Scholar] [CrossRef] [Green Version]
- Clemens, S.; Ma, J.F. Toxic heavy metal and metalloid accumulation in crop plants and foods. Annu. Rev. Plant Biol. 2016, 67, 489–512. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Chen, H.; Kopittke, P.M.; Zhao, F.J. Cadmium contamination in agricultural soils of China and the impact on food safety. Environ. Pollut. 2019, 249, 1038–1048. [Google Scholar] [CrossRef]
- Rani, A.; Kumar, A.; Lal, A.; Pant, M. Cellular mechanisms of cadmium-induced toxicity: A review. Int. J. Environ. Health Res. 2014, 24, 378–399. [Google Scholar] [CrossRef]
- Zhang, L.; Pei, Y.; Wang, H.; Jin, Z.; Liu, Z.; Qiao, Z.; Fang, H.; Zhang, Y. Hydrogen sulfide alleviates cadmium-induced cell death through restraining ROS accumulation in roots of Brassica rapa L. Ssp. Pekinensis. Oxid. Med. Cell. Longev. 2015, 2015, 804603. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.K.; Pena, L.B.; Romero-Puertas, M.C.; Hernandez, A.; Inouhe, M.; Sandalio, L.M. NADPH oxidases differentially regulate ROS metabolism and nutrient uptake under cadmium toxicity. Plant Cell Environ. 2016, 40, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Ali, S.; Abbas, T.; Zia-Ur-Rehman, M.; Hannan, F.; Keller, C.; Al-Wabel, M.I.; Ok, Y.S. Cadmium minimization in wheat: A critical review. Ecotoxicol. Environ. Saf. 2016, 130, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Aarts, M.G.M. The molecular mechanism of zinc and cadmium stress response in plants. Cell. Mol. Life Sci. 2012, 69, 3187–3206. [Google Scholar] [CrossRef]
- Tao, Q.; Jupa, R.; Liu, Y.; Luo, J.; Li, J.; Kováč, J.; Ki, B.; Li, Q.; Wu, K.; Liang, Y.; et al. Abscisic acid-mediated modifications of radial apoplastic transport pathway play a key role in cadmium uptake in hyperaccumulator Sedum alfredii. Plant Cell Environ. 2019, 42, 1425–1440. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Ishimaru, Y.; Igura, M.; Kuramata, M.; Abe, T.; Senoura, T.; Hase, Y.; Arao, T.; Nishizawa, N.K.; Nakanishi, H. Ion-beam irradiation, gene identification, and marker-assisted breeding in the development of low-cadmium rice. Proc. Natl. Acad. Sci. USA 2012, 109, 19166–19171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.S.; Huang, J.; Zeng, D.L.; Peng, J.S.; Zhang, G.B.; Ma, H.L.; Guan, Y.; Yi, H.Y.; Fu, Y.L.; Han, B.; et al. A defensin-like protein drives cadmium efflux and allocation in rice. Nat. Commun. 2018, 9, 645. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, P.; Zanella, L.; De Paolis, A.; Di Litta, D.; Cecchetti, V.; Falasca, G.; Barbieri, M.; Altamura, M.M.; Costantino, P.; Cardarelli, M. Cadmium-inducible expression of the ABC-type transporter AtABCC3 increases phytochelatin-mediated cadmium tolerance in Arabidopsis. J. Exp. Bot. 2015, 66, 3815–3829. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Yuan, S.; Wen, X.; Xie, Z.; Lou, L.; Hu, B.; Cai, Q.; Xu, B. Transcriptome analysis of Cd-treated switchgrass root revealed novel transcripts and the importance of HSF/HSP network in switchgrass Cd tolerance. Plant Cell Rep. 2018, 37, 1485–1497. [Google Scholar] [CrossRef]
