Liver Iron Retention Estimated from Utilization of Oral and Intravenous Radioiron in Various Anemias and Hemochromatosis in Humans


Abstract
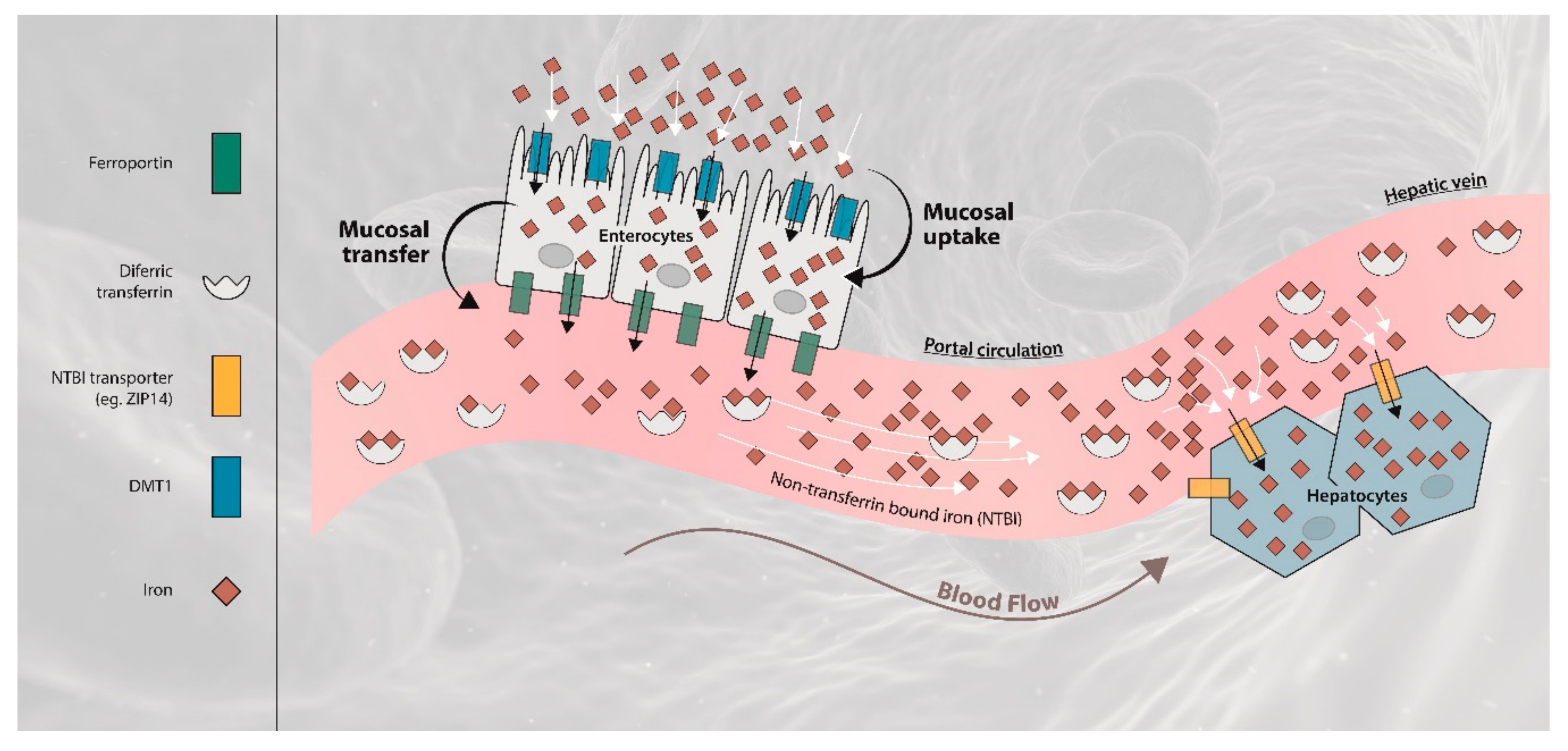
:1. Introduction
2. Results
2.1. Body Iron Retention is Significantly Increased in patients with Non-Transfusion-Dependent Hereditary Anemia and Hereditary Hemochromatosis
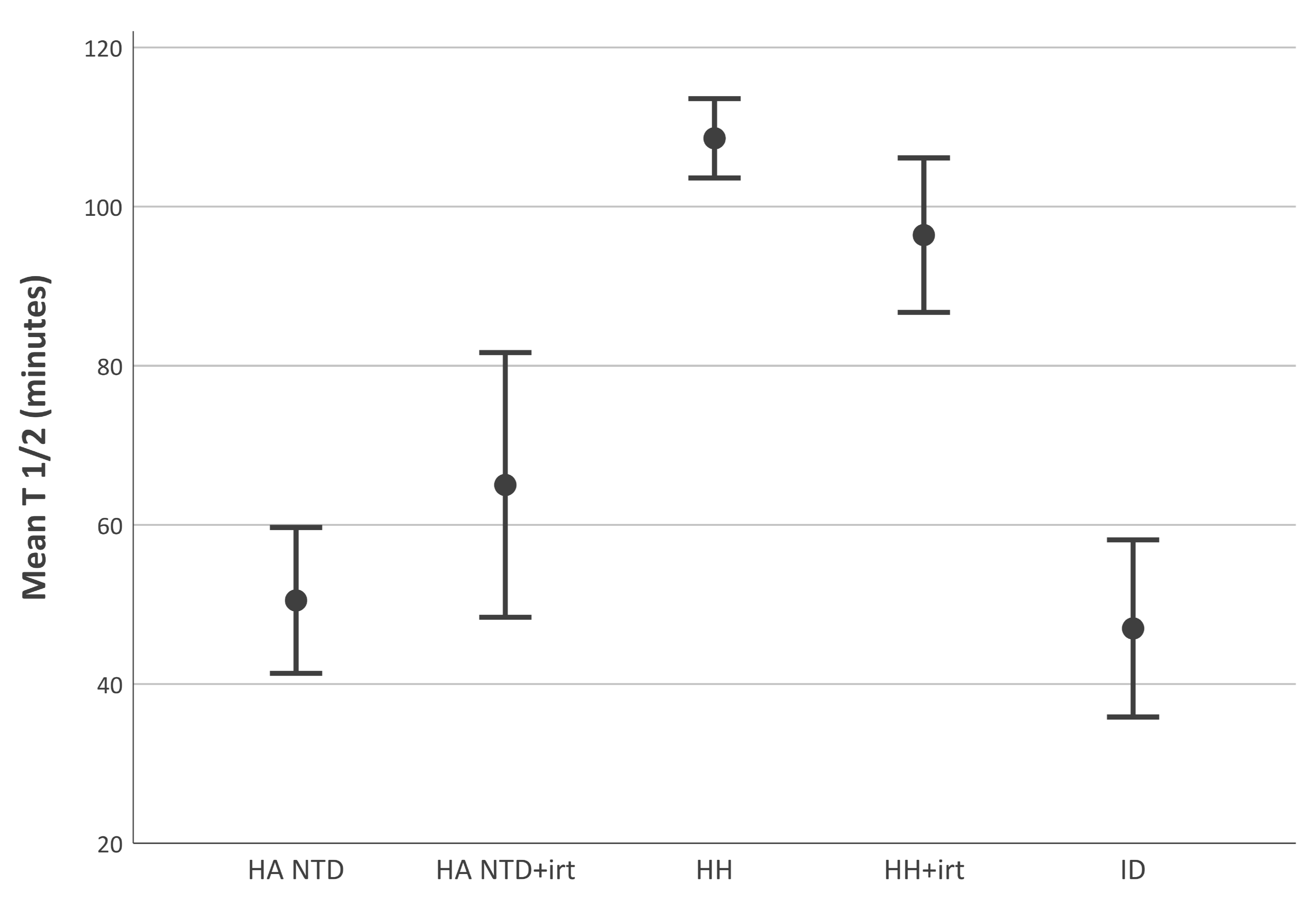
2.2. Disappearance Half-Life of Transferrin-Bound is Shorter in Non-Transfusion-Dependent Hereditary Anemia than in Hereditary Hemochromatosis
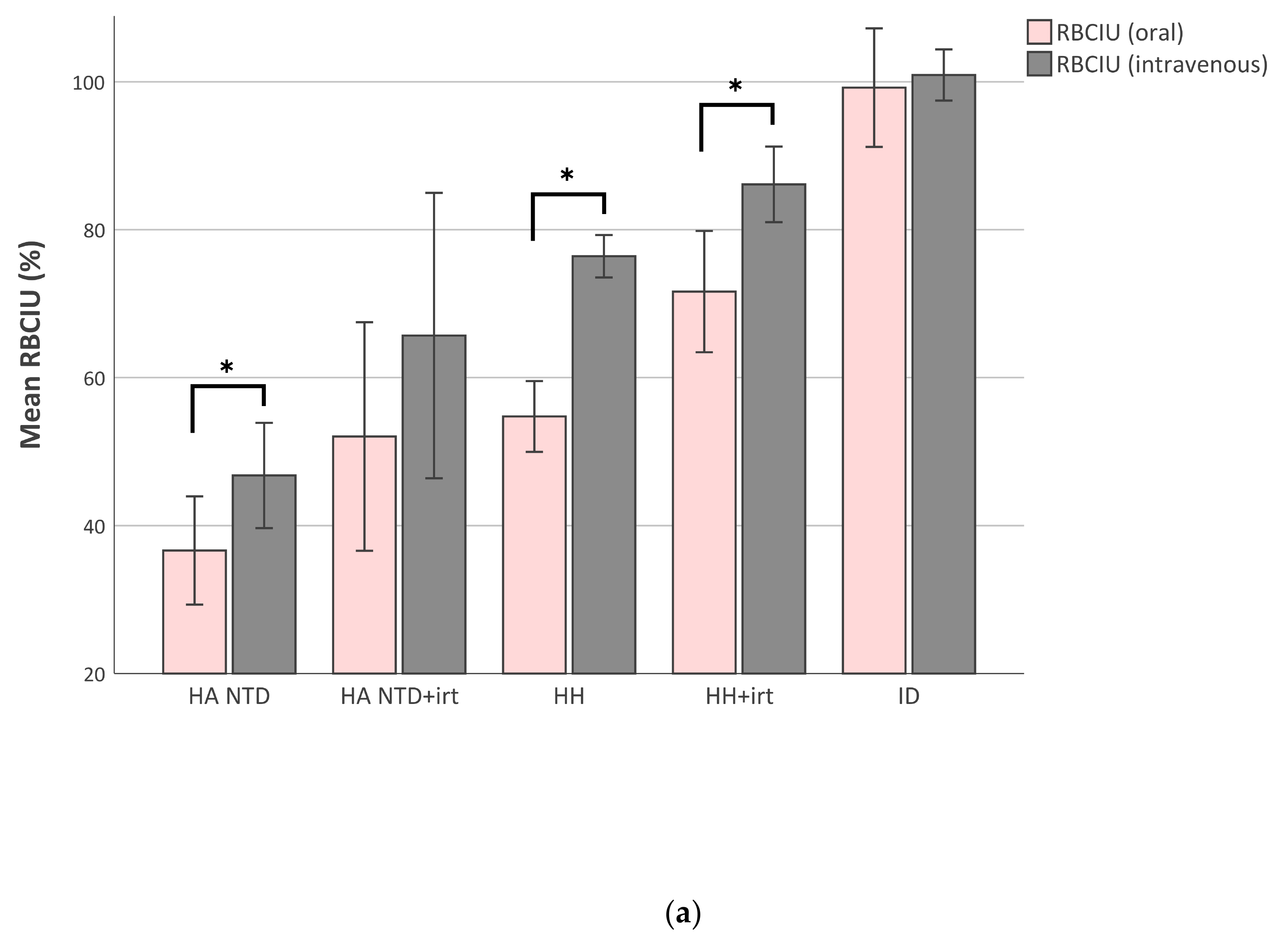
2.3. Red Blood Cell Iron Utilization Decreases When Iron-Overload Increases
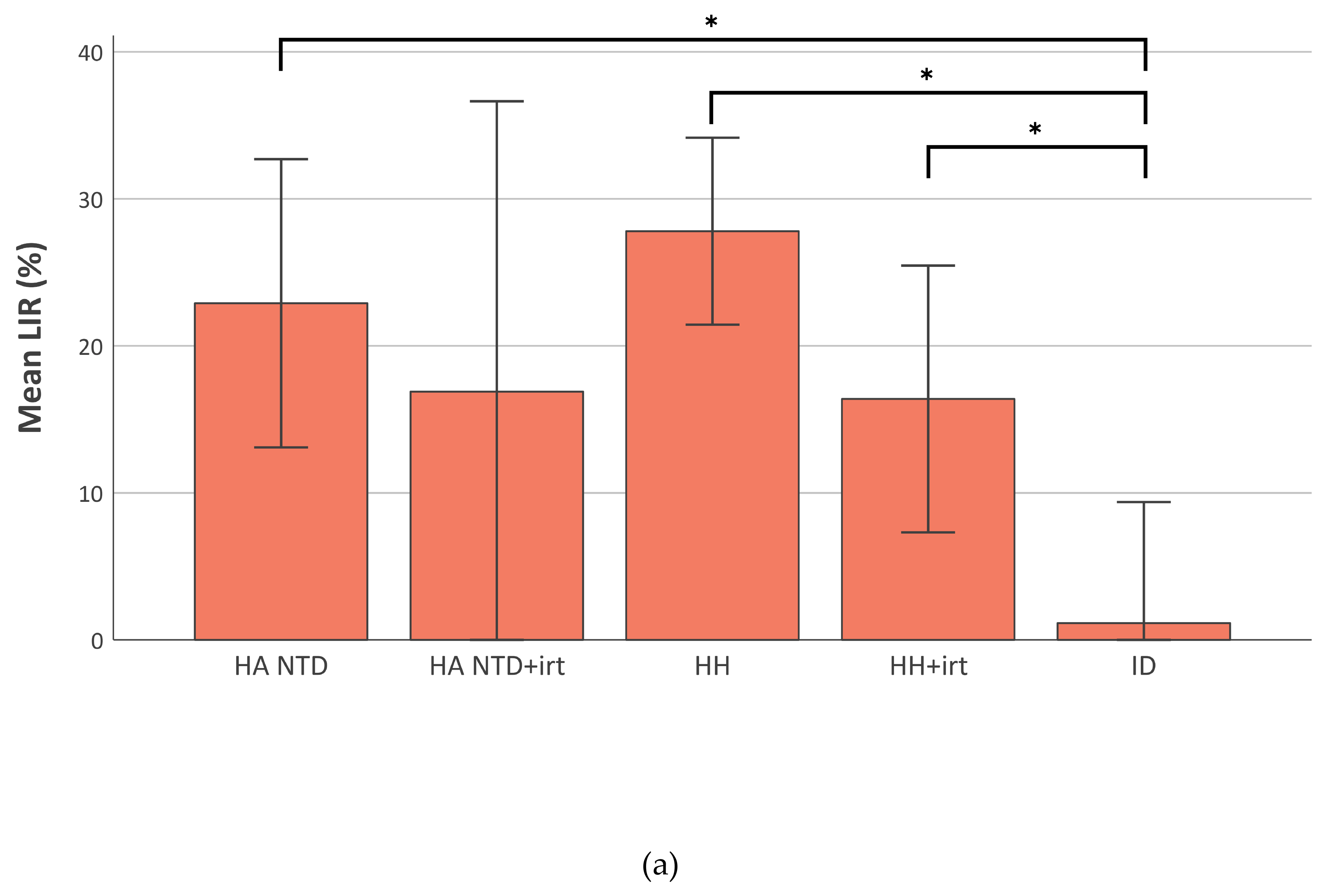
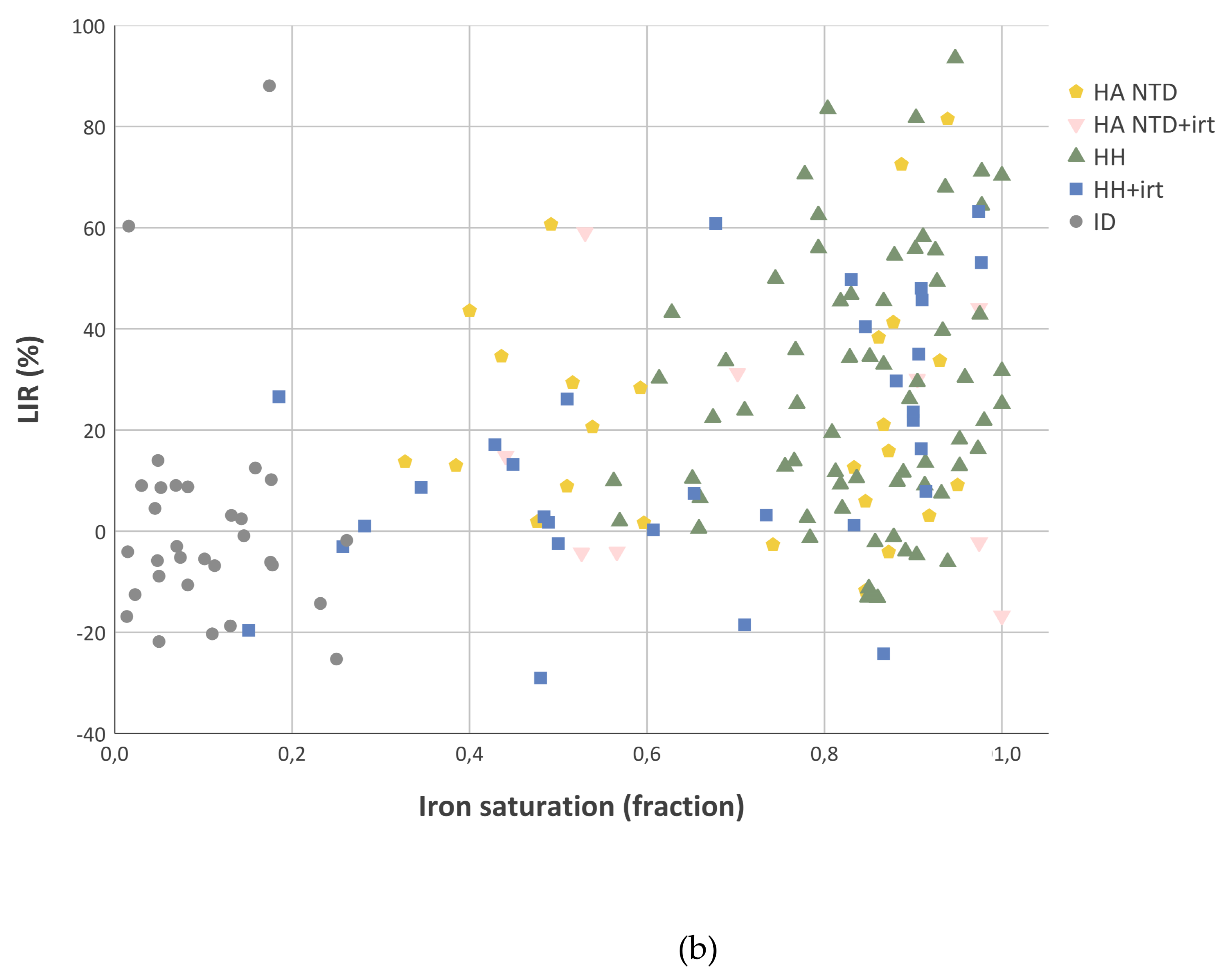
2.4. Liver Iron Retention is Increased in Iron-Overload
3. Discussion
4. Materials and Methods
4.1. Test Subjects
4.2. Patient Selection
4.3. Iron Test Doses
4.4. Measurement of Radioactivity
4.5. Iron Absorption Studies
4.6. Ferrokinetic Studies
4.6.1. Disappearance half-life of iron
4.6.2. Iron incorporation in RBCs
4.6.3. Liver iron retention
4.7. Laboratory Assays
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DMT1 | Divalent metal transporter 1 |
| HH | Hereditary haemochromatosis |
| LIR | Liver iron retention |
| LPI | Labile plasma iron |
| NTBI | Non-transferrin-bound iron |
| RBC | Red blood cell |
| RBCIU | Red blood cell iron utilization |
| TfR1 | Transferrin receptor 1 |
| TSAT | Transferrin saturation |
References
- Marx, J.J. Iron absorption and its regulation. A review. Haematologica 1979, 64, 479–493. [Google Scholar]
- Ricketts, C.; Cavill, I.; Napier, J.A.; Jacobs, A. Ferrokinetics and erythropoiesis in man: An evaluation of ferrokinetic measurements. Br. J. Haematol. 1977, 35, 41–47. [Google Scholar] [CrossRef]
- Ricketts, C.; Jacobs, A.; Cavill, I. Ferrokinetics and erythropoiesis in man: The measurement of effective erythropoiesis, ineffective erythropoiesis and red cell lifespan using 59Fe. Br. J. Haematol. 1975, 31, 65–75. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar] [CrossRef] [Green Version]
