Flexible Data Trimming Improves Performance of Global Machine Learning Methods in Omics-Based Personalized Oncology





Abstract
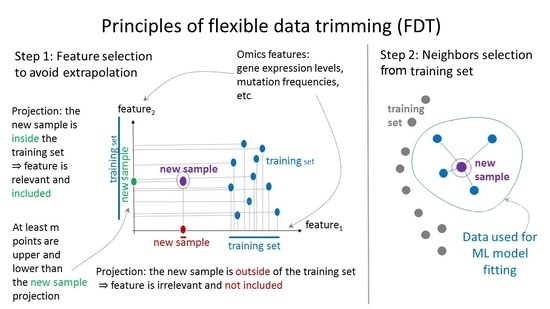
:
1. Introduction
2. Results
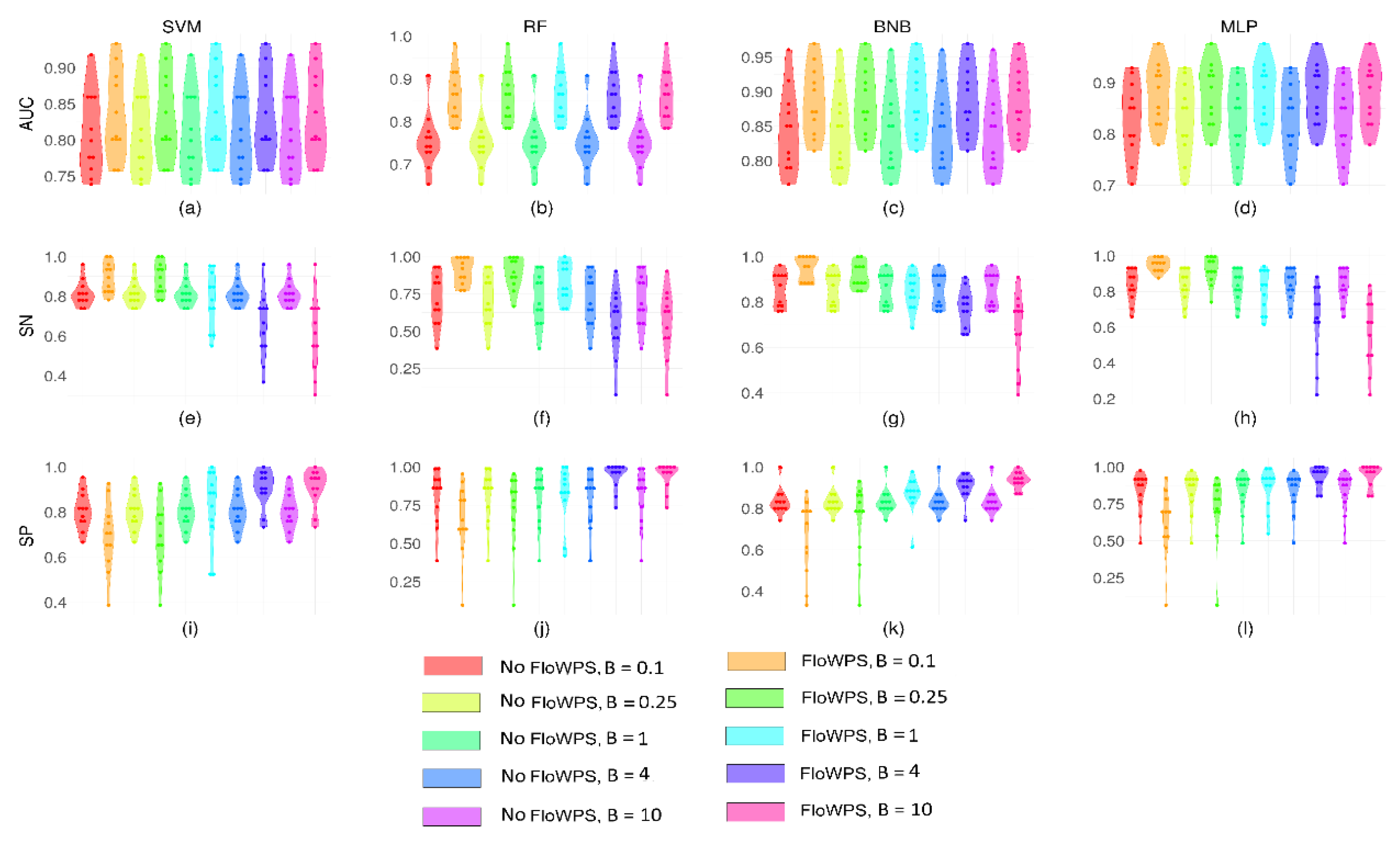
2.1. Performance of FloWPS for Equalized Datasets Using All ML Methods with Default Settings
2.2. Performance of FloWPS for Equalized Datasets Using BNB, MLP and RF Methods with the Advanced Settings
2.3. Performance of FloWPS for Non-Equalized Datasets Using BNB, MLP, RF and SVM Methods with the Advanced Settings
2.4. Correlation Study Between Different ML Methods at the Level of Feature Importance
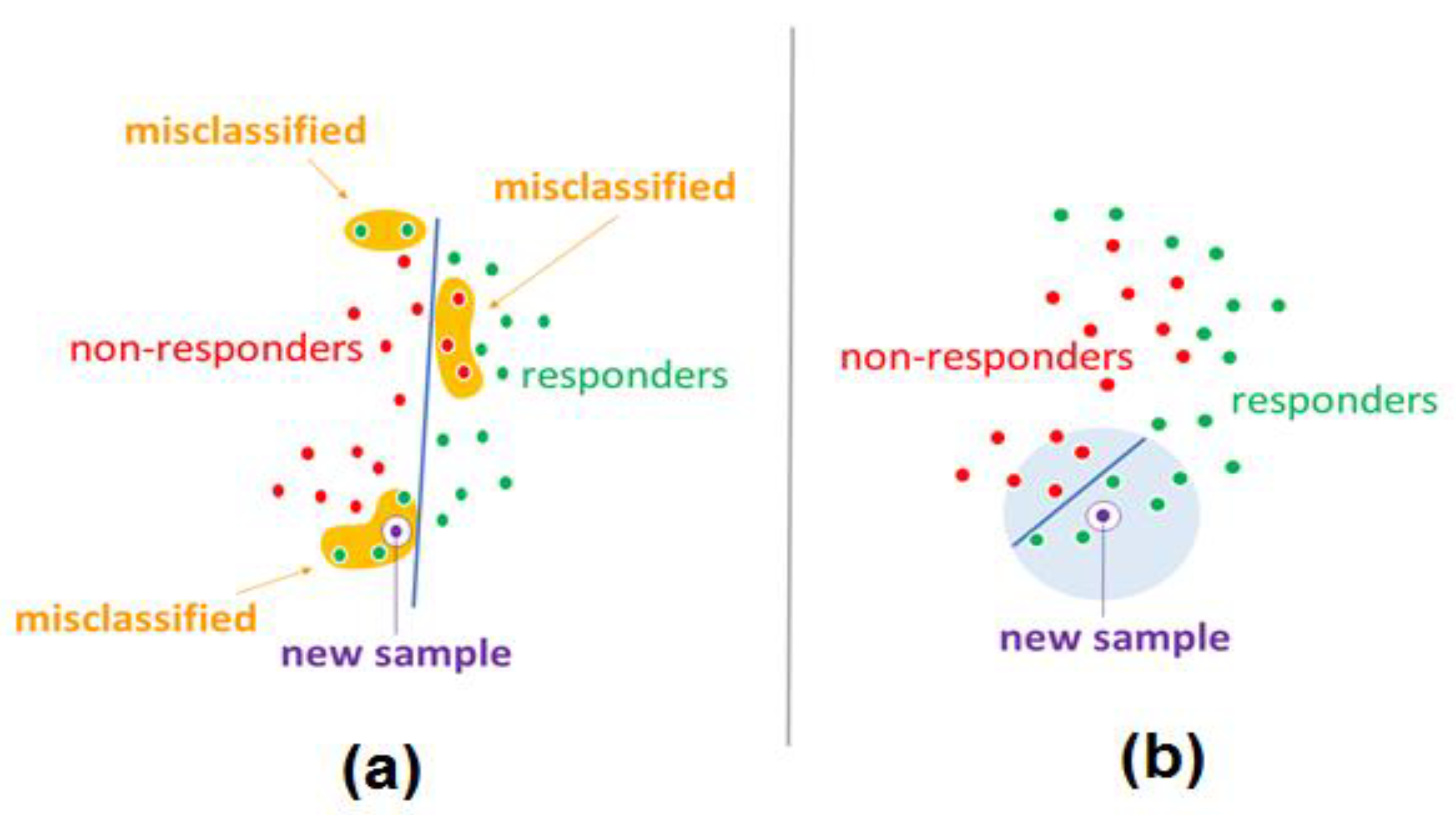
3. Discussion
4. Materials and Methods
4.1. Clinically Annotated Molecular Datasets
- Labelling each patient as either responder or non-responder on the therapy used;
- For each dataset, finding top marker genes having the highest AUC values for distinguishing responder and non-responder classes;
- Performing the leave-one-out (LOO) cross-validation procedure to complete the robust core marker gene set used for building the ML model.
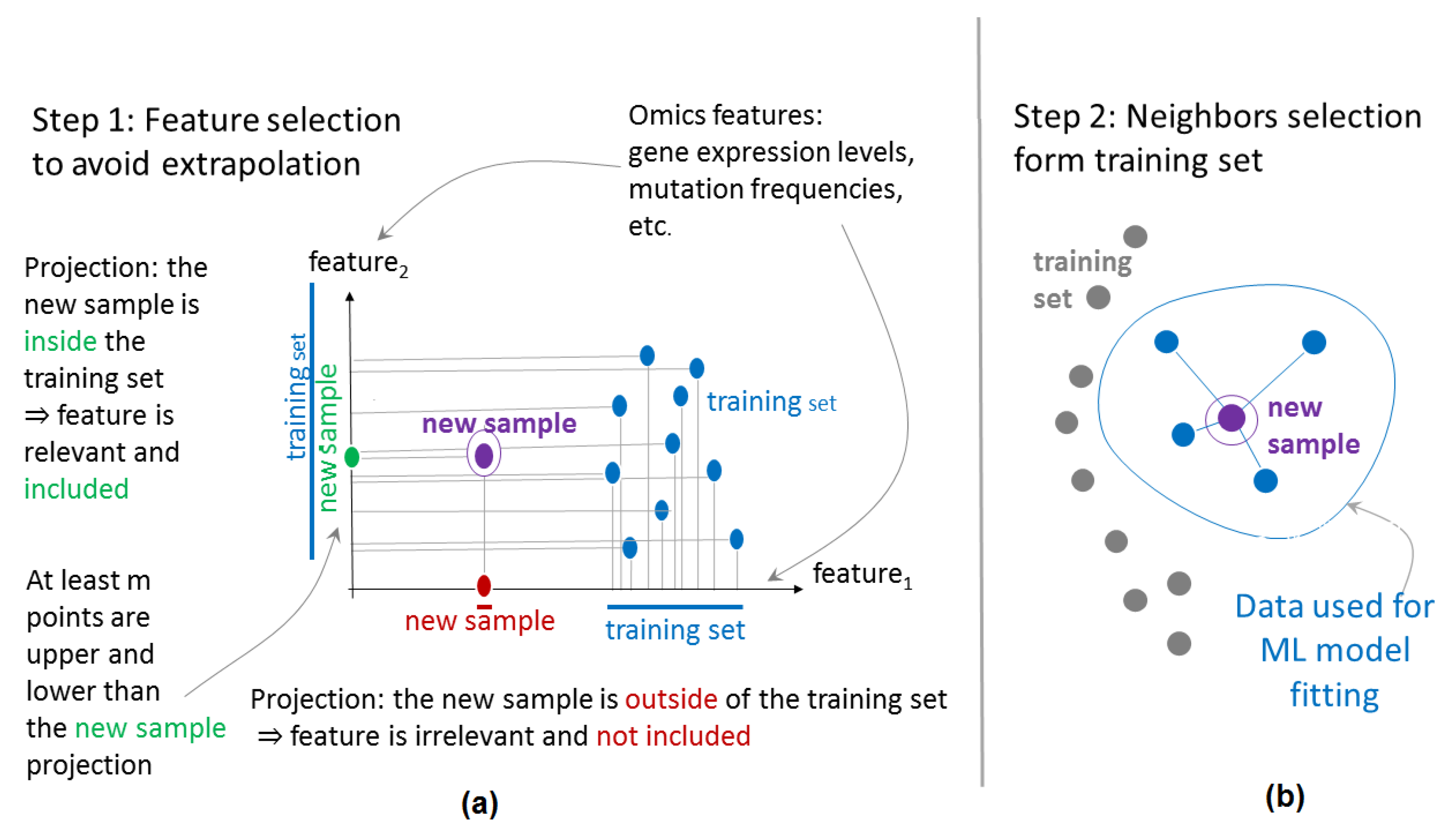
4.2. Principles of Flexible Data Trimming
- First, it helped us to specify the core marker gene sets (see Materials and Methods), which form the feature space F = (f1,…,fS) for subsequent application of data trimming;
- Second, it was applied for every ML prediction act for the wide range of data trimming parameters, m and k;
- Third, it was used for the final prediction of the treatment response for every patient and optimized (for all remaining patients) values of parameters m and k.
4.3. Application of ML Methods
4.4. False Positive Vs. False Negative Error Balance
4.5. Feature Importance Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADA | Adaptive boosting |
| AML | Acute myelogenous leukemia |
| ASCT | Allogeneic stem cell transplantation |
| AUC | Area under curve |
| BNB | Binomial naïve Bayes |
| FloWPS | Floating window projective separator |
| FN | False negative |
| FP | False positive |
| GEO | Gene expression omnibus |
| GSE | GEO series |
| HER2 | Human epidermal growth factor receptor 2 |
| kNN | k nearest neighbors |
| LOO | Leave-one-out |
| ML | Machine learning |
| MLP | Multi-layer perceptron |
| RF | Random forest |
| ROC | Receiver operating characteristic |
| RR | Ridge regression |
| SN | Sensitivity |
| SP | Specificity |
| SVM | Support vector machine |
| TP | True positive |
| TN | True negative |
References
- Buzdin, A.; Sorokin, M.; Garazha, A.; Sekacheva, M.; Kim, E.; Zhukov, N.; Wang, Y.; Li, X.; Kar, S.; Hartmann, C.; et al. Molecular pathway activation—New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin. Cancer Biol. 2018, 53, 110–124. [Google Scholar] [CrossRef]
- Zhukov, N.V.; Tjulandin, S.A. Targeted therapy in the treatment of solid tumors: Practice contradicts theory. Biochem. Biokhimiia 2008, 73, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Glusker, A.; Aleshin, A.; Poddubskaya, E.; Sekacheva, M.; Kim, E.; Gaifullin, N.; Giese, A.; et al. RNA sequencing for research and diagnostics in clinical oncology. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Artemov, A.; Aliper, A.; Korzinkin, M.; Lezhnina, K.; Jellen, L.; Zhukov, N.; Roumiantsev, S.; Gaifullin, N.; Zhavoronkov, A.; Borisov, N.; et al. A method for predicting target drug efficiency in cancer based on the analysis of signaling pathway activation. Oncotarget 2015, 6, 29347–29356. [Google Scholar] [CrossRef] [PubMed]