- He, F.; Liu, Q.; Zheng, L.; Cui, Y.; Shen, Z.; Zheng, L. RNA-Seq analysis of rice roots reveals the involvement of post-transcriptional regulation in response to cadmium stress. Front. Plant Sci. 2015, 6, 1136. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Sun, L.; Yang, X.; Liu, J.X. Transcriptomic analysis of cadmium stress response in the heavy metal hyperaccumulator Sedum alfredii Hance. PLoS ONE 2014, 8, e64643. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Sun, M.; Wang, J.; Lei, M.; Li, C.; Zhao, D.; Huang, J.; Li, W.; Li, S.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2018, 97, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Gao, Y.; Wu, L.; Wang, L.; Zhang, T.; Dai, C.; Xu, W.; Feng, L.; Ma, M.; Zhu, Y.G.; et al. Potential use of the Pteris vittata arsenic hyperaccumulation-regulation network for phytoremediation. J. Hazard. Mater. 2019, 368, 386–396. [Google Scholar] [CrossRef]
- Wang, J.; Deng, Y.; Zhou, Y.; Liu, D.; Yu, H.; Zhou, Y.; Lv, J.; Ou, L.; Li, X.; Ma, Y.; et al. Full-length mRNA sequencing and gene expression profiling reveal broad involvement of natural antisense transcript gene pairs in pepper development and response to stresses. Plant J. 2019. [Google Scholar] [CrossRef]
- Yuan, H.; Yu, H.; Huang, T.; Shen, X.; Xia, J.; Pang, F.; Wang, J.; Zhao, M. The complexity of the Fragaria x ananassa (octoploid) transcriptome by single-molecule long-read sequencing. Hortic. Res. 2019, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Anvar, S.Y.; Allard, G.; Tseng, E.; Sheynkman, G.M.; de Klerk, E.; Vermaat, M.; Yin, R.H.; Johansson, H.E.; Ariyurek, Y.; den Dunnen, J.T.; et al. Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing. Genome Biol. 2018, 19, 46. [Google Scholar] [CrossRef] [Green Version]
- Steijger, T.; Abril, J.F.; Engstrom, P.G.; Kokocinski, F.; Hubbard, T.; Guigo, R.; Harrow, J.; Bertone, P. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 2013, 10, 1177–1184. [Google Scholar] [CrossRef] [Green Version]
- Hirata, M.; Cai, H.; Inoue, M.; Yuyama, N.; Miura, Y.; Komatsu, T.; Takamizo, T.; Fujimori, M. Development of simple sequence repeat (SSR) markers and construction of an SSR-based linkage map in Italian ryegrass (Lolium multiflorum Lam.). Theor. Appl. Genet. 2006, 113, 270–279. [Google Scholar] [CrossRef]
- Lee, D.K.; Aberle, E.; Anderson, E.K.; Anderson, W.; Baldwin, B.S.; Baltensperger, D.; Barrett, M.; Blumenthal, J.; Bonos, S.; Bouton, J. Biomass production of herbaceous energy crops in the United States: Field trial results and yield potential maps from the multiyear regional feedstock partnership. GCB Bioenergy 2018, 10, 698–716. [Google Scholar] [CrossRef]
- Duhoux, A.; Carrere, S.; Gouzy, J.; Bonin, L.; Delye, C. RNA-Seq analysis of rye-grass transcriptomic response to an herbicide inhibiting acetolactate-synthase identifies transcripts linked to non-target-site-based resistance. Plant Mol. Biol. 2015, 87, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Knorst, V.; Byrne, S.; Yates, S.; Asp, T.; Widmer, F.; Studer, B.; Kolliker, R. Pooled DNA sequencing to identify SNPs associated with a major QTL for bacterial wilt resistance in Italian ryegrass (Lolium multiflorum Lam.). Theor. Appl. Genet. 2019, 132, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M. A next-generation approach to the characterization of a non-model plant transcriptome. Curr. Sci. 2011, 101, 1435–1439. [Google Scholar]
- Chmielowska-Bąk, J.; Gzyl, J.A.; Rucińska-Sobkowiak, R.; Arasimowicz-Jelonek, M.; Deckert, J. The new insights into cadmium sensing. Front. Plant Sci. 2014, 5, 245. [Google Scholar] [CrossRef]
- Braconi, D.; Bernardini, G.; Santucci, A. Linking protein oxidation to environmental pollutants: Redox proteomic approaches. J. Proteom. 2011, 74, 2324–2337. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef]
