Effects of Dietary Anaplerotic and Ketogenic Energy Sources on Renal Fatty Acid Oxidation Induced by Clofibrate in Suckling Neonatal Pigs

 and
and
Abstract
:1. Introduction
2. Results
2.1. Animal Growth Performance
2.2. Renal Fatty Acid Profile
2.3. Renal β-hydroxybutyrate and Acetate Concentrations
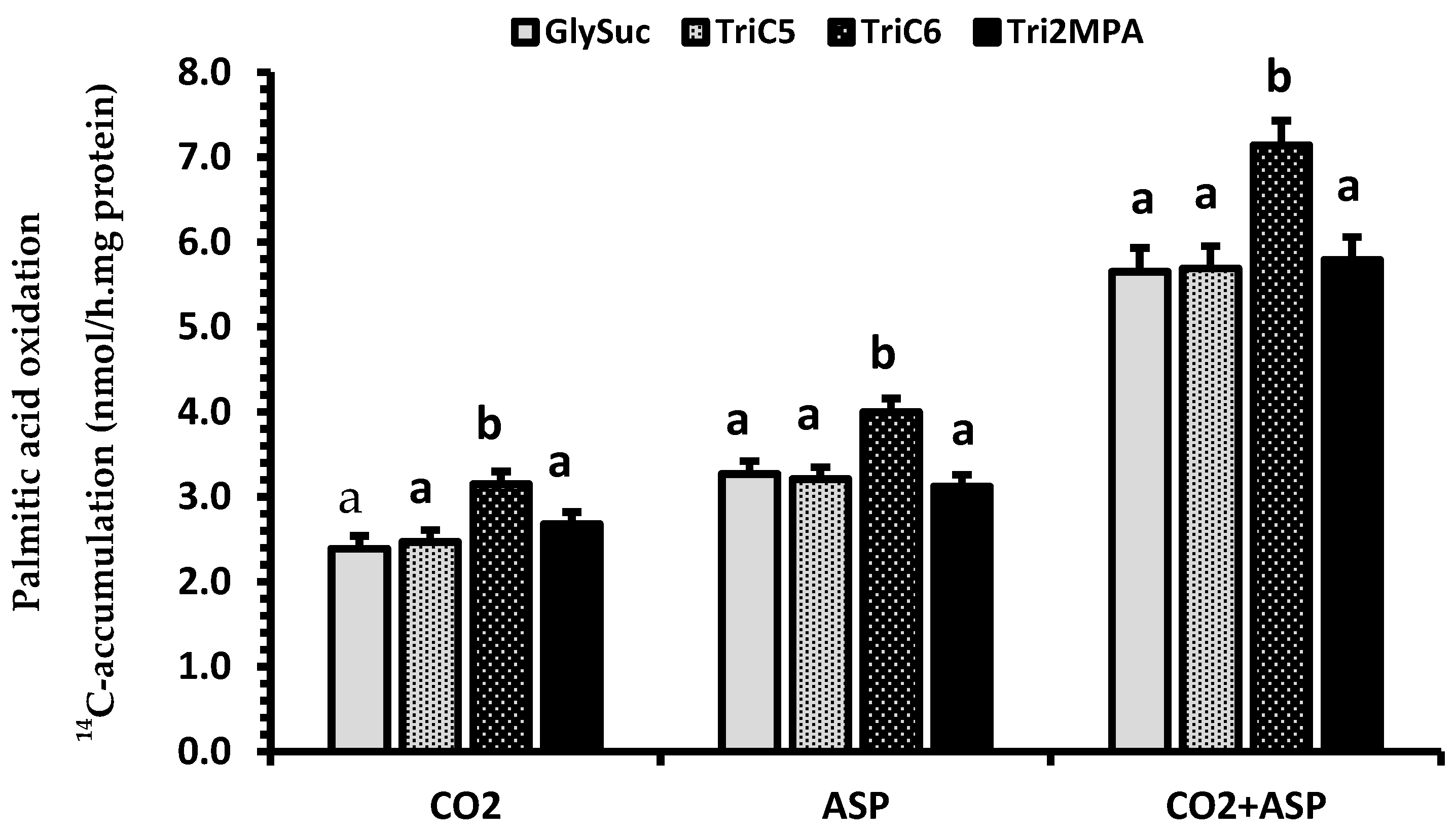
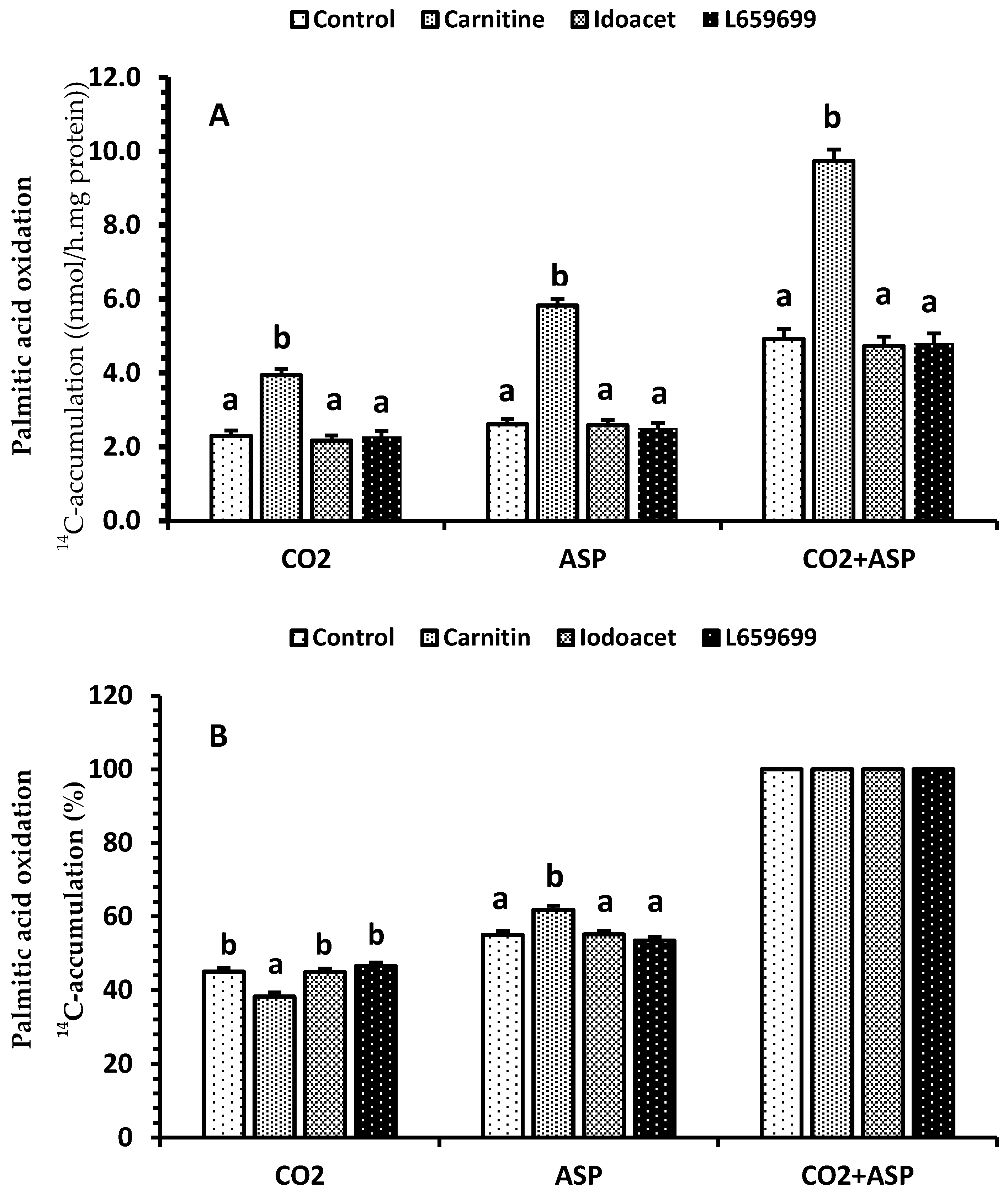
2.4. Renal Palmitic Acid Oxidation
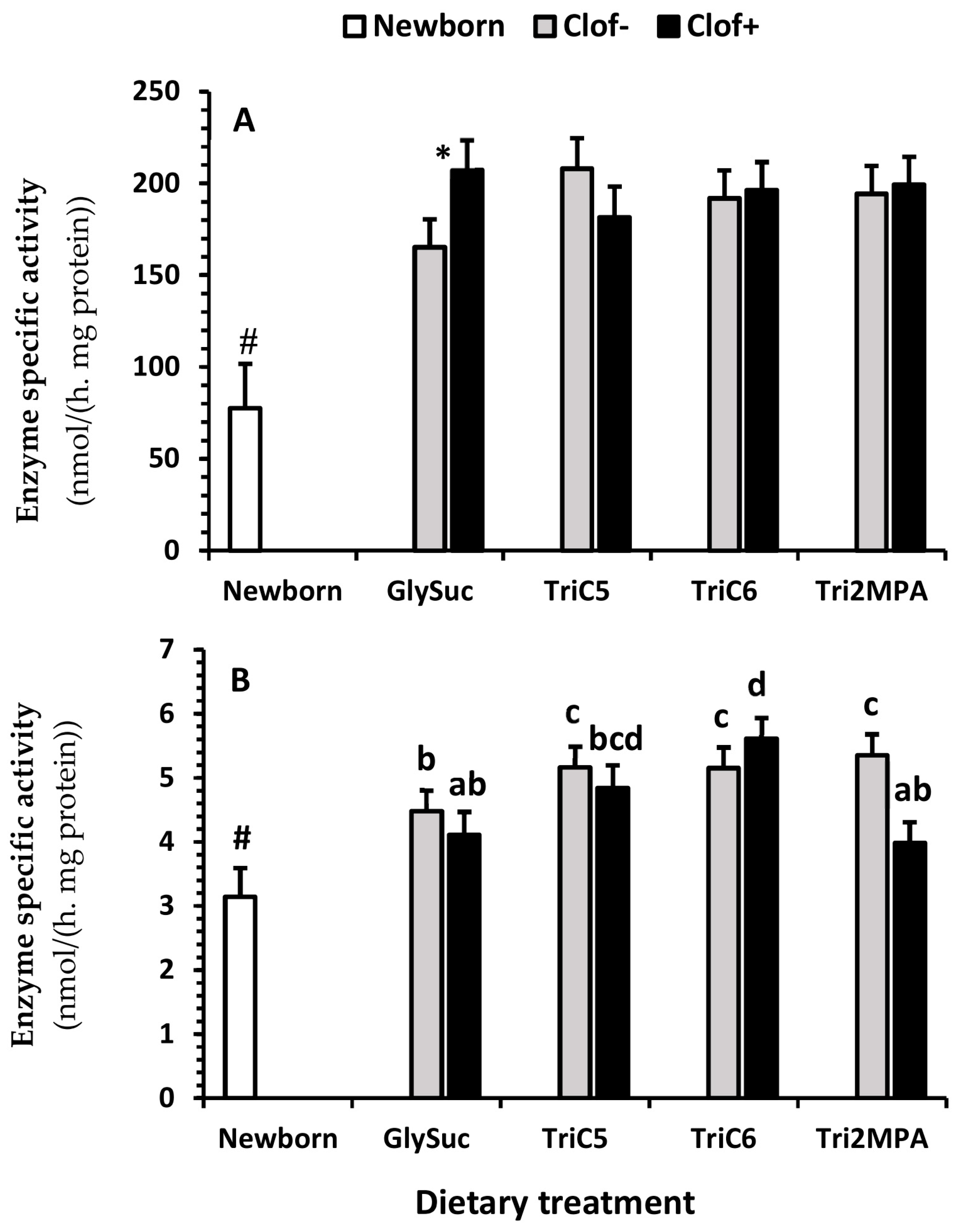
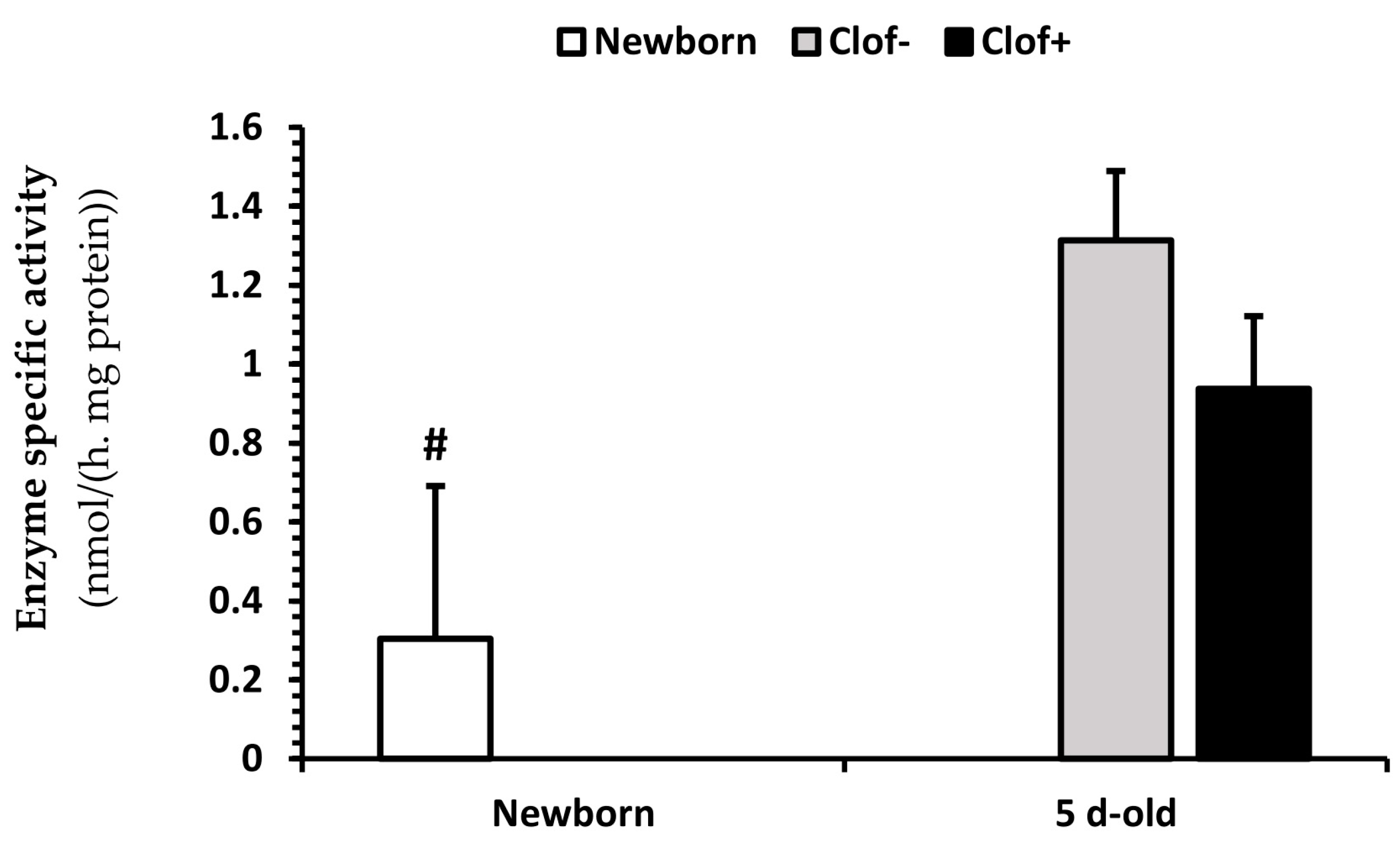
2.5. Enzyme Activity
2.6. Gene Expression
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Fatty Acid Oxidation Measurement In Vitro
4.3. Metabolite Assays
4.4. Enzyme Assays
4.5. Gene Expression
4.6. Chemicals
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAC | Citric acid cycle |
| GlySuc | Glycerol-succinate |
| TriC5 | Triglycerides of valeric acid |
| TriC6 | Triglycerides of hexanoic acid |
| TriMPA | Triglycerides of 2-methylpentonoic acid |
| ASP | Acid soluble products |
| PPARα | Peroxisome proliferator-activated receptor α |
| CKD | Chronic kidney disease |
| mHMGCS | Mitochondrial 3-hydroxy-3-methyl-glutaryl-CoA synthase |
| MCFA | Medium-chain fatty acids |
| AACD | Acetoacetyl-CoA deacylase |
References
- Takahashi, K.; Kamijo, Y.; Hora, K.; Hashimoto, K.; Higuchi, M.; Nakajima, T.; Ehara, T.; Shigematsu, H.; Gonzalez, F.J.; Aoyama, T. Pretreatment by low-dose fibrates protects against acute free fatty acid-induced renal tubule toxicity by counteracting PPARα deterioration. Toxicol. Appl. Pharmacol. 2011, 252, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Muroya, Y.; Ito, O. Effect of clofibrate on fatty acid metabolism in the kidney of puromycin-induced nephrotic rats. Clin. Exp. Nephrol. 2016, 20, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Chuppa, S.; Liang, M.; Liu, P.; Liu, Y.; Casati, M.C.; Cowley, A.W.; Patullo, L.; Kriegel, A.J. MicroRNA-21 regulates peroxisome proliferator-activated receptor alpha, a molecular mechanism of cardiac pathology in Cardiorenal Syndrome Type 4. Kidney Int. 2018, 93, 375–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fall, C.H. Fetal malnutrition and long-term outcomes. Nestle Nutr. Inst. Workshop Ser. 2013, 74, 11–25. [Google Scholar]