- Shepelin, D.; Korzinkin, M.; Vanyushina, A.; Aliper, A.; Borisov, N.; Vasilov, R.; Zhukov, N.; Sokov, D.; Prassolov, V.; Gaifullin, N.; et al. Molecular pathway activation features linked with transition from normal skin to primary and metastatic melanomas in human. Oncotarget 2016, 7, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Zolotovskaia, M.A.; Sorokin, M.I.; Emelianova, A.A.; Borisov, N.M.; Kuzmin, D.V.; Borger, P.; Garazha, A.V.; Buzdin, A.A. Pathway Based Analysis of Mutation Data Is Efficient for Scoring Target Cancer Drugs. Front. Pharmacol. 2019, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Buzdin, A.; Sorokin, M.; Poddubskaya, E.; Borisov, N. High-Throughput Mutation Data Now Complement Transcriptomic Profiling: Advances in Molecular Pathway Activation Analysis Approach in Cancer Biology. Cancer Inf. 2019, 18, 1176935119838844. [Google Scholar] [CrossRef] [Green Version]
- Tkachev, V.; Sorokin, M.; Mescheryakov, A.; Simonov, A.; Garazha, A.; Buzdin, A.; Muchnik, I.; Borisov, N. FLOating-Window Projective Separator (FloWPS): A Data Trimming Tool for Support Vector Machines (SVM) to Improve Robustness of the Classifier. Front. Genet. 2019, 9, 717. [Google Scholar] [CrossRef]
- Bartlett, P.; Shawe-Taylor, J. Generalization performance of support vector machines and other pattern classifiers. In Advances in Kernel Methods: Support Vector Learning; MIT Press: Cambridge, MA, USA, 1999; pp. 43–54. ISBN 0262194163. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Lisacek, F.; Sanchez, J.-C.; Müller, M. Bioinformatics for protein biomarker panel classification: What is needed to bring biomarker panels into in vitro diagnostics? Expert Rev. Proteomics 2009, 6, 675–689. [Google Scholar] [CrossRef] [Green Version]
- Toloşi, L.; Lengauer, T. Classification with correlated features: Unreliability of feature ranking and solutions. Bioinformatics 2011, 27, 1986–1994. [Google Scholar] [CrossRef]
- Stigler, S.M. The History of Statistics: The Measurement of Uncertainty Before 1900; Belknap Press of Harvard University Press: Cambridge, MA, USA, 1986; ISBN 978-0-674-40340-6. [Google Scholar]
- Cramer, J.S. The Origins of Logistic Regression; Tinbergen Institute Working Paper No. 2002-119/4; Tinbergen Institute: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Santosa, F.; Symes, W.W. Linear Inversion of Band-Limited Reflection Seismograms. SIAM J. Sci. Stat. Comput. 1986, 7, 1307–1330. [Google Scholar] [CrossRef]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Tikhonov, A.N.; Arsenin, V.I. Solutions of Ill-Posed Problems; Scripta series in mathematics; Winston: Washington, DC, USA; Halsted Press: New York, NY, USA, 1977; ISBN 978-0-470-99124-4. [Google Scholar]
- Minsky, M.L.; Papert, S.A. Perceptrons—Expanded Edition: An Introduction to Computational Geometry; MIT Press: Boston, MA, USA, 1987; pp. 152–245. [Google Scholar]
- Prados, J.; Kalousis, A.; Sanchez, J.-C.; Allard, L.; Carrette, O.; Hilario, M. Mining mass spectra for diagnosis and biomarker discovery of cerebral accidents. Proteomics 2004, 4, 2320–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuna, E.; Freund, R.; Girosi, F. An improved training algorithm for support vector machines. In Neural Networks for Signal Processing VII, Proceedings of the 1997 IEEE Signal Processing Society Workshop, Amelia Island, FL, USA, 24–26 September 1997; IEEE: Piscataway, NJ, USA, 1997; pp. 276–285. [Google Scholar]
- Turki, T.; Wang, J.T.L. Clinical intelligence: New machine learning techniques for predicting clinical drug response. Comput. Biol. Med. 2019, 107, 302–322. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, H.; Wu, Z.; Wang, T.; Li, W.; Tang, Y.; Liu, G. In Silico Prediction of Blood-Brain Barrier Permeability of Compounds by Machine Learning and Resampling Methods. ChemMedChem 2018, 13, 2189–2201. [Google Scholar] [CrossRef]