- Shim, D.; Hwang, J.U.; Lee, J.; Lee, S.; Choi, Y.; An, G.; Martinoia, E.; Lee, Y. Orthologs of the class A4 heat shock transcription factor HsfA4a confer cadmium tolerance in wheat and rice. Plant Cell 2009, 21, 4031–4043. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.Y.; Zhang, Y.; Xu, Y.P.; Qi, Z.Y.; Li, M.Q.; Ahammed, G.J.; Xia, X.J.; Shi, K.; Zhou, Y.H.; Reiter, R.J. HsfA1a upregulates melatonin biosynthesis to confer cadmium tolerance in tomato plants. J. Pineal Res. 2017, 62, e12387. [Google Scholar] [CrossRef]
- Tamás, M.; Sharma, S.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 2014, 4, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Cui, F.; Li, Q.; Yin, B.; Zhang, H.; Lin, B.; Wu, Y.; Xia, R.; Tang, S.; Xie, Q. The endoplasmic reticulum-associated degradation is necessary for plant salt tolerance. Cell Res. 2011, 21, 957. [Google Scholar] [CrossRef] [Green Version]
- Piper, P.; Truman, A.; Millson, S.; Nuttall, J. Hsp90 chaperone control over transcriptional regulation by the yeast Slt2(Mpk1)p and human ERK5 mitogen-activated protein kinases (MAPKs). Biochem. Soc. Trans. 2006, 34, 783–785. [Google Scholar] [CrossRef]
- Rodriguez, F.; Arsène-Ploetze, F.; Rist, W.; Rüdiger, S.; Schneider-Mergener, J.; Mayer, M.P.; Bukau, B. Molecular basis for regulation of the heat shock transcription factor σ32 by the DnaK and DnaJ chaperones. Mol. Cell 2008, 32, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The functions of metallothionein and ZIP and ZnT transporters: An overview and perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cao, Q.; Jiang, Q.; Li, J.; Yu, R.; Shi, G. Comparative transcriptome analysis reveals gene network regulating cadmium uptake and translocation in peanut roots under iron deficiency. BMC Plant Biol. 2019, 19, 35. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Trinh, N.N.; Fu, S.F.; Hsiung, Y.C.; Chia, L.C.; Lin, C.W.; Huang, H.J. Comparison of early transcriptome responses to copper and cadmium in rice roots. Plant Mol. Biol. 2013, 81, 507–522. [Google Scholar] [CrossRef]
- Yuan, H.M.; Huang, X. Inhibition of root meristem growth by cadmium involves nitric oxide-mediated repression of auxin accumulation and signalling in Arabidopsis. Plant Cell Environ. 2016, 39, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.F.; Zhou, G.; Na, X.F.; Yang, L.; Nan, W.B.; Liu, X.; Zhang, Y.Q.; Li, J.L.; Bi, Y.R. Cadmium interferes with maintenance of auxin homeostasis in Arabidopsis seedlings. J. Plant Physiol. 2013, 170, 965–975. [Google Scholar] [CrossRef]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape. Arabidopsis Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Xia, Z.; Luo, Y.; Jiang, X.; Qian, B.; Xie, H.; Zhu, J.K.; Xiong, L.; Zhu, J.; Wang, Z.Y. Spliceosomal protein U1A is involved in alternative splicing and salt stress tolerance in Arabidopsis thaliana. Nucleic Acids Res. 2018, 46, 1777–1792. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; He, F.; Berkowitz, O.; Liu, J.; Cao, P.; Tang, M.; Shi, H.; Wang, W.; Li, Q.; Shen, Z.; et al. Alternative splicing plays a critical role in maintaining mineral nutrient homeostasis in rice (Oryza sativa). Plant Cell 2018, 30, 2267–2285. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef]
- Zuo, C.; Blow, M.; Sreedasyam, A.; Kuo, R.C.; Ramamoorthy, G.K.; Torres-Jerez, I.; Li, G.; Wang, M.; Dilworth, D.; Barry, K.; et al. Revealing the transcriptomic complexity of switchgrass by PacBio long-read sequencing. Biotechnol. Biofuels 2018, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.J.; Liu, X.; Pires, J.C.; Soltis, P.S.; Soltis, D.E. Nonadditive gene expression in polyploids. Annu. Rev. Genet. 2014, 48, 485–517. [Google Scholar] [CrossRef]