- Sampaio, L.S.; da Silva, P.A.; Ribeiro, V.S.; Castro-Chaves, C.; Lara, L.S.; Vieyra, A.; Einicker-Lamas, M. Bioactive lipids are altered in the kidney of chronic undernourished rats: Is there any correlation with the progression of prevalent nephropathies? Lipids Health Dis. 2017, 16, 245. [Google Scholar] [CrossRef] [Green Version]
- Drożdż, D.; Kwinta, P.; Sztefko, K.; Kordon, Z.; Drożdż, T.; Łątka, M.; Miklaszewska, M.; Zachwieja, K.; Rudziński, A.; Pietrzyk, J.A. Oxidative Stress Biomarkers and Left Ventricular Hypertrophy in Children with Chronic Kidney Disease. Oxid. Med. Cell Longev. 2016, 2016, 7520231. [Google Scholar] [CrossRef] [Green Version]
- Portilla, D.; Dai, G.; Peters, J.M.; Gonzalez, F.J.; Crew, M.D.; Proia, A.D. Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am. J. Physiol. Renal Physiol. 2000, 278, F667–F675. [Google Scholar] [CrossRef] [Green Version]
- Veerkamp, J.H.; van Moerkerk, H.T. Peroxisomal fatty acid oxidation in rat and human tissues. Effect of nutritional state, clofibrate treatment and postnatal development in the rat. Biochim. Biophys. Acta 1986, 875, 301–310. [Google Scholar] [CrossRef]
- Ouali, F.; Djouadi, F.; Merlet-Bénichou, C.; Bastin, J. Dietary lipids regulate beta-oxidation enzyme gene expression in the developing rat kidney. Am. J. Physiol. 1998, 275, F777–F784. [Google Scholar]
- Vamecq, J.; Draye, J.P. Pathophysiology of peroxisomal beta-oxidation. Essays Biochem. 1989, 24, 115–225. [Google Scholar]
- Palmer, C.N.; Hsu, M.H.; Griffin, K.J.; Raucy, J.L.; Johnson, E.F. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol. Pharmacol. 1998, 53, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odle, J.; Lin, X.; Jacobi, S.K.; Kim, S.W.; Stahl, C.H. The suckling piglet as an agrimedical model for the study of pediatric nutrition and metabolism. Annu. Rev. Anim. Biosci. 