- Camaschella, C.; Pagani, A. Advances in understanding iron metabolism and its crosstalk with erythropoiesis. Br. J. Haematol. 2018, 182, 481–494. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Pietrangelo, A. Iron and the liver. Liv. Int. 2016, 36 (Suppl. S1), 116–123. [Google Scholar] [CrossRef]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [Green Version]
- Fleming, R.E.; Sly, W.S. Hepcidin: A putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8160–8162. [Google Scholar] [CrossRef] [Green Version]
- Krause, A.; Neitz, S.; Magert, H.J.; Schulz, A.; Forssmann, W.G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Brissot, P.; Troadec, M.B.; Loreal, O.; Brissot, E. Pathophysiology and classification of iron overload diseases; update. Transfus. Clin. Biol. 2018 2019, 26, 80–88. [Google Scholar] [CrossRef]
- Rivella, S. Ineffective erythropoiesis and thalassemias. Curr. Opin. Hematol. 2009, 16, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Core, A.B.; Canali, S.; Zumbrennen-Bullough, K.B.; Ozer, S.; Umans, L.; Zwijsen, A.; Babitt, J.L. Smad1/5 is required for erythropoietin-mediated suppression of hepcidin in mice. Blood 2017, 130, 73–83. [Google Scholar] [CrossRef]
- Origa, R.; Galanello, R.; Ganz, T.; Giagu, N.; Maccioni, L.; Faa, G.; Nemeth, E. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica 2007, 92, 583–588. [Google Scholar] [CrossRef]
- Kearney, S.L.; Nemeth, E.; Neufeld, E.J.; Thapa, D.; Ganz, T.; Weinstein, D.A.; Cunningham, M.J. Urinary hepcidin in congenital chronic anemias. Pediatr. Blood Cancer 2007, 48, 57–63. [Google Scholar] [CrossRef]
- Karafin, M.S.; Koch, K.L.; Rankin, A.B.; Nischik, D.; Rahhal, G.; Simpson, P.; Field, J.J. Erythropoietic drive is the strongest predictor of hepcidin level in adults with sickle cell disease. Blood Cell. Mol. Dis. 2015, 55, 304–307. [Google Scholar] [CrossRef] [Green Version]
- Mojzikova, R.; Koralkova, P.; Holub, D.; Zidova, Z.; Pospisilova, D.; Cermak, J.; Striezencova Laluhova, Z.; Indrak, K.; Sukova, M.; Partschova, M.; et al. Iron status in patients with pyruvate kinase deficiency: Neonatal hyperferritinaemia associated with a novel frameshift deletion in the PKLR gene (p.Arg518fs), and low hepcidin to ferritin ratios. Br. J. Haematol. 2014, 165, 556–563. [Google Scholar]