- Yosipof, A.; Guedes, R.C.; García-Sosa, A.T. Data Mining and Machine Learning Models for Predicting Drug Likeness and Their Disease or Organ Category. Front. Chem. 2018, 6, 162. [Google Scholar] [CrossRef] [Green Version]
- Azarkhalili, B.; Saberi, A.; Chitsaz, H.; Sharifi-Zarchi, A. DeePathology: Deep Multi-Task Learning for Inferring Molecular Pathology from Cancer Transcriptome. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Turki, T.; Wei, Z. A link prediction approach to cancer drug sensitivity prediction. BMC Syst. Biol. 2017, 11, 94. [Google Scholar] [CrossRef]
- Turki, T.; Wei, Z.; Wang, J.T.L. Transfer Learning Approaches to Improve Drug Sensitivity Prediction in Multiple Myeloma Patients. IEEE Access 2017, 5, 7381–7393. [Google Scholar] [CrossRef]
- Turki, T.; Wei, Z.; Wang, J.T.L. A transfer learning approach via procrustes analysis and mean shift for cancer drug sensitivity prediction. J. Bioinform. Comput. Biol. 2018, 16, 1840014. [Google Scholar] [CrossRef]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007, 109, 3177–3188. [Google Scholar] [CrossRef] [PubMed]
- Bishop, C.M. Pattern Recognition and Machine Learning; Information science and statistics; Corrected at 8th printing 2009; Springer: New York, NY, USA, 2009; ISBN 978-0-387-31073-2. [Google Scholar]
- Borisov, N.; Buzdin, A. New Paradigm of Machine Learning (ML) in Personalized Oncology: Data Trimming for Squeezing More Biomarkers from Clinical Datasets. Front. Oncol. 2019, 9, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabl, A.A.; Alkhateeb, A.; ElMaraghy, W.; Rueda, L.; Ngom, A. A Machine Learning Approach for Identifying Gene Biomarkers Guiding the Treatment of Breast Cancer. Front. Genet. 2019, 10, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potamias, G.; Koumakis, L.; Moustakis, V. Gene Selection via Discretized Gene-Expression Profiles and Greedy Feature-Elimination. In Methods and Applications of Artificial Intelligence; Vouros, G.A., Panayiotopoulos, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; Volume 3025, pp. 256–266. ISBN 978-3-540-21937-8. [Google Scholar]
- Allen, M. Data Trimming. In The SAGE Encyclopedia of Communication Research Methods; SAGE Publications Inc.: Thousand Oaks, CA, USA, 2017; p. 130. ISBN 978-1-4833-8143-5. [Google Scholar]
- Borisov, N.; Tkachev, V.; Muchnik, I.; Buzdin, A. Individual Drug Treatment Prediction in Oncology Based on Machine Learning Using Cell Culture Gene Expression Data; ACM Press: New York, NY, USA, 2017; pp. 1–6. [Google Scholar]
- Borisov, N.; Tkachev, V.; Suntsova, M.; Kovalchuk, O.; Zhavoronkov, A.; Muchnik, I.; Buzdin, A. A method of gene expression data transfer from cell lines to cancer patients for machine-learning prediction of drug efficiency. Cell Cycle 2018, 17, 486–491. [Google Scholar] [CrossRef]
- Borisov, N.; Tkachev, V.; Buzdin, A.; Muchnik, I. Prediction of Drug Efficiency by Transferring Gene Expression Data from Cell Lines to Cancer Patients. In Braverman Readings in Machine Learning. Key Ideas from Inception to Current State; Rozonoer, L., Mirkin, B., Muchnik, I., Eds.; Springer International Publishing: Cham, Switzerland, 2018; Volume 11100, pp. 201–212. ISBN 978-3-319-99491-8. [Google Scholar]
- Arimoto, R.; Prasad, M.-A.; Gifford, E.M. Development of CYP3A4 inhibition models: Comparisons of machine-learning techniques and molecular descriptors. J. Biomol. Screen. 2005, 10, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Balabin, R.M.; Lomakina, E.I. Support vector machine regression (LS-SVM)—An alternative to artificial neural networks (ANNs) for the analysis of quantum chemistry data? Phys. Chem. Chem. Phys. 2011, 13, 11710–11718. [Google Scholar] [CrossRef]
