The Relationship between DNA Methylation and Antidepressant Medications: A Systematic Review


Abstract
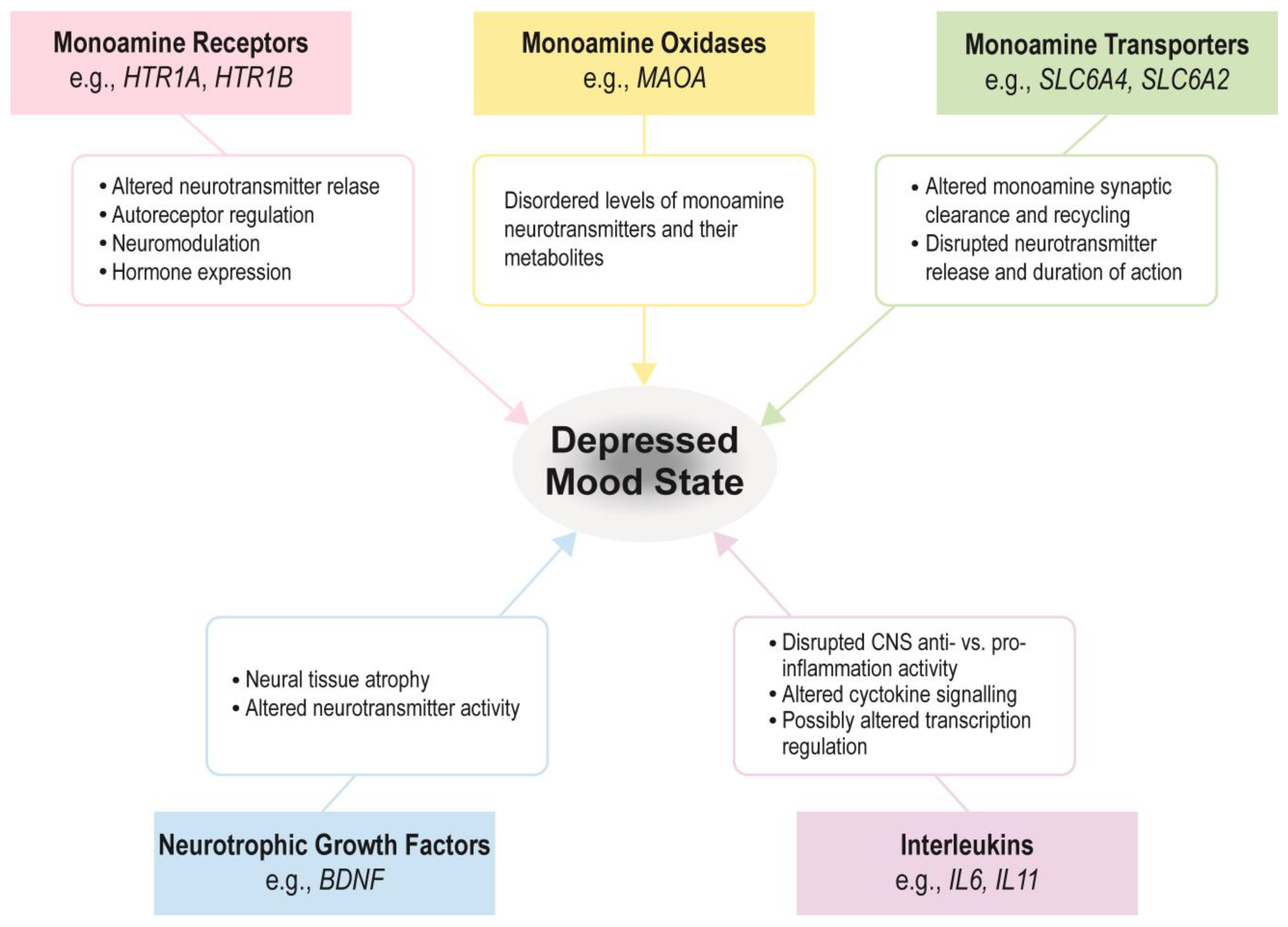
:1. Introduction
2. Results
2.1. BDNF (Brain-Derived Neurotrophic Factor)
2.2. MAOA
2.3. SLC6A4
2.4. SLC6A2
2.5. HTR1A/1B
2.6. IL6 and IL11
2.7. Global DNA Methylation
3. Discussion
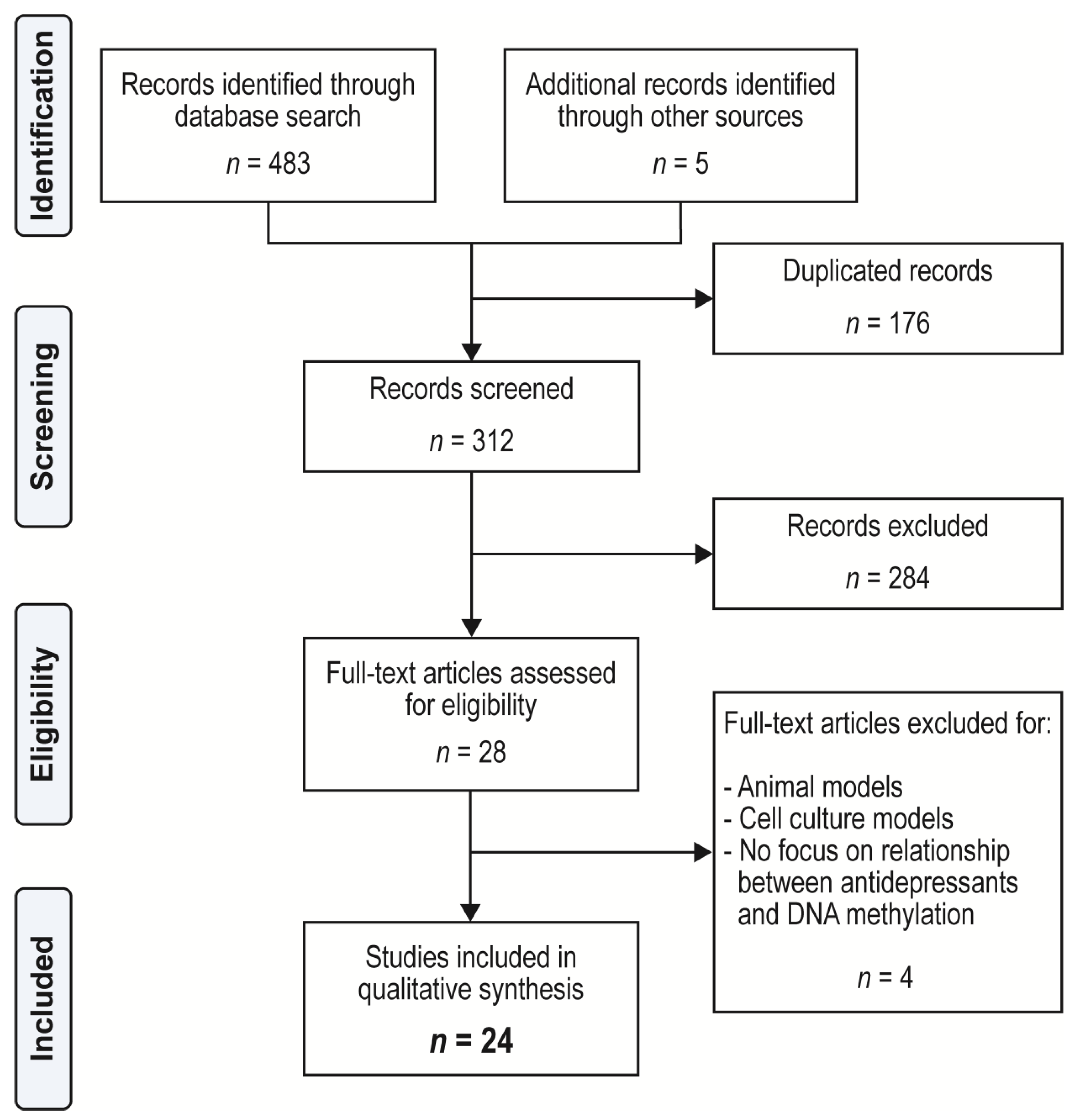
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| WHO | World Health Organization |
| MDD | Major depressive disorder |
| CpG | Cytosine-phosphate-guanine dinucleotides |
| BDNF | Brain-derived neurotrophic factor |
| PBMC | Peripheral blood mononuclear cells |
| CNS | Central nervous system |
| BD1 | Bipolar disorder type 1 |
| BD2 | Bipolar disorder type 2 |
| HC | Healthy controls |
| SSRI | Selective serotonin reuptake inhibitor |
| SNRI | Selective norepinephrine reuptake inhibitor |
| TCA | Tricyclic antidepressant |
| BD | Bipolar disorder |
| SI | Suicidal ideation |
| MAO-I | Monoamine-oxidase inhibitor |
| ACS | Acute coronary syndrome |
| H3K27 | Histone H3, lysine 27 |
| HAM-D | Hamilton Depression Rating Scale |
| MAO-A | Monoamine oxidase A |