- Nam, H.; Wang, C.Y.; Zhang, L.; Zhang, W.; Hojyo, S.; Fukada, T.; Knutson, M.D. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: Implications for tissue iron uptake in iron-related disorders. Haematologica 2013, 98, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Chua, A.C.; Delima, R.D.; Morgan, E.H.; Herbison, C.E.; Tirnitz-Parker, J.E.; Graham, R.M.; Fleming, R.E.; Britton, R.S.; Bacon, B.R.; Olynyk, J.K.; et al. Iron uptake from plasma transferrin by a transferrin receptor 2 mutant mouse model of haemochromatosis. J. Hepatol. 2010, 52, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Wilms, J.W.; Batey, R.G. Effect of iron stores on hepatic metabolism of transferrin-bound iron. Am. J. Physiol. 1983, 244, G138–G144. [Google Scholar] [CrossRef]
- Hosain, F.; Marsaglia, G.; Finch, C.A. Blood ferrokinetics in normal man. J. Clin. Invest. 1967, 46, 1–9. [Google Scholar] [CrossRef]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. 2014, 28, 653–670. [Google Scholar] [CrossRef]
- Marx, J.J.; Dinant, H.J. Ferrokinetics and red cell iron uptake in old age: Evidence for increased liver iron retention? Haematologica 1982, 67, 161–168. [Google Scholar]
- Breuer, W.; Ghoti, H.; Shattat, A.; Goldfarb, A.; Koren, A.; Levin, C.; Rachmilewitz, E.; Cabantchik, Z.I. Non-transferrin bound iron in Thalassemia: Differential detection of redox active forms in children and older patients. Am. J. Hematol. 2012, 87, 55–61. [Google Scholar] [CrossRef]
- Le Lan, C.; Loreal, O.; Cohen, T.; Ropert, M.; Glickstein, H.; Laine, F.; Pouchard, M.; Deugnier, Y.; Le Treut, A.; Breuer, W.; et al. Redox active plasma iron in C282Y/C282Y hemochromatosis. Blood 2005, 105, 4527–4531. [Google Scholar] [CrossRef] [Green Version]
- Pootrakul, P.; Breuer, W.; Sametband, M.; Sirankapracha, P.; Hershko, C.; Cabantchik, Z.I. Labile plasma iron (LPI) as an indicator of chelatable plasma redox activity in iron-overloaded beta-thalassemia/HbE patients treated with an oral chelator. Blood 2004, 104, 1504–1510. [Google Scholar] [CrossRef]