- Balabin, R.M.; Smirnov, S.V. Interpolation and extrapolation problems of multivariate regression in analytical chemistry: Benchmarking the robustness on near-infrared (NIR) spectroscopy data. Analyst 2012, 137, 1604–1610. [Google Scholar] [CrossRef]
- Betrie, G.D.; Tesfamariam, S.; Morin, K.A.; Sadiq, R. Predicting copper concentrations in acid mine drainage: A comparative analysis of five machine learning techniques. Environ. Monit. Assess. 2013, 185, 4171–4182. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-learn: Machine Learning in Python. arXiv 2012, arXiv:1201.0490. [Google Scholar]
- Gent, D.H.; Esker, P.D.; Kriss, A.B. Statistical Power in Plant Pathology Research. Phytopathology 2018, 108, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, J.P.A.; Hozo, I.; Djulbegovic, B. Optimal type I and type II error pairs when the available sample size is fixed. J. Clin. Epidemiol. 2013, 66, 903–910. [Google Scholar] [CrossRef]
- Litière, S.; Alonso, A.; Molenberghs, G. Type I and Type II Error Under Random-Effects Misspecification in Generalized Linear Mixed Models. Biometrics 2007, 63, 1038–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Qiu, Y.; Deng, A. A note on Type S/M errors in hypothesis testing. Br. J. Math. Stat. Psychol. 2019, 72, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetterslev, J.; Jakobsen, J.C.; Gluud, C. Trial Sequential Analysis in systematic reviews with meta-analysis. BMC Med. Res. Methodol. 2017, 17, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, N.; Shabalina, I.; Tkachev, V.; Sorokin, M.; Garazha, A.; Pulin, A.; Eremin, I.I.; Buzdin, A. Shambhala: A platform-agnostic data harmonizer for gene expression data. BMC Bioinf. 2019, 20, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owhadi, H.; Scovel, C. Toward Machine Wald. In Handbook of Uncertainty Quantification; Ghanem, R., Higdon, D., Owhadi, H., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 1–35. ISBN 978-3-319-11259-6. [Google Scholar]
- Owhadi, H.; Scovel, C.; Sullivan, T.J.; McKerns, M.; Ortiz, M. Optimal Uncertainty Quantification. SIAM Rev. 2013, 55, 271–345. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, T.J.; McKerns, M.; Meyer, D.; Theil, F.; Owhadi, H.; Ortiz, M. Optimal uncertainty quantification for legacy data observations of Lipschitz functions. ESAIM Math. Model. Numer. Anal. 2013, 47, 1657–1689. [Google Scholar] [CrossRef] [Green Version]
- Hatzis, C.; Pusztai, L.; Valero, V.; Booser, D.J.; Esserman, L.; Lluch, A.; Vidaurre, T.; Holmes, F.; Souchon, E.; Wang, H.; et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011, 305, 1873–1881. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Iwamoto, T.; Matsuoka, J.; Nogami, T.; Motoki, T.; Shien, T.; Taira, N.; Niikura, N.; Hayashi, N.; Ohtani, S.; et al. Estrogen receptor (ER) mRNA expression and molecular subtype distribution in ER-negative/progesterone receptor-positive breast cancers. Breast Cancer Res. Treat. 2014, 143, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Horak, C.E.; Pusztai, L.; Xing, G.; Trifan, O.C.; Saura, C.; Tseng, L.-M.; Chan, S.; Welcher, R.; Liu, D. Biomarker analysis of neoadjuvant doxorubicin/cyclophosphamide followed by ixabepilone or Paclitaxel in early-stage breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1587–1595. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terragna, C.; Remondini, D.; Martello, M.; Zamagni, E.; Pantani, L.; Patriarca, F.; Pezzi, A.; Levi, G.; Offidani, M.; Proserpio, I.; et al. The genetic and genomic background of multiple myeloma patients achieving complete response after induction therapy with bortezomib, thalidomide and dexamethasone (VTD). Oncotarget 2016, 7, 9666–9679. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.B.; Yip, W.-K.; Minvielle, S.; Broyl, A.; Li, Y.; Hanlon, B.; Swanson, D.; Shah, P.K.; Moreau, P.; van der Holt, B.; et al. Gene expression profile alone is inadequate in predicting complete response in multiple myeloma. Leukemia 2014, 28, 2229–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, M.; Craft, B.; Swatloski, T.; Cline, M.; Morozova, O.; Diekhans, M.; Haussler, D.; Zhu, J. The UCSC Cancer Genomics Browser: Update 2015. Nucleic Acids Res. 2015, 43, D812–D817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, A.L.; Ooms, A.; Gadd, S.; Gerhard, D.S.; Smith, M.A.; Guidry Auvil, J.M.; Meerzaman, D.; Chen, Q.-R.; Hsu, C.H.; Yan, C.; et al. Recurrent DGCR8, DROSHA, and SIX Homeodomain Mutations in Favorable Histology Wilms Tumors. Cancer Cell 2015, 27, 286–297. [Google Scholar] [CrossRef] [Green Version]
- Tricoli, J.V.; Blair, D.G.; Anders, C.K.; Bleyer, W.A.; Boardman, L.A.; Khan, J.; Kummar, S.; Hayes-Lattin, B.; Hunger, S.P.; Merchant, M.; et al. Biologic and clinical characteristics of adolescent and young adult cancers: Acute lymphoblastic leukemia, colorectal cancer, breast cancer, melanoma, and sarcoma: Biology of AYA Cancers. Cancer 2016, 122, 1017–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korde, L.A.; Lusa, L.; McShane, L.; Lebowitz, P.F.; Lukes, L.; Camphausen, K.; Parker, J.S.; Swain, S.M.; Hunter, K.; Zujewski, J.A. Gene expression pathway analysis to predict response to neoadjuvant docetaxel and capecitabine for breast cancer. Breast Cancer Res. Treat. 2010, 119, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R.; Larionov, A. Changes in expression of oestrogen regulated and proliferation genes with neoadjuvant treatment highlight heterogeneity of clinical resistance to the aromatase inhibitor, letrozole. Breast Cancer Res. BCR 2010, 12, R52. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R.; Larionov, A.; Anderson, T.J.; Evans, D.B.; Dixon, J.M. Sequential changes in gene expression profiles in breast cancers during treatment with the aromatase inhibitor, letrozole. Pharmacogenomics J. 2012, 12, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Popovici, V.; Chen, W.; Gallas, B.G.; Hatzis, C.; Shi, W.; Samuelson, F.W.; Nikolsky, Y.; Tsyganova, M.; Ishkin, A.; Nikolskaya, T.; et al. Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. BCR 2010, 12, R5. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, T.; Bianchini, G.; Booser, D.; Qi, Y.; Coutant, C.; Shiang, C.Y.-H.; Santarpia, L.; Matsuoka, J.; Hortobagyi, G.N.; Symmans, W.F.; et al. Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J. Natl. Cancer Inst. 2011, 103, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, T.; Nakayama, T.; Naoi, Y.; Yamamoto, N.; Otani, Y.; Kim, S.J.; Shimazu, K.; Shimomura, A.; Maruyama, N.; Tamaki, Y.; et al. GSTP1 expression predicts poor pathological complete response to neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci. 2012, 103, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Voisin, V.; Bader, G.D.; Deng, T.; Pusztai, L.; Symmans, W.F.; Esteva, F.J.; Egan, S.E.; Zacksenhaus, E. Seventeen-gene signature from enriched Her2/Neu mammary tumor-initiating cells predicts clinical outcome for human HER2+:ERα- breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 5832–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, K.; Qi, Y.; Song, N.; Tian, C.; Rice, S.D.; Gabrin, M.J.; Brower, S.L.; Symmans, W.F.; O’Shaughnessy, J.A.; Holmes, F.A.; et al. Cell line derived multi-gene predictor of pathologic response to neoadjuvant chemotherapy in breast cancer: A validation study on US Oncology 02-103 clinical trial. BMC Med. Genomics 2012, 5, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raponi, M.; Harousseau, J.