| SLC6A4 | Serotonin transporter |
| IR | Improvement ratio |
| SLC6A2 | Sodium:norepinephrine symporter |
| HTR1A | 5-hydroxytryptamine transporter 1A |
| HTR1B | 5-hydroxytryptamine transporter 1B |
| LES | Life Event Score |
| IL6 | Interleukin-6 |
| IL-11 | Interleukin-11 |
| GENDEP | Genome-based Therapeutic Drugs for Depression Project |
| MADRS | Montgomery-Asberg Depression Rating Scale |
| BR | Best response |
| WR | Worst response |
| HS3ST1 | Heparan sulfate-glucosamine 3-sulfotransferase 1 |
| PTSD | Post-traumatic stress disorder |
| MECP2 | Methyl-CpG-binding protein 2 |
| TNF-α | Tumor necrosis factor alpha |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
References
- World Health Organization. Depression and Other Common Mental Disorders Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017; pp. 1–24. [Google Scholar]
- Marcus, M.; Yasamy, M.T.; van Ommeren, M.; Chisholm, D.; Saxena, S. Depression A Global Public Health Concern; WHO Department of Mental Health and Substance Abuse. Available online: https://www.who.int/mental_health/management/depression/who_paper_depression_wfmh_2012.pdf (accessed on 27 January 2020).
- Knol, M.J.; Twisk, J.W.; Beekman, A.T.; Heine, R.J.; Snoek, F.J.; Pouwer, F. Depression as a risk factor for the onset of type 2 diabetes mellitus. A meta-analysis. Diabetologia 2006, 49, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lett, H.S.; Blumenthal, J.A.; Babyak, M.A.; Sherwood, A.; Strauman, T.; Robins, C.; Newman, M.F. Depression as a risk factor for coronary artery disease: Evidence, mechanisms, and treatment. Psychosom. Med. 2004, 66, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.E.; Kessler, R.C.; Birnbaum, H.G.; Leong, S.A.; Lowe, S.W.; Berglund, P.A.; Corey-Lisle, P.K. The Economic Burden of Depression in the United States: How Did It Change Between 1990 and 2000? J. Clin. Psychiatry 2003, 64, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Roser, M.; Ritchie, H.; Ortiz-Ospina, E. World Population Growth. Available online: https://ourworldindata.org/world-population-growth? (accessed on 27 January 2020).
- World Health Organization. Mental Health and Older Adults. Geneva. Available online: https://www.who.int/news-room/fact-sheets/detail/mental-health-of-older-adults (accessed on 27 January 2020).
