DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy



Abstract
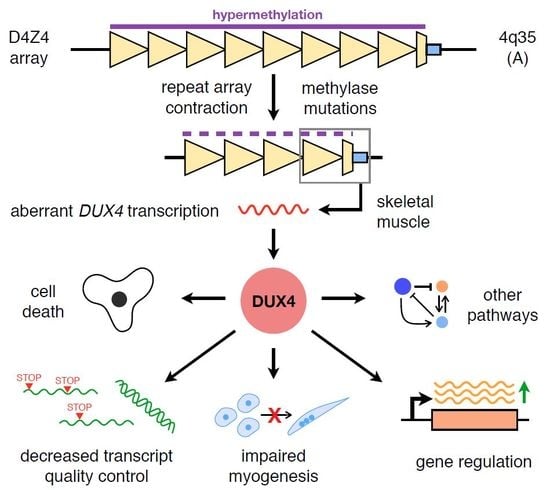
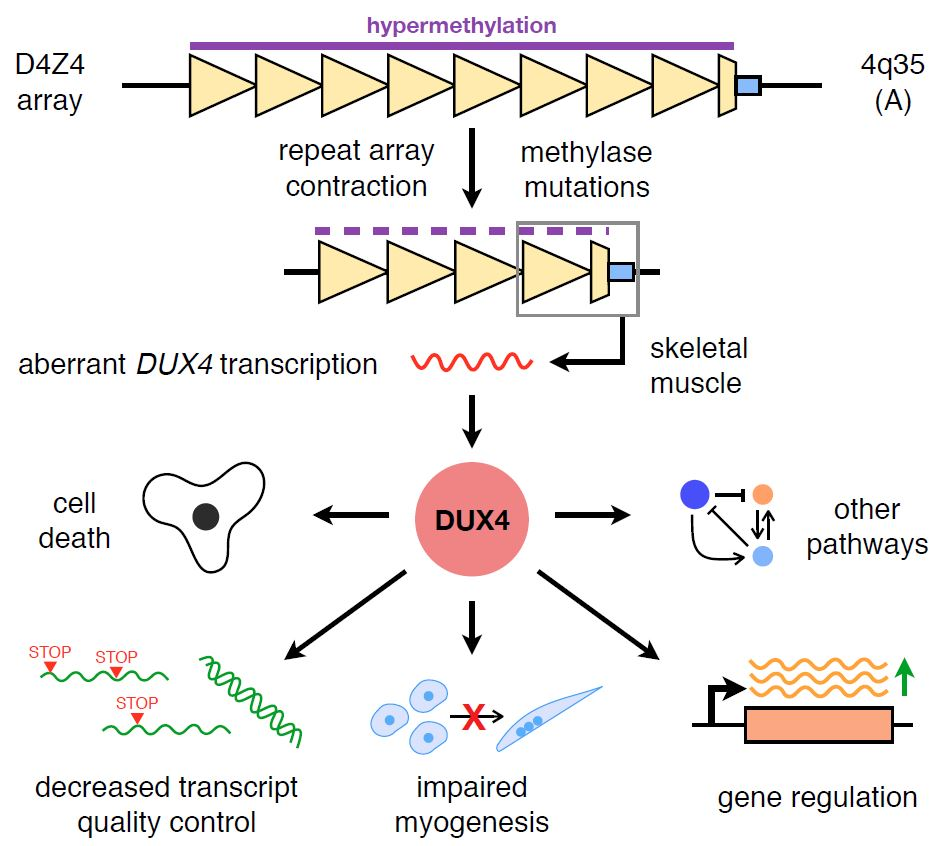
:
{kind=link}
{kind=link}
1. Introduction: Facioscapulohumeral Muscular Dystrophy and DUX4
2. Clinical Characteristics of FSHD
2.1. Skeletal Muscle Manifestations
2.2. Extramuscular Manifestations
2.3. Early-Onset FSHD
3. DUX4 in Skeletal Muscle Signalling, Growth, and Development
3.1. Cell Death
3.2. Oxidative Stress
3.3. Muscle Development
3.4. Transcript Quality Control
3.5. Immune Response Activation
3.6. Gene Regulation
3.7. Other Pathways
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FSHD | facioscapulohumeral muscular dystrophy |
| DUX4 | double homeobox protein 4 |
| ORF | open reading frame |
| AAV | adeno-associated virus |
| ROS | reactive oxygen species |
| RTK | receptor tyrosine kinase |
| NMD | nonsense-mediated decay |
| NADPH | reduced nicotinamide adenine dinucleotide phosphate |
| cAMP | cyclic adenosine monophosphate |
| MaLR | mammalian apparent LTR retrotransposon |
| PML | promyelocytic leukemia |
References
- Wang, L.H.; Tawil, R. Facioscapulohumeral Dystrophy. Curr. Neurol. Neurosci. Rep. 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Pastorello, E.; Cao, M.; Trevisan, C.P. Atypical onset in a series of 122 cases with FacioScapuloHumeral Muscular Dystrophy. Clin. Neurol. Neurosurg. 2012, 114, 230–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mah, J.; Chen, Y.-W. A Pediatric Review of Facioscapulohumeral Muscular Dystrophy. J. Pediatr. Neurol. 2018, 16, 222–231. [Google Scholar] [PubMed]
- Klinge, L.; Eagle, M.; Haggerty, I.D.; Roberts, C.E.; Straub, V.; Bushby, K.M. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2006, 16, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. Facioscapulohumeral dystrophy: The path to consensus on pathophysiology. Skelet. Muscle 2014, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonini, M.M.O.; Passos-Bueno, M.R.; Cerqueira, A.; Matioli, S.R.; Pavanello, R.; Zatz, M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 2004, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.; Coppée, F.; Thomas, N.; Belayew, A.; Upadhyaya, M. Facioscapulohumeral muscular dystrophy (FSHD): An enigma unravelled? Hum. Genet. 2012, 131, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Deenen, J.C.W.; Arnts, H.; van der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.G.M.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.M.; van Engelen, B.G.M. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef] [Green Version]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.-M.; Hofker, M.H.; Moerer, P.; Williamson, R.; et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992, 2, 26–30. [Google Scholar] [CrossRef]
- Deutekom, J.C.T.V.; Wljmenga, C.; Tlenhoven, E.A.E.V.; Gruter, A.-M.; Hewitt, J.E.; Padberg, G.W.; Ommen, G.-J.B.V.; Hofker, M.H.; Fronts, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef]
- Gabriëls, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32. [Google Scholar] [CrossRef]
- Hewitt, J.E.; Lyle, R.; Clark, L.N.; Valleley, E.M.; Wright, T.J.; Wijmenga, C.; van Deutekom, J.C.T.; Francis, F.; Sharpe, P.T.; Hofker, M.; et al. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystropothhy. Hum. Mol. Genet. 1994, 3, 1287–1295. [Google Scholar] [CrossRef]