- De Valk, B.; Addicks, M.A.; Gosriwatana, I.; Lu, S.; Hider, R.C.; Marx, J.J. Non-transferrin-bound iron is present in serum of hereditary haemochromatosis heterozygotes. Eur. J. Clin. Investig. 2000, 30, 248–251. [Google Scholar] [CrossRef]
- Brittenham, G.M.; Andersson, M.; Egli, I.; Foman, J.T.; Zeder, C.; Westerman, M.E.; Hurrell, R.F. Circulating non-transferrin-bound iron after oral administration of supplemental and fortification doses of iron to healthy women: A randomized study. Am. J. Clin. Nutr. 2014, 100, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Schumann, K.; Kroll, S.; Romero-Abal, M.E.; Georgiou, N.A.; Marx, J.J.; Weiss, G.; Solomons, N.W. Impact of oral iron challenges on circulating non-transferrin-bound iron in healthy Guatemalan males. Ann. Nutr. Metab. 2012, 60, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Hider, R.C. Influence of non-enzymatic post-translation modifications on the ability of human serum albumin to bind iron. Implications for non-transferrin-bound iron speciation. Biochim. Biophys. Acta 2009, 1794, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.W.; Rafique, R.; Zarea, A.; Rapisarda, C.; Cammack, R.; Evans, P.J.; Porter, J.B.; Hider, R.C. Nature of non-transferrin-bound iron: Studies on iron citrate complexes and thalassemic sera. J. Biol. Inorg. Chem. 2008, 13, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Esposito, B.P.; Breuer, W.; Sirankapracha, P.; Pootrakul, P.; Hershko, C.; Cabantchik, Z.I. Labile plasma iron in iron overload: Redox activity and susceptibility to chelation. Blood 2003, 102, 2670–2677. [Google Scholar] [CrossRef] [Green Version]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free. Radic. Biol. Med. 2019, 133, 101–111. [Google Scholar] [CrossRef]
- Van der Heul, C.; van Eijk, H.G.; Wiltink, W.F.; Leijnse, B. The binding of iron to transferrin and to other serum components at different degrees of saturation with iron. Clin. Chim. Acta 1972, 38, 347–353. [Google Scholar] [CrossRef]
- Taylor, K.M.; Nicholson, R.I. The LZT proteins; the LIV-1 subfamily of zinc transporters. Biochim. Biophys. Acta 2003, 1611, 16–30. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.M.; Morgan, H.E.; Johnson, A.; Nicholson, R.I. Structure-function analysis of a novel member of the LIV-1 subfamily of zinc transporters, ZIP14. FEBS Lett. 2005, 579, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [Green Version]