2014, 2, 419–444. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Khan, I.; Bai, X.; Odle, J.; Xi, L. Activation of PPARα by Oral Clofibrate Increases Renal Fatty Acid Oxidation in Developing Pigs. Int. J. Mol. Sci. 2017, 18, 2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Shim, K.; Odle, J. Carnitine palmitoyltransferase I control of acetogenesis, the major pathway of fatty acid {beta}-oxidation in liver of neonatal swine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1435–R1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, K.; Jacobi, S.; Odle, J.; Lin, X. Pharmacologic activation of peroxisome proliferator-activating receptor-α accelerates hepatic fatty acid oxidation in neonatal pigs. Oncotarget 2018, 9, 23900–23914. [Google Scholar] [CrossRef] [Green Version]
- Courchesne-Loyer, A.; Fortier, M.; Tremblay-Mercier, J.; Chouinard-Watkins, R.; Roy, M.; Nugent, S.; Castellano, C.A.; Cunnane, S.C. Stimulation of mild, sustained ketonemia by medium-chain triacylglycerols in healthy humans: Estimated potential contribution to brain energy metabolism. Nutrition 2013, 29, 635–640. [Google Scholar] [CrossRef]
- Lowe, D.M.; Tubbs, P.K. Succinylation and inactivation of 3-hydroxy-3-methylglutaryl-CoA synthase by succinyl-CoA and its possible relevance to the control of ketogenesis. Biochem. J. 1985, 232, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Freund, N.; Sedraoui, M.; Geloso, J.P. Fatty acid oxidation by developing rat kidney. Biol. Neonate. 1984, 45, 183–187. [Google Scholar] [CrossRef]
- Bai, X.; Lin, X.; Drayton, J.; Liu, Y.; Ji, C.; Odle, J. Clofibrate increases long-chain fatty acid oxidation by neonatal pigs. J. Nutr. 2014, 144, 1688–1693. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.H.; Alho, C.S.; Asins, G.; Hegardt, F.G.; Marrero, P.F. Gene expression of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in a poorly ketogenic mammal: Effect of starvation during the neonatal period of the piglet. Biochem. J. 1997, 324, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Odle, J. New