- Van Overveld, P.G.M.; Lemmers, R.J.F.L.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.-J.B.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003, 35, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.L.F.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, J.E. Loss of epigenetic silencing of the DUX4 transcription factor gene in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2015, 24, R17–R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Greef, J.C.; Lemmers, R.J.L.F.; Camaño, P.; Day, J.W.; Sacconi, S.; Dunand, M.; van Engelen, B.G.M.; Kiuru-Enari, S.; Padberg, G.W.; Rosa, A.L.; et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 2010, 75, 1548–1554. [Google Scholar] [CrossRef] [Green Version]
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genomics Hum. Genet. 2019, 20, 265–291. [Google Scholar] [CrossRef]
- Lemmers, R.J.L.F.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.E.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Van den Boogaard, M.L.; Lemmers, R.J.L.F.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.L.F.; Goeman, J.J.; van der Vliet, P.J.; van Nieuwenhuizen, M.P.; Balog, J.; Vos-Versteeg, M.; Camano, P.; Ramos Arroyo, M.A.; Jerico, I.; Rogers, M.T.; et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 2015, 24, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Sacconi, S.; Briand-Suleau, A.; Gros, M.; Baudoin, C.; Lemmers, R.J.L.F.; Rondeau, S.; Lagha, N.; Nigumann, P.; Cambieri, C.; Puma, A.; et al. FSHD1 and FSHD2 form a disease continuum. Neurology 2019, 92, e2273–e2285. [Google Scholar] [CrossRef] [PubMed]
- Snider, L.; Geng, L.N.; Lemmers, R.J.L.F.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Chadwick, B.P. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS ONE 2016, 11, e0160022. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, H.; Ishimaru, S.; Homma, S.; Yu, B.; Honma, Y.; Beermann, M.L.; Miller, J.B. Functional domains of the FSHD-associated DUX4 protein. Biol. Open 2018, 7, bio033977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Snider, L.; Balog, J.; Lemmers, R.J.L.F.; Van Der Maarel, S.M.; Tawil, R.; Tapscott, S.J. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 2014, 23, 5342–5352. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, P.G.; Doráis, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved roles for murine DUX and human DUX4 in activating cleavage stage genes and MERVL / HERVL retrotransposons. Nat. Genet. 2017, 49, 925–934. [Google Scholar] [CrossRef]
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017, 49, 941–945. [Google Scholar] [CrossRef]
- Tawil, R. Facioscapulohumeral muscular dystrophy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 148, pp. 541–548. [Google Scholar]
- Tawil, R.; Van Der Maarel, S.M. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006, 34, 1–15. [Google Scholar] [CrossRef]
- Statland, J.; Tawil, R. Facioscapulohumeral muscular dystrophy. Neurol. Clin. 2014, 32, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Hamel, J.; Tawil, R. Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurotherapeutics 2018, 15, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Statland, J.M.; Tawil, R. Facioscapulohumeral Muscular Dystrophy. Continuum (Minneap. Minn). 2016, 22, 1916–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eger, K.; Jordan, B.; Habermann, S.; Zierz, S. Beevor’s sign in facioscapulohumeral muscular dystrophy: An old sign with new implications. J. Neurol. 2010, 257, 436–438. [Google Scholar] [CrossRef]