- Fang, Z.; Lou, L.; Tai, Z.; Wang, Y.; Yang, L.; Hu, Z.; Cai, Q. Comparative study of Cd uptake and tolerance of two Italian ryegrass (Lolium multiflorum) cultivars. Peer J. 2017, 5, e3621. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Feng, S.; Liu, Y.; Zhao, L.; Tian, L.; Hu, Y.; Yang, T.; Wei, A. Single-molecule long-read sequencing of Zanthoxylum bungeanum maxim. Transcriptome: Identification of aroma-related genes. Forests 2018, 9, 765. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Tang, Q.; Wu, M.; Mou, D.; Liu, H.; Wang, S.; Zhang, C.; Ding, L.; Luo, J. Comparative transcriptomics provides novel insights into the mechanisms of selenium tolerance in the hyperaccumulator plant Cardamine hupingshanensis. Sci. Rep. 2018, 8, 2789. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.D.; Reeder, J.; Lawrence, M.; Becker, G.; Brauer, M.J. GMAP and GSNAP for genomic sequence alignment: Enhancements to speed, accuracy, and functionality. In Statistical Genomics; Humana Press: New York, NY, USA, 2016; pp. 283–334. [Google Scholar] [CrossRef]
- Alamancos, G.P.; Pagès, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Annotated Number | Length 0–<1000 | Length 1000–<2000 | Length 2000–<3000 | Length 3000–<6000 | Length ≥6000 |
|---|---|---|---|---|---|---|
| GO | 123,344 | 5759 | 59,645 | 21,204 | 33,834 | 2902 |
| KEGG | 72,725 | 3579 | 38,770 | 11,890 | 16,983 | 1503 |
| KOG | 81,350 | 3319 | 41,539 | 13,234 | 21,480 | 1778 |
| NR | 145,825 | 7126 | 72,604 | 24,372 | 38,526 | 3197 |
| NT | 139,068 | 6697 | 68,837 | 23,560 | 36,922 | 3052 |
| Swiss-Prot | 130,374 | 5837 | 64,719 | 22,006 | 34,869 | 2943 |
| All | 146,545 | 7269 | 72,943 | 24,448 | 38,678 | 3207 |
| Sample Name | Total Reads | Mapped Reads (%) | Unique Mapped Reads (%) | Multi Mapped Reads (%) |
|---|---|---|---|---|
| Control-1 | 96,605,492 | 66,392,568 (68.73%) | 6,831,848 (10.29%) | 59,560,720 (89.71%) |
| Control-2 | 97,407,032 | 67,836,176 (69.64%) | 6,790,096 (10.01%) | 61,046,080 (89.99%) |
| Control-3 | 78,680,946 | 56,203,632 (71.43%) | 5,183,088 (9.22%) | 51,020,544 (90.78%) |
| Cd-1 | 102,165,716 | 71,120,258 (69.61%) | 7,203,654 (10.13%) | 63,916,604 (89.87%) |
| Cd-2 | 88,241,902 | 61,102,730 (69.24%) | 6,008,134 (9.83%) | 55,094,596 (90.17%) |
| Cd-3 | 89,834,568 | 63,034,550 (70.17%) | 6,166,310 (9.78%) | 56,868,240 (90.22%) |
| NO. | GO.ID | Term | Significant |
|---|---|---|---|
| 1 | GO:0055114 | oxidation-reduction process | 221 |
| 2 | GO:0006457 | Protein folding | 103 |
| 3 | GO:0009813 | Flavonoid biosynthetic process | 38 |
| 4 | GO:0042744 | Hydrogen peroxide catabolic process | 19 |
| 5 | GO:0052696 | Flavonoid glucuronidation | 32 |
| 6 | GO:0009992 | Cellular water homeostasis | 17 |
| 7 | GO:0015793 | Glycerol transport | 17 |
| 8 | GO:0006833 | Water transport | 18 |
| 9 | GO:0006979 | Response to oxidative stress | 82 |
| 10 | GO:0010041 | Response to iron(III) ion | 14 |
| 11 | GO:0006098 | pentose-phosphate shunt | 8 |
| 12 | GO:0009414 | Response to water deprivation | 14 |
| 13 | GO:0009651 | Response to salt stress | 34 |
| 14 | GO:0006024 | Glycosaminoglycan biosynthetic process | 5 |
| 15 | GO:0006065 | UDP-glucuronate biosynthetic process | 5 |
| 16 | GO:0009408 | Response to heat | 154 |
| 17 | GO:0044550 | Secondary metabolite biosynthetic process | 41 |
| 18 | GO:0019521 | D-gluconate metabolic process | 7 |
| 19 | GO:0006559 | L-phenylalanine catabolic process | 17 |
| 20 | GO:0006857 | Oligopeptide transport | 1 |