insights into the utilization of medium-chain triglycerides by the neonate: Observations from a piglet model. J. Nutr. 1997, 127, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Owen, O.E.; Felig, P.; Morgan, A.P.; Wahren, J.; Cahill, G.F., Jr. Liver and kidney metabolism during prolonged starvation. J. Clin. Investig. 1969, 48, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Smalley, K.J.; D’Alessio, D.A.; Mozzoli, M.A.; Dawson, E.K. Protein, fat, and carbohydrate requirements during starvation: Anaplerosis and cataplerosis. Am. J. Clin. Nutr. 1998, 68, 12–34. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reszko, A.E.; Kasumov, T.; Pierce, B.A.; David, F.; Hoppel, C.L.; Stanley, W.C.; Des Rosiers, C.; Brunengraber, H. Assessing the reversibility of the anaplerotic reactions of the propionyl-CoA pathway in heart and liver. J. Biol. Chem. 2003, 278, 34959–34965. [Google Scholar] [CrossRef] [Green Version]
- Tian, Q.; Grzemski, F.A.; Panagiotopoulos, S.; Ahokas, J.T. Peroxisome proliferator-activated receptor alpha agonist, clofibrate, has profound influence on myocardial fatty acid composition. Chem. Biol. Interact. 2006, 160, 241–251. [Google Scholar] [CrossRef]
- Craig, S.R.; Gatlin, D.M. 3rd. Coconut oil and beef tallow, but not tricaprylin, can replace menhaden oil in the diet of red drum (Sciaenops ocellatus) without adversely affecting growth or fatty acid composition. J. Nutr. 1995, 125, 3041–3048. [Google Scholar]
- Garcia Caraballo, S.C.; Comhair, T.M.; Houten, S.M.; Dejong, C.H.; Lamers, W.H.; Koehler, S.E. High-protein diets prevent steatosis and induce hepatic accumulation of monomethyl branched-chain fatty acids. J. Nutr. Biochem. 2014, 25, 1263–1274. [Google Scholar] [CrossRef]
- Fischer, M.; Varady, J.; Hirche, F.; Kluge, H.; Eder, K. Supplementation of L-carnitine in pigs: Absorption of carnitine and effect on plasma and tissue carnitine concentrations. Arch. Anim. Nutr. 2009, 63, 1–15. [Google Scholar] [CrossRef]