- Statland, J.M.; Shah, B.; Henderson, D.; Van Der Maarel, S.; Tapscott, S.J.; Tawil, R. Muscle pathology grade for facioscapulohumeral muscular dystrophy biopsies. Muscle Nerve 2015, 52, 521–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, M.A.; Eichinger, K.J.; Donlin-Smith, C.M.; Tawil, R.; Statland, J.M. Restrictive lung involvement in facioscapulohumeral muscular dystrophy. Muscle Nerve 2014, 50, 739–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlgemuth, M.; van der Kooi, E.L.; van Kesteren, R.G.; van der Maarel, S.M.; Padberg, G.W. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology 2004, 63, 176–178. [Google Scholar] [CrossRef]
- Laforêt, P.; de Toma, C.; Eymard, B.; Becane, H.M.; Jeanpierre, M.; Fardeau, M.; Duboc, D. Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology 1998, 51, 1454–1456. [Google Scholar] [CrossRef]
- Van Dijk, G.P.; van der Kooi, E.; Behin, A.; Smeets, J.; Timmermans, J.; van der Maarel, S.; Padberg, G.; Voermans, N.; van Engelen, B. High prevalence of incomplete right bundle branch block in facioscapulohumeral muscular dystrophy without cardiac symptoms. Funct. Neurol. 2014, 29, 159–165. [Google Scholar]
- Fitzsimons, R.B.; Gurwin, E.B.; Bird, A.C. Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy. A general association with genetic and therapeutic implications. Brain 1987, 110 Pt 3, 631–648. [Google Scholar] [CrossRef]
- Padberg, G.W.; Brouwer, O.F.; de Keizer, R.J.; Dijkman, G.; Wijmenga, C.; Grote, J.J.; Frants, R.R. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve. Suppl. 1995, S73–S80. [Google Scholar] [CrossRef] [Green Version]
- Lutz, K.L.; Holte, L.; Kliethermes, S.A.; Stephan, C.; Mathews, K.D. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology 2013, 81, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Statland, J.M.; Sacconi, S.; Farmakidis, C.; Donlin-Smith, C.M.; Chung, M.; Tawil, R. Coats syndrome in facioscapulohumeral dystrophy type 1: Frequency and D4Z4 contraction size. Neurology 2013, 80, 1247–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwer, O.F.; Padberg, G.W.; Wijmenga, C.; Frants, R.R. Facioscapulohumeral muscular dystrophy in early childhood. Arch. Neurol. 1994, 51, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Goselink, R.J.M.; Voermans, N.C.; Okkersen, K.; Brouwer, O.F.; Padberg, G.W.; Nikolic, A.; Tupler, R.; Dorobek, M.; Mah, J.K.; van Engelen, B.G.M.; et al. Early onset facioscapulohumeral dystrophy - a systematic review using individual patient data. Neuromuscul. Disord. 2017, 27, 1077–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.-H.; Lai, Y.-H.; Lee, P.-L.; Hsu, J.-H.; Goto, K.; Hayashi, Y.K.; Nishino, I.; Lin, C.-W.; Shih, H.-H.; Huang, C.-C.; et al. Infantile facioscapulohumeral muscular dystrophy revisited: Expansion of clinical phenotypes in patients with a very short EcoRI fragment. Neuromuscul. Disord. 2013, 23, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.-W.; Mercier, J.; Coppée, F.; Belayew, A. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013, 17, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Mattéotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowaljow, V.; Marcowycz, A.; Ansseau, E.; Conde, C.B.; Sauvage, S.; Mattéotti, C.; Arias, C.; Corona, E.D.; Nuñez, N.G.; Leo, O.; et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007, 17, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Wallace, L.M.; Garwick, S.E.; Mei, W.; Belayew, A.; Coppee, F.; Ladner, K.J.; Guttridge, D.; Yang, J.; Harper, S.Q. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 2011, 69, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- Block, G.J.; Narayanan, D.; Amell, A.M.; Petek, L.M.; Davidson, K.C.; Bird, T.D.; Tawil, R.; Moon, R.T.; Miller, D.G. Wnt/β-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum. Mol. Genet. 2013, 22, 4661–4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosnakovski, D.; Gearhart, M.D.; Toso, E.A.; Recht, O.O.; Cucak, A.; Jain, A.K.; Barton, M.C.; Kyba, M. p53-independent DUX4 pathology in cell and animal models of facioscapulohumeral muscular dystrophy. Dis. Model. Mech. 2017, 10, 1211–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadle, S.C.; Zhong, J.W.; Campbell, A.E.; Conerly, M.L.; Jagannathan, S.; Wong, C.-J.; Morello, T.D.; van der Maarel, S.M.; Tapscott, S.J. DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLOS Genet. 2017, 13, e1006658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosnakovski, D.; Xu, Z.; Gang, E.J.; Galindo, C.L.; Liu, M.; Simsek, T.; Garner, H.R.; Agha-Mohammadi, S.; Tassin, A.; Coppée, F.; et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008, 27, 2766–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar] [PubMed]
- Xu, H.; Wang, Z.; Jin, S.; Hao, H.; Zheng, L.; Zhou, B.; Zhang, W.; Lv, H.; Yuan, Y. Dux4 induces cell cycle arrest at G1 phase through upregulation of p21 expression. Biochem. Biophys. Res. Commun. 2014, 446, 235–240. [Google Scholar] [CrossRef]
- Porter, J.R.; Fisher, B.E.; Baranello, L.; Liu, J.C.; Kambach, D.M.; Nie, Z.; Koh, W.S.; Luo, J.; Stommel, J.M.; Levens, D.; et al. Global Inhibition with Specific Activation: How p53 and MYC Redistribute the Transcriptome in the DNA Double-Strand Break Response. Mol. Cell 2017, 67, 1013–1025.e9. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.B. MYC and the Control of Apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [Green Version]
- Banerji, C.R.S.; Knopp, P.; Moyle, L.A.; Severini, S.; Orrell, R.W.; Teschendorff, A.E.; Zammit, P.S. β-catenin is central to DUX4-driven network rewiring in facioscapulohumeral muscular dystrophy. J. R. Soc. Interface 2015, 12, 20140797. [Google Scholar] [CrossRef] [Green Version]
- Pećina-Šlaus, N. Wnt signal transduction pathway and apoptosis: A review. Cancer Cell Int. 2010, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Von Maltzahn, J.; Chang, N.C.; Bentzinger, C.F.; Rudnicki, M.A. Wnt signaling in myogenesis. Trends Cell Biol. 2012, 22, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denny, A.P.; Heather, A.K. Are Antioxidants a Potential Therapy for FSHD? A Review of the Literature. Oxid. Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turki, A.; Hayot, M.; Carnac, G.; Pillard, F.; Passerieux, E.; Bommart, S.; de Mauverger, E.R.; Hugon, G.; Pincemail, J.; Pietri, S.; et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic. Biol. Med. 2012, 53, 1068–1079. [Google Scholar] [CrossRef]
- Musarò, A.; Fulle, S.; Fanò, G. Oxidative stress and muscle homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 236–242. [Google Scholar] [CrossRef]
- Dmitriev, P.; Bou Saada, Y.; Dib, C.; Ansseau, E.; Barat, A.; Hamade, A.; Dessen, P.; Robert, T.; Lazar, V.; Louzada, R.A.N.; et al. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radic. Biol. Med. 2016, 99, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Winokur, S.T.; Barrett, K.; Martin, J.H.; Forrester, J.R.; Simon, M.; Tawil, R.; Chung, S.-A.; Masny, P.S.; Figlewicz, D.A. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul. Disord. 2003, 13, 322–333. [Google Scholar] [CrossRef]