- Pinilla-Tenas, J.J.; Sparkman, B.K.; Shawki, A.; Illing, A.C.; Mitchell, C.J.; Zhao, N.; Liuzzi, J.P.; Cousins, R.J.; Knutson, M.D.; Mackenzie, B. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell. Physiol. 2011, 301, C862–C871. [Google Scholar] [CrossRef] [Green Version]
- Jenkitkasemwong, S.; Wang, C.Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell. Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydemir, T.B.; Cousins, R.J. The Multiple Faces of the Metal Transporter ZIP14 (SLC39A14). J. Nutr. 2018, 148, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.L.; Fitz, J.G.; Weisiger, R.A. Non-transferrin-bound iron uptake by rat liver. Role of membrane potential difference. J. Biol. Chem. 1988, 263, 1842–1847. [Google Scholar] [PubMed]
- Brissot, P.; Wright, T.L.; Ma, W.L.; Weisiger, R.A. Efficient clearance of non-transferrin-bound iron by rat liver. Implications for hepatic iron loading in iron overload states. J. Clin. Investig. 1985, 76, 1463–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, T.L.; Brissot, P.; Ma, W.L.; Weisiger, R.A. Characterization of non-transferrin-bound iron clearance by rat liver. J. Biol. Chem. 1986, 261, 10909–10914. [Google Scholar]
- Cortes-Puch, I.; Wang, D.; Sun, J.; Solomon, S.B.; Remy, K.E.; Fernandez, M.; Feng, J.; Kanias, T.; Bellavia, L.; Sinchar, D.; et al. Washing older blood units before transfusion reduces plasma iron and improves outcomes in experimental canine pneumonia. Blood 2014, 123, 1403–1411. [Google Scholar] [CrossRef] [Green Version]
- Collard, K.J.; White, D.L. On the source of the non-transferrin-bound iron which accumulates in packed red blood cell units during storage. Blood Transfus. 2014, 12, 527–532. [Google Scholar]
- Hod, E.A.; Zhang, N.; Sokol, S.A.; Wojczyk, B.S.; Francis, R.O.; Ansaldi, D.; Francis, K.P.; Della-Latta, P.; Whittier, S.; Sheth, S.; et al. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood 2010, 115, 4284–4292. [Google Scholar] [CrossRef] [Green Version]