- Broderick, T.L.; Cusimano, F.A.; Carlson, C.; Tamura, L.K. Acute Exercise Stimulates Carnitine Biosynthesis and OCTN2 Expression in Mouse Kidney. Kidney Blood Press Res. 2017, 42, 398–405. [Google Scholar] [CrossRef]
- Ringseis, R.; Keller, J.; Eder, K. Basic mechanisms of the regulation of L-carnitine status in monogastrics and efficacy of L-carnitine as a feed additive in pigs and poultry. J. Anim. Physiol. Anim. Nutr. 2018, 102, 1686–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.R.; Goodband, R.D.; Tokach, M.D.; Dritz, S.S.; Nelssen, J.L.; Minton, J.E.; Higgins, J.J.; Lin, X.; Odle, J.; Woodworth, J.C.; et al. Effects of feeding L-carnitine to gilts through day 70 of gestation on litter traits and the expression of insulin-like growth factor system components and L-carnitine concentration in foetal tissues. J. Anim. Physiol. Anim. Nutr. 2008, 92, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Van Kempen, T.A.; Odle, J. Medium-chain fatty acid oxidation in colostrum-deprived newborn piglets: Stimulative effect of L-carnitine supplementation. J. Nutr. 1993, 123, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Marín, V.B.; Azocar, M.; Molina, M.; Guerrero, J.L.; Ratner, R.; Cano, F. Total carnitine and acylated carnitine ratio: Relationship of free carnitine with lipid parameters in pediatric dialysis patients. Adv. Perit. Dial. 2006, 22, 130–135. [Google Scholar] [PubMed]
- Heo, K.N.; Lin, X.; Han, I.K.; Odle, J. Medium-chain fatty acids but not L-carnitine accelerate the kinetics of [14C]triacylglycerol utilization by colostrum-deprived newborn pigs. J. Nutr. 2002, 132, 1989–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corl, B.A.; Mathews Oliver, S.A.; Lin, X.; Oliver, W.T.; Ma, Y.; Harrell, R.J.; Odle, J. Conjugated linoleic acid reduces body fat accretion and lipogenic gene expression in neonatal pigs fed low- or high-fat formulas. J. Nutr. 2008, 138, 449–454. [Google Scholar] [CrossRef]