- Bosnakovski, D.; Choi, S.; Strasser, J.M.; Toso, E.A.; Walters, M.A.; Kyba, M. High-throughput screening identifies inhibitors of DUX4-induced myoblast toxicity. Skelet. Muscle 2014, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Harafuji, N.; Belayew, A.; Chen, Y.-W. DUX4 Differentially Regulates Transcriptomes of Human Rhabdomyosarcoma and Mouse C2C12 Cells. PLoS ONE 2013, 8, e64691. [Google Scholar] [CrossRef] [Green Version]
- Delhalle, S.; Deregowski, V.; Benoit, V.; Merville, M.-P.; Bours, V. NF-κB-dependent MnSOD expression protects adenocarcinoma cells from TNF-α-induced apoptosis. Oncogene 2002, 21, 3917–3924. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhou, H.-M. The Role of Manganese Superoxide Dismutase in Inflammation Defense. Enzyme Res. 2011, 2011, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Sasaki-Honda, M.; Jonouchi, T.; Arai, M.; Hotta, A.; Mitsuhashi, S.; Nishino, I.; Matsuda, R.; Sakurai, H. A patient-derived iPSC model revealed oxidative stress increases facioscapulohumeral muscular dystrophy-causative DUX4. Hum. Mol. Genet. 2018, 27, 4024–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerji, C.R.S.; Panamarova, M.; Pruller, J.; Figeac, N.; Hebaishi, H.; Fidanis, E.; Saxena, A.; Contet, J.; Sacconi, S.; Severini, S.; et al. Dynamic transcriptomic analysis reveals suppression of PGC1 α/ERR α drives perturbed myogenesis in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2019, 28, 1244–1259. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Foo, S.-Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bou Saada, Y.; Dib, C.; Dmitriev, P.; Hamade, A.; Carnac, G.; Laoudj-Chenivesse, D.; Lipinski, M.; Vassetzky, Y.S. Facioscapulohumeral dystrophy myoblasts efficiently repair moderate levels of oxidative DNA damage. Histochem. Cell Biol. 2016, 145, 475–483. [Google Scholar] [CrossRef]
- Bosnakovski, D.; Gearhart, M.D.; Toso, E.A.; Ener, E.T.; Choi, S.H.; Kyba, M. Low level DUX4 expression disrupts myogenesis through deregulation of myogenic gene expression. Sci. Rep. 2018, 8, 16957. [Google Scholar] [CrossRef] [Green Version]
- Knopp, P.; Krom, Y.D.; Banerji, C.R.S.; Panamarova, M.; Moyle, L.A.; den Hamer, B.; van der Maarel, S.M.; Zammit, P.S. DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J. Cell Sci. 2016, 129, 3816–3831. [Google Scholar] [CrossRef] [Green Version]
- Bosnakovski, D.; Toso, E.A.; Hartweck, L.M.; Magli, A.; Lee, H.A.; Thompson, E.R.; Dandapat, A.; Perlingeiro, R.C.R.; Kyba, M. The DUX4 homeodomains mediate inhibition of myogenesis and are functionally exchangeable with the Pax7 homeodomain. J. Cell Sci. 2017, 130, 3685–3697. [Google Scholar] [CrossRef] [Green Version]
- Banerji, C.R.S.; Panamarova, M.; Hebaishi, H.; White, R.B.; Relaix, F.; Severini, S.; Zammit, P.S. PAX7 target genes are globally repressed in facioscapulohumeral muscular dystrophy skeletal muscle. Nat. Commun. 2017, 8, 2152. [Google Scholar] [CrossRef] [Green Version]
- Banerji, C.R.S.; Zammit, P.S. PAX7 target gene repression is a superior FSHD biomarker than DUX4 target gene activation, associating with pathological severity and identifying FSHD at the single-cell level. Hum. Mol. Genet. 2019, 28, 2224–2236. [Google Scholar] [CrossRef] [Green Version]