| NO. | GO.ID | Term | Significant |
|---|---|---|---|
| 1 | GO:0051082 | Unfolded protein binding | 65 |
| 2 | GO:0020037 | Heme binding | 52 |
| 3 | GO:0003924 | GTPase activity | 36 |
| 4 | GO:0015250 | Water channel activity | 17 |
| 5 | GO:0015254 | Glycerol channel activity | 17 |
| 6 | GO:0004601 | Peroxidase activity | 23 |
| 7 | GO:0005525 | GTP binding | 39 |
| 8 | GO:0005200 | Structural constituent of cytoskeleton | 20 |
| 9 | GO:0004197 | cysteine-type endopeptidase activity | 15 |
| 10 | GO:0080043 | quercetin_3-O-glucosyltransferase activity | 27 |
| 11 | GO:0080044 | quercetin_7-O-glucosyltransferase activity | 27 |
| 12 | GO:0033760 | 2’-deoxymugineic-acid_2’-dioxygenase activity | 14 |
| 13 | GO:0003979 | UDP-glucose_6-dehydrogenase activity | 5 |
| 14 | GO:0004623 | phospholipase_A2 activity | 7 |
| 15 | GO:0019904 | protein domain specific binding | 6 |
| 16 | GO:0004497 | monooxygenase activity | 36 |
| 17 | GO:0016709 | oxidoreductase activity, acting on paired donors, with incorporation or reduction | 17 |
| 18 | GO:0045548 | Phenylalanine ammonia-lyase activity | 17 |
| 19 | GO:0004616 | phosphogluconate dehydrogenase (decarboxylating) activity | 8 |
| 20 | GO:0003700 | Transcription factor activity, sequence-specific DNA binding | 19 |
| NO. | GO.ID | Term | Significant |
|---|---|---|---|
| 1 | GO:0005576 | Extracellular region | 79 |
| 2 | GO:0005615 | Extracellular space | 15 |
| 3 | GO:0005764 | lysosome | 15 |
| 4 | GO:0005773 | vacuole | 67 |
| 5 | GO:0005788 | Endoplasmic reticulum lumen | 21 |
| 6 | GO:0009505 | plant-type cell wall | 24 |
| 7 | GO:0009941 | Chloroplast envelope | 12 |
| 8 | GO:0005874 | microtubule | 17 |
| 9 | GO:0048046 | apoplast | 24 |
| 10 | GO:0009570 | Chloroplast stroma | 1 |
| 11 | GO:0046658 | Anchored component of plasma membrane | 4 |
| 12 | GO:0005774 | Vacuolar membrane | 25 |
| 13 | GO:0009535 | Chloroplast thylakoid membrane | 5 |
| 14 | GO:0005777 | peroxisome | 3 |
| 15 | GO:0009506 | plasmodesma | 37 |
| 16 | GO:0005730 | nucleolus | 8 |
| 17 | GO:0048226 | Casparian strip | 5 |
| 18 | GO:0008287 | Protein serine/threonine phosphatase complex | 1 |
| 19 | GO:0005618 | Cell wall | 41 |
| 20 | GO:0031201 | SNARE complex | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Zhang, Y.; He, Y.; Cao, Q.; Zhang, T.; Lou, L.; Cai, Q. Full-Length Transcriptome Assembly of Italian Ryegrass Root Integrated with RNA-Seq to Identify Genes in Response to Plant Cadmium Stress. Int. J. Mol. Sci. 2020, 21, 1067. https://doi.org/10.3390/ijms21031067
Hu Z, Zhang Y, He Y, Cao Q, Zhang T, Lou L, Cai Q. Full-Length Transcriptome Assembly of Italian Ryegrass Root Integrated with RNA-Seq to Identify Genes in Response to Plant Cadmium Stress. International Journal of Molecular Sciences. 2020; 21(3):1067. https://doi.org/10.3390/ijms21031067
Chicago/Turabian StyleHu, Zhaoyang, Yufei Zhang, Yue He, Qingqing Cao, Ting Zhang, Laiqing Lou, and Qingsheng Cai. 2020. "Full-Length Transcriptome Assembly of Italian Ryegrass Root Integrated with RNA-Seq to Identify Genes in Response to Plant Cadmium Stress" International Journal of Molecular Sciences 21, no. 3: 1067. https://doi.org/10.3390/ijms21031067
APA StyleHu, Z., Zhang, Y., He, Y., Cao, Q., Zhang, T., Lou, L., & Cai, Q. (2020). Full-Length Transcriptome Assembly of Italian Ryegrass Root Integrated with RNA-Seq to Identify Genes in Response to Plant Cadmium Stress. International Journal of Molecular Sciences, 21(3), 1067. https://doi.org/10.3390/ijms21031067