- Greenspan, M.D.; Yudkovitz, J.B.; Lo, C.Y.; Chen, J.S.; Alberts, A.W.; Hunt, V.M.; Chang, M.N.; Yang, S.S.; Thompson, K.L.; Chiang, Y.C. Inhibition of hydroxymethylglutaryl-coenzyme A synthase by L-659,699. Proc. Natl. Acad. Sci. USA 1987, 84, 7488–7492. [Google Scholar] [CrossRef] [Green Version]
- Burch, R.E.; Wertheim, A.R. Subcellular localization of acetoacetyl-CoA deacylase and its role in acetoacetate synthesis. Am. J. Clin. Nutr. 1973, 26, 814–822. [Google Scholar] [CrossRef]
- Walter, K.R.; Lin, X.; Jacobi, S.K.; Käser, T.; Esposito, D.; Odle, J. Dietary arachidonate in milk replacer triggers dual benefits of PGE2 signaling in LPS-challenged piglet alveolar macrophages. J. Anim. Sci. Biotechnol. 2019. [Google Scholar] [CrossRef]
- Hügler, M.; Krieger, R.S.; Jahn, M.; Fuchs, G. Characterization of acetyl-CoA/propionyl-CoA carboxylase in Metallosphaera sedula. Carboxylating enzyme in the 3-hydroxypropionate cycle for autotrophic carbon fixation. Eur. J. Biochem. 2003, 270, 736–744. [Google Scholar] [CrossRef]
- Peffer, P.L.; Lin, X.; Odle, J. Hepatic beta-oxidation and carnitine palmitoyltransferase I in neonatal pigs after dietary treatments of clofibric acid, isoproterenol, and medium-chain triglycerides. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1518–R1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Effects @ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clofibrate | Medium-Chain Fatty Acid | |||||||||||
| FA | NB † | Clof− | Clo+ | SEM | p-V | GlySuc | TriC5 | TriC6 | Tri2MPA | SEM | p-V | |
| µg/100 mg Tissue | ||||||||||||
| C14:0 | 2.49 | 0.46 | 0.89 | 0.18 | 0.087 | 0.48 | 0.81 | 0.90 | 0.52 | 0.25 | 0.538 | |
| C15:0 | 1.26 | 0.25 | 0.37 | 0.06 | 0.201 | 0.09 a | 0.38 b | 0.11 a | 0.68 c | 0.08 | 0.001 | |
| C16:0 | 149 | 122 | 123 | 47.0 | 0.742 | 127 | 124 | 132 | 108 | 6.56 | 0.081 | |
| C16:1n9 | 14.5 | 2.79 | 4.72 * | 0.50 | 0.001 | 3.17 | 4.41 | 3.94 | 3.40 | 0.71 | 0.605 | |
| C17:1 | 0.68 | 0.38 | 0.46 | 0.10 | 0.637 | 0.23 | 0.39 | 0.42 | 0.60 | 0.14 | 0.288 | |
| C18:0 | 93.3 | 102 | 95.5 | 3.19 | 0.834 | 99.3 | 99.5 | 103 | 94.0 | 4.59 | 0.533 | |
| C18:1n9 | 175 | 113 | 136 * | 7.00 | 0.041 | 102 | 125 | 137 | 132 | 9.84 | 0.087 | |