-L.; Lancet, J.E.; Löwenberg, B.; Stone, R.; Zhang, Y.; Rackoff, W.; Wang, Y.; Atkins, D. Identification of molecular predictors of response in a study of tipifarnib treatment in relapsed and refractory acute myelogenous leukemia. Clin. Cancer Res. 2007, 13, 2254–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, A.K.; Arthur, L.M.; Renshaw, L.; Larionov, A.A.; Kay, C.; Dunbier, A.K.; Thomas, J.S.; Dowsett, M.; Sims, A.H.; Dixon, J.M. Accurate Prediction and Validation of Response to Endocrine Therapy in Breast Cancer. J. Clin. Oncol. 2015, 33, 2270–2278. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Kim, H.-Y. Statistical notes for clinical researchers: Type I and type II errors in statistical decision. Restor. Dent. Endod. 2015, 40, 249. [Google Scholar] [CrossRef]
- Cummins, R.O.; Hazinski, M.F. Guidelines based on fear of type II (false-negative) errors: Why we dropped the pulse check for lay rescuers. Circulation 2000, 102, I377–I379. [Google Scholar] [CrossRef]
- Rodriguez, P.; Maestre, Z.; Martinez-Madrid, M.; Reynoldson, T.B. Evaluating the Type II error rate in a sediment toxicity classification using the Reference Condition Approach. Aquat. Toxicol. 2011, 101, 207–213. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ML Method | Method Type | Median AUC without FloWPS | Median AUC with FloWPS | Paired t-Test p-Value for AUC with-vs.-w/o FloWPS | Advantage of FloWPS | Median SN at B = 4 | Median SP at B = 0.25 |
|---|---|---|---|---|---|---|---|
| SVM | Global | 0.74 | 0.80 | 1.3 × 10−5 | Yes | 0.45 | 0.42 |
| kNN | Local | 0.76 | 0.75 | 0.53 | No | 0.25 | 0.34 |
| RF | Global | 0.74 | 0.82 | 1.3 × 10-5 | Yes | 0.45 | 0.42 |
| RR | Local | 0.80 | 0.79 | 0.16 | No | 0.36 | 0.41 |
| BNB | Global | 0.77 | 0.82 | 2.7 × 10−4 | Yes | 0.51 | 0.58 |
| ADA | Global | 0.70 | 0.76 | 2.4 × 10−4 | Yes | 0.32 | 0.41 |
| MLP | Global | 0.73 | 0.82 | 6.4 × 10−5 | Yes | 0.53 | 0.53 |
| ML Method | Median AUC without FloWPS | Median AUC with FloWPS | Paired t-Test p-Value for AUC with-vs.-w/o FloWPS | Median SN at B = 4 | Median SP at B = 0.25 |
|---|---|---|---|---|---|
| RF | 0.75 | 0.83 | 3.5 × 10−6 | 0.50 | 0.56 |
| BNB | 0.78 | 0.83 | 6.7 × 10−4 | 0.50 | 0.60 |
| MLP | 0.77 | 0.84 | 2.4 × 10−4 | 0.50 | 0.51 |
| Method | Median AUC without FloWPS | Median AUC with FloWPS | Paired t-Test p-Value for AUC with-vs.-w/o FloWPS | Median SN at B = 4 | Median SP at B = 0.25 |
|---|---|---|---|---|---|
| SVM | 0.81 | 0.83 | 0.013 | 0.65 | 0.70 |
| RF | 0.76 | 0.86 | 4.9 × 10−6 | 0.56 | 0.71 |
| BNB | 0.84 | 0.89 | 7.5 × 10−4 | 0.78 | 0.75 |
| MLP | 0.83 | 0.88 | 1.0 × 10−4 | 0.63 | 0.71 |
| SVM | RF | RR | BNB | MLP | |
|---|---|---|---|---|---|
| SVM | 1 | 0.53/0.55 | 0.40/0.39 | 0.37/0.34 | 0.46/0.46 |
| RF | 0.34/0.40 | 1 | 0.51/0.32 | 0.48/0.31 | 0.59/0.38 |
| RR | 0.19/0.14 | 0.35/0.04 | 1 | 0.93/0.79 | 0.89/0.52 |
| BNB | 0.24/0.14 | 0.33/0.09 | 0.88/0.64 | 1 | 0.81/0.46 |
| MLP | 0.33/0.30 | 0.40/0.17 | 0.76/0.06 | 0.61/0.12 | 1 |
| Dataset # | Dataset ID | Min | Median | Mean | Max |
|---|---|---|---|---|---|
| 1 | GSE25066 | 0.41/0.28 | 0.72/0.44 | 0.67/0.46 | 0.93/0.81 |
| 2 | GSE41998 | −0.02/−0.10 | 0.55/0.39 | 0.49/0.35 | 0.87/0.83 |
| 3 | GSE9782 | 0.37/0.19 | 0.58/0.41 | 0.62/0.41 | 0.97/0.88 |