- Hod, E.A.; Brittenham, G.M.; Billote, G.B.; Francis, R.O.; Ginzburg, Y.Z.; Hendrickson, J.E.; Jhang, J.; Schwartz, J.; Sharma, S.; Sheth, S.; et al. Transfusion of human volunteers with older, stored red blood cells produces extravascular hemolysis and circulating non-transferrin-bound iron. Blood 2011, 118, 6675–6682. [Google Scholar] [CrossRef]
- Piga, A.; Longo, F.; Duca, L.; Roggero, S.; Vinciguerra, T.; Calabrese, R.; Hershko, C.; Cappellini, M.D. High nontransferrin bound iron levels and heart disease in thalassemia major. Am. J. Hematol. 2009, 84, 29–33. [Google Scholar] [CrossRef]
- Li, H.; Rybicki, A.C.; Suzuka, S.M.; von Bonsdorff, L.; Breuer, W.; Hall, C.B.; Cabantchik, Z.I.; Bouhassira, E.E.; Fabry, M.E.; Ginzburg, Y.Z. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat. Med. 2010, 16, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring, D.; Minana, B.; Bell, O.; Janser, H.G.; Muckenthaler, M.; Schumann, K.; Hentze, M.W. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood 2005, 106, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.; Liu, L.; Nemeth, E.; Cherian, S.; Ganz, T.; Abkowitz, J.L. Evidence that the expression of transferrin receptor 1 on erythroid marrow cells mediates hepcidin suppression in the liver. Exp. Hematol. 2015, 43, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Slotki, I.; Cabantchik, Z.I. The Labile Side of Iron Supplementation in CKD. J. Am. Soc. Nephrol. 2015, 26, 2612–2619. [Google Scholar] [CrossRef] [Green Version]
- Kooistra, M.P.; Kersting, S.; Gosriwatana, I.; Lu, S.; Nijhoff-Schutte, J.; Hider, R.C.; Marx, J.J. Nontransferrin-bound iron in the plasma of haemodialysis patients after intravenous iron saccharate infusion. Eur. J. Clin. Investig. 2002, 32, 36–41. [Google Scholar] [CrossRef]
- Kartikasari, A.E.; Georgiou, N.A.; Visseren, F.L.; van Kats-Renaud, H.; van Asbeck, B.S.; Marx, J.J. Intracellular labile iron modulates adhesion of human monocytes to human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2257–2262. [Google Scholar] [CrossRef] [Green Version]