| C18:2n6 | 64.2 | 117 | 132 | 6.39 | 0.101 | 140 b | 127 b | 136 b | 92.5 a | 8.96 | 0.002 | |
| C18:3n6 | 0.63 | 0.46 | 0.71 * | 0.06 | 0.005 | 0.53 | 0.58 | 0.49 | 0.73 | 0.09 | 0.228 | |
| C18:3n3 | 0.36 | 2.20 | 3.06 * | 0.23 | 0.013 | 3.16 | 2.82 | 2.07 | 2.48 | 0.34 | 0.124 | |
| C20:0 | 4.46 | 3.91 | 3.74 | 0.19 | 0.570 | 3.88 | 3.80 | 3.80 | 3.85 | 0.28 | 0.995 | |
| C20:1n9 | 1.98 | 0.83 | 0.96 | 0.06 | 0.190 | 1.02 | 0.98 | 0.86 | 0.71 | 0.09 | 0.091 | |
| C20:2n7 | 6.90 | 6.02 | 6.58 | 0.45 | 0.481 | 7.76 b | 6.63 ab | 6.17 ab | 4.84 a | 0.07 | 0.022 | |
| C20:3n6 | 6.81 | 4.65 | 5.54 | 0.35 | 0.091 | 4.70 ab | 5.88 b | 5.78 b | 3.97 a | 0.06 | 0.028 | |
| C20:4n6 | 155 | 120 | 106 | 5.55 | 0.088 | 115 | 120 | 117 | 100 | 8.12 | 0.303 | |
| C20:3n3 | 0.48 | 3.20 | 2.50 | 0.48 | 0.168 | 2.99 | 2.45 | 3.86 | 2.09 | 0.68 | 0.292 | |
| C20:5n3 | 1.28 | 1.52 | 1.75 | 0.11 | 0.223 | 1.53 | 1.68 | 1.80 | 1.47 | 0.16 | 0.492 | |
| C22:0 | 3.51 | 4.20 | 3.67 | 0.20 | 0.068 | 4.10 | 4.02 | 3.97 | 3.67 | 0.28 | 0.953 | |
| C22:1n9 | 0.30 | 0.26 | 0.30 | 0.07 | 0.709 | 0.31 | 0.38 | 0.22 | 0.19 | 0.09 | 0.516 | |
| C22:2 | 0.29 | 0.36 | 0.43 | 0.07 | 0.508 | 0.55 | 0.36 | 0.21 | 0.46 | 0.09 | 0.079 | |
| C23:0 | 1.62 | 1.50 | 1.76 | 0.15 | 0.298 | 1.46 | 1.50 | 1.62 | 1.88 | 0.24 | 0.494 | |
| C22:5n3 | 11.8 | 7.94 | 8.54 | 0.55 | 0.388 | 8.83 | 8.30 | 8.08 | 7.36 | 0.78 | 0.342 | |
| C24:0 | 4.43 | 5.74 | 5.58 | 0.28 | 0.662 | 6.14 b | 5.85 b | 6.03 b | 4.56 a | 0.39 | 0.022 | |
| C22:6n3 | 8.35 | 10.6 | 8.13 * | 0.66 | 0.009 | 9.94 | 10.0 | 9.55 | 7.87 | 0.93 | 0.324 | |
| C24:1 | 10.3 | 8.89 | 8.10 | 0.54 | 0.260 | 8.84 | 9.22 | 8.80 | 6.95 | 0.77 | 0.164 | |
| Sum | 789 | 697 | 722 | 21.1 | 0.424 | 731 b | 732 b | 759 b | 617 a | 30.3 | 0.009 | |
| MUFA | 203 | 127 | 149 * | 7.46 | 0.043 | 139 | 151 | 145 | 114 | 10.8 | 0.080 | |
| PUFA | 255 | 275 | 274 | 11.3 | 0.964 | 296 b | 286 b | 291 b | 223 a | 16.6 | 0.009 | |