| 4 | GSE39754 | 0.34/0.28 | 0.50/0.37 | 0.54/0.41 | 0.84/0.72 |
| 5 | GSE68871 | 0.50/0.43 | 0.62/0.60 | 0.68/0.64 | 0.95/0.93 |
| 6 | GSE55145 | 0.32/0.29 | 0.57/0.42 | 0.60/0.45 | 0.85/0.70 |
| 7 | TARGET50 | 0.34/0.57 | 0.69/0.74 | 0.66/0.72 | 0.95/0.82 |
| 8 | TARGET10 | 0.32/0.30 | 0.50/0.45 | 0.58/0.48 | 0.90/0.77 |
| 9 | TARGET20 busulfan | 0.63/0.55 | 0.70/0.66 | 0.76/0.70 | 0.97/0.89 |
| 10 | TARGET20 no busulfan | 0.16/0.35 | 0.63/0.53 | 0.60/0.55 | 0.92/0.79 |
| 11 | GSE18728 | 0.38/0.21 | 0.54/0.46 | 0.62/0.45 | 0.95/0.79 |
| 12 | GSE20181 | 0.33/0.17 | 0.43/0.43 | 0.56/0.43 | 0.96/0.79 |
| 13 | GSE20194 | 0.06/0.04 | 0.50/0.30 | 0.49/0.34 | 0.93/0.80 |
| 14 | GSE23988 | 0.28/0.18 | 0.46/0.35 | 0.55/0.39 | 0.96/0.82 |
| 15 | GSE32646 | 0.23/0.11 | 0.37/0.28 | 0.49/0.32 | 0.95/0.74 |
| 16 | GSE37946 | 0.40/0.26 | 0.62/0.45 | 0.62/0.44 | 0.92/0.69 |
| 17 | GSE42822 | 0.34/0.03 | 0.52/0.40 | 0.58/0.38 | 0.89/0.82 |
| 18 | GSE5122 | 0.12/−0.06 | 0.40/0.20 | 0.46/0.25 | 0.93/0.79 |
| 19 | GSE59515 | 0.37/0.26 | 0.47/0.47 | 0.59/0.49 | 0.96/0.74 |
| 20 | TCGA-LGG | 0.27/0.13 | 0.64/0.47 | 0.63/0.42 | 0.94/0.76 |
| 21 | TCGA-LC | 0.44/0.23 | 0.62/0.55 | 0.66/0.53 | 0.95/0.90 |
| Reference | Dataset ID | Disease Type | Treatment | Experimental Platform | Number NC of Cases (R vs. NR) | Number S of Core Marker Genes |
|---|---|---|---|---|---|---|
| [50,51] | GSE25066 | Breast cancer with different hormonal and HER2 status | Neoadjuvant taxane + anthracycline | Affymetrix Human Genome U133 Array | 235 (118 R: Complete response + partial response; 117 NR: Residual disease + progressive disease) | 20 |
| [52] | GSE41998 | Breast cancer with different hormonal and HER2 status | Neoadjuvant doxorubicin + cyclophosphamide, followed by paclitaxel | Affymetrix Human Genome U133 Array | 68 (34 R: Complete response + partial response; 34 NR: Residual disease + progressive disease) | 11 |
| [27] | GSE9782 | Multiple myeloma | Bortezomib monotherapy | Affymetrix Human Genome U133 Array | 169 (85 R: Complete response + partial response; 84 NR: No change + progressive disease) | 18 |
| [53] | GSE39754 | Multiple myeloma | Vincristine + adriamycin + dexamethasone followed by autologous stem cell transplantation (ASCT) | Affymetrix Human Exon 1.0 ST Array | 124 (62 R: Complete, near-complete and very good partial responders, 62 NR: Partial, minor and worse) | 16 |
| [54] | GSE68871 | Multiple myeloma | Bortezomib-thalido-mide-dexamethasone | Affymetrix Human Genome U133 Plus | 98 (49 R: Complete, near-complete and very good partial responders, 49 NR: Partial, minor and worse) | 12 |
| [55] | GSE55145 | Multiple myeloma | Bortezomib followed by ASCT | Affymetrix Human Exon 1.0 ST Array | 56 (28 R: Complete, near-complete and very good partial responders, 28 NR: Partial, minor and worse) | 14 |
| [56,57] | TARGET-50 | Pediatric kidney Wilms tumor | Vincristine sulfate + cyclosporine, cytarabine, daunorubicin + conventional surgery + radiation therapy | Illumina HiSeq 2000 | 72 (36 R, 36 NR: See Reference [8]) | 14 |
| [56,58] | TARGET-10 | Pediatric acute lymphoblastic leukemia | Vincristine sulfate + carboplatin, cyclophosphamide, doxorubicin | Illumina HiSeq 2000 | 60 (30 R, 30 NR: See Reference [8]) | 14 |
| [56] | TARGET-20 | Pediatric acute myeloid leukemia | Non-target drugs (asparaginase, cyclosporine, cytarabine, daunorubicin, etoposide; methotrexate, mitoxantrone), including busulfan and cyclophosphamide | Illumina HiSeq 2000 | 46 (23 R, 23 NR: See Reference [8]) | 10 |
| [56] | TARGET-20 | Pediatric acute myeloid leukemia | Same non-target drugs, but excluding busulfan and cyclophosphamide | Illumina HiSeq 2000 | 124 (62 R, 62 NR: See Reference [8]) | 16 |