- Sackeim, H.A. The Definition and Meaning of Treatment-Resistant Depression. J. Clin. Psychiatry 2001, 62, 10–17. [Google Scholar]
- Berton, O.; Nestler, E.J. New approaches to antidepressant drug discovery: Beyond monoamines. Nat. Rev. Neurosci. 2006, 7, 137–151. [Google Scholar] [CrossRef]
- Mathew, S.J.; Manji, H.K.; Charney, D.S. Novel Drugs and Therapeutic Targets for Severe Mood Disorders. Neuropsychopharmacology 2008, 33, 2080–2092. [Google Scholar] [CrossRef] [Green Version]
- Hyman, S.E. Revitalizing Psychiatric Therapeutics. Neuropsychopharmacology 2014, 39, 220–229. [Google Scholar] [CrossRef]
- Nestler, E.J. Antidepressant treatments in the 21st century. Biol. Psychiatry 1998, 44, 526–533. [Google Scholar] [CrossRef]
- Wray, N.R.; Pergadia, M.L.; Blackwood, D.H.; Penninx, B.W.; Gordon, S.D.; Nyholt, D.R.; Ripke, S.; MacIntyre, D.J.; McGhee, K.A.; Maclean, A.W.; et al. Genome-wide association study of major depressive disorder: New results, meta-analysis, and lessons learned. Mol. Psychiatry 2012, 17, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Lisoway, A.J.; Zai, C.C.; Tiwari, A.K.; Kennedy, J.L. DNA methylation and clinical response to antidepressant medication in major depressive disorder: A review and recommendations. Neurosci. Lett. 2018, 669, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, R.C.; Bromet, E.J. The Epidemiology of Depression Across Cultures. Annu. Rev. Public Health 2013, 34, 119–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, T.B.; Russig, H.; Weiss, I.C.; Gräff, J.; Linder, N.; Michalon, A.; Vizi, S.; Mansuy, I.M. Epigenetic Transmission of the Impact of Early Stress Across Generations. Biol. Psychiatry 2010, 68, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J. Epigenetic Mechanisms of Depression. JAMA Psychiatry 2014, 71, 454–456. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.L.; Lubin, F.D.; Funk, A.J.; Sweatt, J.D. Lasting Epigenetic Influence of Early-Life Adversity on the BDNF Gene. Biol. Psychiatry 2009, 65, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.C.; Bharadwaj, R.; Whittle, C.; Krueger, W.; Mirnics, K.; Hurd, Y.; Rasmussen, T.; Akbarian, S. The Genome In Three Dimensions: A New Fronteir in Human Brain Research. Biol. Psychiatry 2014, 75, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Merkl, A.; Neumann, W.J.; Huebl, J.; Aust, S.; Horn, A.; Krauss, J.K.; Dziobek, I.; Kuhn, J.; Schneider, G.H.; Bajbouj, M.; et al. Modulation of Beta-Band Activity in the Subgenual Anterior Cingulate Cortex during Emotional Empathy in Treatment-Resistant Depression. Cereb. Cortex 2016, 26, 2626–2638. [Google Scholar] [CrossRef] [Green Version]
- Dolinoy, D.C.; Weidman, J.R.; Jirtle, R.L. Epigenetic gene regulation: Linking early developmental environment to adult disease. Reprod. Toxicol. 2007, 23, 297–307. [Google Scholar] [CrossRef]
- Boks, M.P.; de Jong, N.M.; Kas, M.J.; Vinkers, C.H.; Fernandes, C.; Kahn, R.S.; Mill, J.; Ophoff, R.A. Current status and future prospects for epigenetic psychopharmacology. Epigenetics 2012, 7, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Ptak, C.; Petronis, A. Epigenetics and Complex Disease: From Etiology to New Therapeutics. Annu. Rev. Pharm. Toxicol. 2008, 48, 257–276. [Google Scholar] [CrossRef]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, V.S.; Kolshus, E.; McLoughlin, D.M. Epigenetics and depression: Return of the repressed. J. Affect. Disord 2014, 155, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyf, M. Targeting DNA methylation in cancer. Bull. Cancer 2006, 93, 961–972. [Google Scholar] [CrossRef]
- Weber, M.; Schübeler, D. Genomic patterns of DNA methylation: Targets and function of an epigenetic mark. Curr. Opin. Cell Biol. 2007, 19, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics 2009, 4, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Putiri, E.L.; Robertson, K.D. Epigenetic mechanisms and genome stability. Clin. Epigenetics 2011, 2, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Mukamel, E.A. Turning over DNA methylation in the mind. Front. Neurosci. 2015, 9, 252. [Google Scholar] [CrossRef] [Green Version]
- Alberini, C.M. The role of protein synthesis during the labile phases of memory: Revisiting the skepticism. Neurobiol. Learn. Mem. 2008, 89, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef]