| FAn3 | 21.2 | 25.6 | 23.7 | 1.56 | 0.774 | 26.5 | 25.3 | 25.4 | 21.3 | 2.24 | 0.343 | |
| FAn6 | 227 | 243 | 243 | 10.1 | 0.971 | 260 b | 253 b | 259 b | 197 a | 14.6 | 0.008 | |
| Fan6/n3 | 11.5 | 10.5 | 10.9 | 0.55 | 0.556 | 10.6 | 10.9 | 11.2 | 10.1 | 0.81 | 0.447 | |
| β-Hydroxybutyrate | Acetate | |||
|---|---|---|---|---|
| Main Effects @ | LSmean | SEM | LSmean | SEM |
| µmol/g Tissue | µmol/g Tissue | |||
| Clofibrate† | ||||
| Clof− | 1.68 | 0.12 | 197 | 23.8 |
| Clof+ | 1.94 | 0.12 | 210 | 24.7 |
| p-Value | 0.13 | 0.70 | ||
| Dietary medium-chain fatty acid * | ||||
| GlySuc | 1.42 a | 0.17 | 194 | 35.1 |
| TriC5 | 1.78 ab | 0.17 | 224 | 37.5 |
| TriC6 | 1.89 b | 0.16 | 204 | 32.1 |
| Tri2MPA | 2.14 b | 0.16 | 192 | 32.1 |
| p-Value | 0.023 | 0.91 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Pike, B.; Zhao, J.; Fan, Y.; Zhu, Y.; Zhang, Y.; Wang, F.; Odle, J. Effects of Dietary Anaplerotic and Ketogenic Energy Sources on Renal Fatty Acid Oxidation Induced by Clofibrate in Suckling Neonatal Pigs. Int. J. Mol. Sci. 2020, 21, 726. https://doi.org/10.3390/ijms21030726
Lin X, Pike B, Zhao J, Fan Y, Zhu Y, Zhang Y, Wang F, Odle J. Effects of Dietary Anaplerotic and Ketogenic Energy Sources on Renal Fatty Acid Oxidation Induced by Clofibrate in Suckling Neonatal Pigs. International Journal of Molecular Sciences. 2020; 21(3):726. https://doi.org/10.3390/ijms21030726
Chicago/Turabian StyleLin, Xi, Brandon Pike, Jinan Zhao, Yu Fan, Yongwen Zhu, Yong Zhang, Feng Wang, and Jack Odle. 2020. "Effects of Dietary Anaplerotic and Ketogenic Energy Sources on Renal Fatty Acid Oxidation Induced by Clofibrate in Suckling Neonatal Pigs" International Journal of Molecular Sciences 21, no. 3: 726. https://doi.org/10.3390/ijms21030726
APA StyleLin, X., Pike, B., Zhao, J., Fan, Y., Zhu, Y., Zhang, Y., Wang, F., & Odle, J. (2020). Effects of Dietary Anaplerotic and Ketogenic Energy Sources on Renal Fatty Acid Oxidation Induced by Clofibrate in Suckling Neonatal Pigs. International Journal of Molecular Sciences, 21(3), 726. https://doi.org/10.3390/ijms21030726