- Pinto, J.P.; Arezes, J.; Dias, V.; Oliveira, S.; Vieira, I.; Costa, M.; Vos, M.; Carlsson, A.; Rikers, Y.; Rangel, M.; et al. Physiological implications of NTBI uptake by T lymphocytes. Front. Pharmacol. 2014, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Zhang, S.; Sun, X.; Liu, J.; Wu, Y.; Guo, W.; Wang, F.; Ou, X.; Cong, M.; Jin, E.; et al. Distinct Iron Deposition Profiles of Liver Zones in Various Models with Iron Homeostasis Disorders. Adv. Sci. 2018, 5, 1800866. [Google Scholar] [CrossRef]
- Marx, J.J. Mucosal uptake, mucosal transfer and retention of iron, measured by whole-body counting. Scand. J. Haematol. 1979, 23, 293–302. [Google Scholar] [CrossRef]
- Marx, J.J. Normal iron absorption and decreased red cell iron uptake in the aged. Blood 1979, 53, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, J.J.; van den Beld, B.; van Dongen, R.; Strackee, L.H. Simultaneous measurement of 59Fe and 51Cr in iron absorption studies using a whole-body scanner with mobile shielding. Nuklearmedizin 1980, 19, 140–145. [Google Scholar] [PubMed]
- Cavill, I. The preparation of 59 Fe-labelled transferrin for ferrokinetic studies. J. Clin. Pathol. 1971, 24, 472–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.D.; Marsaglia, G.; Eschbach, J.W.; Funk, D.D.; Finch, C.A. Ferrokinetics: A biologic model for plasma iron exchange in man. J. Clin. Investig. 1970, 49, 197–205. [Google Scholar] [CrossRef]
- Marx, J.; Verzijlbergen, J. Radionuclide Techniques in Haematology. In Nuclear Techniques in Diagnostic Medicine; van Rijk, P., Ed.; Martinus Nijhoff Publishers: Dordrecht, The Netherlands, 1986; pp. 459–517. [Google Scholar]
- Bland, J.M.; Altman, D.G. Statistics Notes: Bootstrap resampling methods. BMJ 2015, 350, h2622. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | HA NTD | HA NTD-irt | HH | HH-irt | ID | Normal |
| No. of patients | 29 | 11 | 79 | 42 | 47 | 70 |
| Male, % | 79 | 82 | 66 | 86 | 26 | 56 |
| Age, years | 29 (22–37) | 26 (15–39) | 44 (35–56) | 44 (37–54) | 38 (23–47) | 67 (26–70) |
| Laboratory parameters | ||||||
| Hb, g/dL | 10.5 (10.5–12.4) | 12.4 (9.5–15.5) | 15.5 (14.8–16.4) | 16.0 (14.6–16.8) | 11.1 (10.2–12.6) | 15.5 (14.6–16.3) |
| Reticulocytes, × 109/L | 15 (12–39) | 8 (5–243) | 11 (7–17) | 13 (7–26) | 12 (8–15) | 10 (6–15) |
| Ferritin, μg/L | 407 (215–1800) | 222 (109–367) | 953 (440–1350) | 70 (36–170) | 7 (5–9) | NA |