- Haynes, P.; Kernan, K.; Zhou, S.-L.; Miller, D.G. Expression patterns of FSHD-causing DUX4 and myogenic transcription factors PAX3 and PAX7 are spatially distinct in differentiating human stem cell cultures. Skelet. Muscle 2017, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Teveroni, E.; Pellegrino, M.; Sacconi, S.; Calandra, P.; Cascino, I.; Farioli-Vecchioli, S.; Puma, A.; Garibaldi, M.; Morosetti, R.; Tasca, G.; et al. Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J. Clin. Investig. 2017, 127, 1531–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderplanck, C.; Ansseau, E.; Charron, S.; Stricwant, N.; Tassin, A.; Laoudj-Chenivesse, D.; Wilton, S.D.; Coppée, F.; Belayew, A. The FSHD Atrophic Myotube Phenotype Is Caused by DUX4 Expression. PLoS ONE 2011, 6, e26820. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.E.; Oliva, J.; Yates, M.P.; Zhong, J.W.; Shadle, S.C.; Snider, L.; Singh, N.; Tai, S.; Hiramuki, Y.; Tawil, R.; et al. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet. Muscle 2017, 7, 16. [Google Scholar] [CrossRef]
- Seko, D.; Ogawa, S.; Li, T.-S.; Taimura, A.; Ono, Y. μ-Crystallin controls muscle function through thyroid hormone action. FASEB J. 2016, 30, 1733–1740. [Google Scholar] [CrossRef] [Green Version]
- Oshima, A. CRYM mutations cause deafness through thyroid hormone binding properties in the fibrocytes of the cochlea. J. Med. Genet. 2006, 43, e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyle, L.A.; Blanc, E.; Jaka, O.; Prueller, J.; Banerji, C.R.; Tedesco, F.S.; Harridge, S.D.R.; Knight, R.D.; Zammit, P.S. Ret function in muscle stem cells points to tyrosine kinase inhibitor therapy for facioscapulohumeral muscular dystrophy. Elife 2016, 5, 1–35. [Google Scholar] [CrossRef]
- Shadle, S.C.; Bennett, S.R.; Wong, C.-J.; Karreman, N.A.; Campbell, A.E.; van der Maarel, S.M.; Bass, B.L.; Tapscott, S.J. DUX4-induced bidirectional HSATII satellite repeat transcripts form intranuclear double stranded RNA foci in human cell models of FSHD. Hum. Mol. Genet. 2019, 1–52. [Google Scholar] [CrossRef]
- Fiorini, F.; Bagchi, D.; Le Hir, H.; Croquette, V. Human Upf1 is a highly processive RNA helicase and translocase with RNP remodelling activities. Nat. Commun. 2015, 6, 7581. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Snider, L.; Jagannathan, S.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J.; Bradley, R.K. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. Elife 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of Protein Kinase PKR in Cell Biology: From Antiviral to Antiproliferative Action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; Van Der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tapscott, S.J. DUX4 activates germline genes, retroelements and immune-mediators: Implications for facioscapulohumeral dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhople, V.; Krukemeyer, A.; Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta-Biomembr. 2006, 1758, 1499–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016, 44, 5161–5173. [Google Scholar] [CrossRef] [Green Version]
- Dancy, B.M.; Cole, P.A. Protein Lysine Acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar]
- Fauquier, L.; Azzag, K.; Parra, M.A.M.; Quillien, A.; Boulet, M.; Diouf, S.; Carnac, G.; Waltzer, L.; Gronemeyer, H.; Vandel, L. CBP and P300 regulate distinct gene networks required for human primary myoblast differentiation and muscle integrity. Sci. Rep. 2018, 8, 12629. [Google Scholar] [CrossRef]