| [59] | GSE18728 | Breast cancer | Docetaxel, capecitabine | Affymetrix Human Genome U133 Plus 2.0 Array | 61 (23R: Complete response + partial response; 38 NR: Residual disease + progressive disease) | 16 |
| [60,61] | GSE20181 | Breast cancer | Letrozole | Affymetrix Human Genome U133A Array | 52 (37 R: Complete response + partial response; 15 NR: Residual disease + progressive disease) | 11 |
| [62] | GSE20194 | Breast cancer | Paclitaxel; (tri)fluoroacetyl chloride; 5-fluorouracil, epirubicin, cyclophosphamide | Affymetrix Human Genome U133A Array | 52 (11 R: Complete response + partial response; 41 NR: Residual disease + progressive disease) | 10 |
| [63] | GSE23988 | Breast cancer | Docetaxel, capecitabine | Affymetrix Human Genome U133A Array | 61 (20 R: Complete response + partial response; 41 NR: Residual disease + progressive disease) | 18 |
| [64] | GSE32646 | Breast cancer | Paclitaxel, 5-fluorouracil, epirubicin, cyclophosphamide | Affymetrix Human Genome U133 Plus 2.0 Array | 115 (27 R: Complete response + partial response; 88 NR: Residual disease + progressive disease) | 17 |
| [65] | GSE37946 | Breast cancer | Trastuzumab | Affymetrix Human Genome U133A Array | 50 (27 R: Complete response + partial response; 23 NR: Residual disease + progressive disease) | 14 |
| [66] | GSE42822 | Breast cancer | Docetaxel, 5-fluorouracil, epirubicin, cyclophosphamide, capecitabine | Affymetrix Human Genome U133A Array | 91 (38 R: Complete response + partial response; 53 NR: Residual disease + progressive disease) | 13 |
| [67] | GSE5122 | Acute myeloid leukemia | Tipifarnib | Affymetrix Human Genome U133A Array | 57 (13 R: Complete response + partial response + stable disease; 44 R: Progressive disease) | 10 |
| [68] | GSE59515 | Breast cancer | Letrozole | Illumina HumanHT-12 V4.0 expression beadchip | 75 (51 R: Complete response + partial response; 24 NR: Residual disease + progressive disease) | 15 |
| [69] | TCGA-LGG | Low-grade glioma | Temozolomide + (optionally) mibefradil | Illumina HiSeq 2000 | 131 (100 R: Complete response + partial response + stable disease; 31 NR: Progressive disease) | 9 |
| [69] | TCGA-LC | Lung cancer | Paclitaxel + (optionally),cisplatin/carboplatin, reolysin | Illumina HiSeq 2000 | 41 (24 R: Complete response + partial response + stable disease; 17 NR: Progressive disease) | 7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tkachev, V.; Sorokin, M.; Borisov, C.; Garazha, A.; Buzdin, A.; Borisov, N. Flexible Data Trimming Improves Performance of Global Machine Learning Methods in Omics-Based Personalized Oncology. Int. J. Mol. Sci. 2020, 21, 713. https://doi.org/10.3390/ijms21030713
Tkachev V, Sorokin M, Borisov C, Garazha A, Buzdin A, Borisov N. Flexible Data Trimming Improves Performance of Global Machine Learning Methods in Omics-Based Personalized Oncology. International Journal of Molecular Sciences. 2020; 21(3):713. https://doi.org/10.3390/ijms21030713
Chicago/Turabian StyleTkachev, Victor, Maxim Sorokin, Constantin Borisov, Andrew Garazha, Anton Buzdin, and Nicolas Borisov. 2020. "Flexible Data Trimming Improves Performance of Global Machine Learning Methods in Omics-Based Personalized Oncology" International Journal of Molecular Sciences 21, no. 3: 713. https://doi.org/10.3390/ijms21030713
APA StyleTkachev, V., Sorokin, M., Borisov, C., Garazha, A., Buzdin, A., & Borisov, N. (2020). Flexible Data Trimming Improves Performance of Global Machine Learning Methods in Omics-Based Personalized Oncology. International Journal of Molecular Sciences, 21(3), 713. https://doi.org/10.3390/ijms21030713