| Iron saturation, fraction | 0.74 (0.50–0.87) | 0.76 (0.53–0.97) | 0.85 (0.78–0.92) | 0.69 (0.44–0.90) | 0.08 (0.05–0.15) | 0.36 (0.29–0.44) |
| Serum iron, μmol/L | 33 (27–41) | 35 (30–38) | 38 (33–43) | 34 (22–41) | 7 (4–10) | 21 (18–26) |
| AST, U/L | 22 (16–42) | 18 (13–27) | 31 (20–44) | 22 (15–26) | 17 (15–23) | NA |
| LD, U/L | 350 (277–644) | 395 (311–593) | 400 (325–473) | 416 (323–459) | 408 (372–455) | NA |
| Bilirubin, mg/dL | 18 (8–44) | 9 (7–28) | 10 (8–16) | 12 (9–18) | 6 (4–9) | NA |
| Iron absorption studies | ||||||
| Mucosal iron uptake, % | 60 (54–77) | 78 (55–87) | 55 (47–72) | 88 (81–93) | 88 (75–96) | 46 (29–55) |
| Mucosal iron transfer, fraction | 0.76 (0.63–0.91) | 0.90 (0.80–0.96) | 0.84 (0.71–0.90) | 0.97 (0.94–0.98) | 0.96 (0.91–0.98) | 0.62 (0.53–0.78) |
| Iron retention, % | 42 (33–63) | 66 (55–74) | 45 (33–63) | 85 (73–91) | 81 (69–91) | 25 (17–36) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Vuren, A.J.; van Wijk, R.; van Beers, E.J.; Marx, J.J.M. Liver Iron Retention Estimated from Utilization of Oral and Intravenous Radioiron in Various Anemias and Hemochromatosis in Humans. Int. J. Mol. Sci. 2020, 21, 1077. https://doi.org/10.3390/ijms21031077
van Vuren AJ, van Wijk R, van Beers EJ, Marx JJM. Liver Iron Retention Estimated from Utilization of Oral and Intravenous Radioiron in Various Anemias and Hemochromatosis in Humans. International Journal of Molecular Sciences. 2020; 21(3):1077. https://doi.org/10.3390/ijms21031077
Chicago/Turabian Stylevan Vuren, Annelies J., Richard van Wijk, Eduard J. van Beers, and Joannes J.M. Marx. 2020. "Liver Iron Retention Estimated from Utilization of Oral and Intravenous Radioiron in Various Anemias and Hemochromatosis in Humans" International Journal of Molecular Sciences 21, no. 3: 1077. https://doi.org/10.3390/ijms21031077
APA Stylevan Vuren, A. J., van Wijk, R., van Beers, E. J., & Marx, J. J. M. (2020). Liver Iron Retention Estimated from Utilization of Oral and Intravenous Radioiron in Various Anemias and Hemochromatosis in Humans. International Journal of Molecular Sciences, 21(3), 1077. https://doi.org/10.3390/ijms21031077