- Bosnakovski, D.; da Silva, M.T.; Sunny, S.T.; Ener, E.T.; Toso, E.A.; Yuan, C.; Cui, Z.; Walters, M.A.; Jadhav, A.; Kyba, M. A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci. Adv. 2019, 5, eaaw7781. [Google Scholar] [CrossRef] [Green Version]
- Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2019, 370, 219–230. [Google Scholar] [CrossRef]
- Cruz, J.M.; Hupper, N.; Wilson, L.S.; Concannon, J.B.; Wang, Y.; Oberhauser, B.; Patora-Komisarska, K.; Zhang, Y.; Glass, D.J.; Trendelenburg, A.-U.; et al. Protein kinase A activation inhibits DUX4 gene expression in myotubes from patients with facioscapulohumeral muscular dystrophy. J. Biol. Chem. 2018, 293, 11837–11849. [Google Scholar] [CrossRef] [Green Version]
- Resnick, R.; Wong, C.-J.; Hamm, D.C.; Bennett, S.R.; Skene, P.J.; Hake, S.B.; Henikoff, S.; van der Maarel, S.M.; Tapscott, S.J. DUX4-Induced Histone Variants H3.X and H3.Y Mark DUX4 Target Genes for Expression. Cell Rep. 2019, 29, 1812–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, A.E.; Shadle, S.C.; Jagannathan, S.; Lim, J.-W.; Resnick, R.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. Elife 2018, 7, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Young, J.M.; Whiddon, J.L.; Yao, Z.; Kasinathan, B.; Snider, L.; Geng, L.N.; Balog, J.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. DUX4 Binding to Retroelements Creates Promoters That Are Active in FSHD Muscle and Testis. PLoS Genet. 2013, 9, e1003947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.-W.; Wong, C.-J.; Yao, Z.; Tawil, R.; van der Maarel, S.M.; Miller, D.G.; Tapscott, S.J.; Filippova, G.N. Small noncoding RNAs in FSHD2 muscle cells reveal both DUX4- and SMCHD1-specific signatures. Hum. Mol. Genet. 2018, 27, 2644–2657. [Google Scholar] [CrossRef]
- Homma, S.; Beermann, M.L.; Yu, B.; Boyce, F.M.; Miller, J.B. Nuclear bodies reorganize during myogenesis in vitro and are differentially disrupted by expression of FSHD-associated DUX4. Skelet. Muscle 2016, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.S.; Zhang, B.; Spector, D.L. Biogenesis and function of nuclear bodies. Trends Genet. 2011, 27, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Dellaire, G.; Bazett-Jones, D.P. PML nuclear bodies: Dynamic sensors of DNA damage and cellular stress. BioEssays 2004, 26, 963–977. [Google Scholar] [CrossRef]
- Homma, S.; Beermann, M.L.; Boyce, F.M.; Miller, J.B. Expression of FSHD-related DUX4-FL alters proteostasis and induces TDP-43 aggregation. Ann. Clin. Transl. Neurol. 2015, 2, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Ansseau, E.; Eidahl, J.O.; Lancelot, C.; Tassin, A.; Matteotti, C.; Yip, C.; Liu, J.; Leroy, B.; Hubeau, C.; Gerbaux, C.; et al. Homologous Transcription Factors DUX4 and DUX4c Associate with Cytoplasmic Proteins during Muscle Differentiation. PLoS ONE 2016, 11, e0146893. [Google Scholar] [CrossRef]
- Jagannathan, S.; Ogata, Y.; Gafken, P.R.; Tapscott, S.J.; Bradley, R.K. Quantitative proteomics reveals key roles for post-transcriptional gene regulation in the molecular pathology of facioscapulohumeral muscular dystrophy. Elife 2019, 8, 1–16. [Google Scholar] [CrossRef]
- Rickard, A.M.; Petek, L.M.; Miller, D.G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015, 24, 5901–5914. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.N.; Cabotage, J.; Shi, R.; Dixit, M.; Sutherland, M.; Liu, J.; Muger, S.; Harper, S.Q.; Nagaraju, K.; Chen, Y.-W. Conditional over-expression of PITX1 causes skeletal muscle dystrophy in mice. Biol. Open 2012, 1, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcil, A. Pitx1 and Pitx2 are required for development of hindlimb buds. Development 2003, 130, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broucqsault, N.; Morere, J.; Gaillard, M.-C.; Dumonceaux, J.; Torrents, J.; Salort-Campana, E.; Maues De Paula, A.; Bartoli, M.; Fernandez, C.; Chesnais, A.L.; et al. Dysregulation of 4q35- and muscle-specific genes in fetuses with a short D4Z4 array linked to facio-scapulo-humeral dystrophy. Hum. Mol. Genet. 2013, 22, 4206–4214. [Google Scholar] [CrossRef] [Green Version]
- Ferreboeuf, M.; Mariot, V.; Bessières, B.; Vasiljevic, A.; Attié-Bitach, T.; Collardeau, S.; Morere, J.; Roche, S.; Magdinier, F.; Robin-Ducellier, J.; et al. DUX4 and DUX4 downstream target genes are expressed in fetal FSHD muscles. Hum. Mol. Genet. 2014, 23, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Mariot, V.; Roche, S.; Hourdé, C.; Portilho, D.; Sacconi, S.; Puppo, F.; Duguez, S.; Rameau, P.; Caruso, N.; Delezoide, A.-L.; et al. Correlation between low FAT1 expression and early affected muscle in facioscapulohumeral muscular dystrophy. Ann. Neurol. 2015, 78, 387–400. [Google Scholar] [CrossRef] [Green Version]
- Van den Heuvel, A.; Mahfouz, A.; Kloet, S.L.; Balog, J.; van Engelen, B.G.M.; Tawil, R.; Tapscott, S.J.; van der Maarel, S.M. Single-cell RNA sequencing in facioscapulohumeral muscular dystrophy disease etiology and development. Hum. Mol. Genet. 2019, 28, 1064–1075. [Google Scholar] [CrossRef]
- Klooster, R.; Straasheijm, K.; Shah, B.; Sowden, J.; Frants, R.; Thornton, C.; Tawil, R.; van der Maarel, S. Comprehensive expression analysis of FSHD candidate genes at the mRNA and protein level. Eur. J. Hum. Genet. 2009, 17, 1615–1624. [Google Scholar] [CrossRef]
- Thijssen, P.E.; Balog, J.; Yao, Z.; Pham, T.; Tawil, R.; Tapscott, S.J.; Van der Maarel, S.M. DUX4 promotes transcription of FRG2 by directly activating its promoter in facioscapulohumeral muscular dystrophy. Skelet. Muscle 2014, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Ferri, G.; Huichalaf, C.H.; Caccia, R.; Gabellini, D. Direct interplay between two candidate genes in FSHD muscular dystrophy. Hum. Mol. Genet. 2015, 24, 1256–1266. [Google Scholar] [CrossRef] [Green Version]
- Bodega, B.; Ramirez, G.D.C.; Grasser, F.; Cheli, S.; Brunelli, S.; Mora, M.; Meneveri, R.; Marozzi, A.; Mueller, S.; Battaglioli, E.; et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, A.E.; Belleville, A.E.; Resnick, R.; Shadle, S.C.; Tapscott, S.J. Facioscapulohumeral dystrophy: Activating an early embryonic transcriptional program in human skeletal muscle. Hum. Mol. Genet. 2018, 27, R153–R162. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 729. https://doi.org/10.3390/ijms21030729
Lim KRQ, Nguyen Q, Yokota T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. International Journal of Molecular Sciences. 2020; 21(3):729. https://doi.org/10.3390/ijms21030729
Chicago/Turabian StyleLim, Kenji Rowel Q., Quynh Nguyen, and Toshifumi Yokota. 2020. "DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy" International Journal of Molecular Sciences 21, no. 3: 729. https://doi.org/10.3390/ijms21030729
APA StyleLim, K. R. Q., Nguyen, Q., & Yokota, T. (2020). DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. International Journal of Molecular Sciences, 21(3), 729. https://doi.org/10.3390/ijms21030729