- Covington III, H.E.; Vialou, V.; Nestler, E.J. From synapse to nucleus: Novel targets for treating depression. Neuropharmacology 2010, 58, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Petronis, A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010, 465, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Vaillancourt, K.; Turecki, G. A role for activity-dependent epigenetics in the development and treatment of major depressive disorder. Genes Brain Behav. 2018, 17, e12446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habano, W.; Kawamura, K.; Iizuka, N.; Terashima, J.; Sugai, T.; Ozawa, S. Analysis of DNA methylation landscape reveals the roles of DNA methylation in the regulation of drug metabolizing enzymes. Clin. Epigenetics 2015, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiili, E.M.; Antikainen, M.S.; Mitiushkina, N.V.; Sukhovskaya, O.A.; Imyanitov, E.N.; Hirvonen, A.P. Effect of genotype and methylation of CYP2D6 on smoking behaviour. Pharm. Genom. 2015, 25, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Manji, H.; Lu, B. New insights into BDNF function in depression and anxiety. Nat. Neurosci. 2007, 10, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, Z.Y. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharm. Sin. 2011, 32, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Rasmusson, A.M.; Shi, L.; Duman, R. Downregulation of BDNF mRNA in the Hippocampal Dentate Gyrus after Re-exposure to Cues Previously Associated with Footshock. Neuropsychopharmacology 2002, 27, 133–142. [Google Scholar] [CrossRef]
- Roceri, M.; Hendriks, W.; Racagni, G.; Ellenbroek, B.A.; Riva, M.A. Early maternal deprivation reduces the expression of BDNF and NMDA receptor subunits in rat hippocampus. Mol. Psychiatry 2002, 7, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Blaze, J.; Asok, A.; Borrelli, K.; Tulbert, C.; Bollinger, J.; Ronca, A.E.; Roth, T.L. Intrauterine exposure to maternal stress alters Bdnf IV DNA methylation and telomere length in the brain of adult rat offspring. Int. J. Dev. Neurosci. 2017, 62, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Duman, R.S. Pathophysiology of depression: The concept of synaptic plasticity. Eur. Psychiatry 2002, 17 (Suppl. 3), 306–310. [Google Scholar] [CrossRef]
- Chen, B.; Dowlatshahi, D.; MacQueen, G.M.; Wang, J.F.; Young, L.T. Increased hippocampal bdnf immunoreactivity in subjects treated with antidepressant medication. Biol. Psychiatry 2001, 50, 260–265. [Google Scholar] [CrossRef]
- Fujimura, H.; Altar, C.A.; Chen, R.; Nakamura, T.; Nakahashi, T.; Kambayashi, J.; Sun, B.; Tandon, N.N. Brain-derived Neurotrophic Factor is Stored in Human Platelets and Released by Agonist Stimulation. Thromb. Haemost. 2002, 87, 728–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, P.; Benito, E.; Fischer, A. MicroRNAs as biomarkers for CNS disease. Front. Mol. Neurosci. 2013, 6, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.P. Blood chromatin as a biosensor of the epigenetic milieu: A tool for studies in living psychiatric patients. Epigenomics 2012, 4, 551–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Addario, C.; Dell’Osso, B.; Palazzo, M.C.; Benatti, B.; Lietti, L.; Cattaneo, E.; Galimberti, D.; Fenoglio, C.; Cortini, F.; Scarpini, E.; et al. Selective DNA Methylation of BDNF Promoter in Bipolar Disorder: Differences Among Patients with BDI and BDII. Neuropsychopharmacology 2012, 37, 1647–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Addario, C.; Dell’Osso, B.; Galimberti, D.; Palazzo, M.C.; Benatti, B.; Di Francesco, A.; Scarpini, E.; Altamura, A.C.; Maccarrone, M. Epigenetic Modulation of BDNF Gene in Patients with Major Depressive Disorder. Biol. Psychiatry 2013, 73, e6–e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlberg, L.; Scheibelreiter, J.; Hassler, M.R.; Schloegelhofer, M.; Schmoeger, M.; Ludwig, B.; Kasper, S.; Aschauer, H.; Egger, G.; Schosser, A. Brain-derived neurotrophic factor (BDNF)-Epigenetic regulation in unipolar and bipolar affective disorder. J. Affect. Disord. 2014, 168, 399–406. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, C.; Lv, Q.; Bao, C.; Sun, H.; Ma, G.; Fang, Y.; Yi, Z.; Cai, W. Association of DNA methylation in BDNF with escitalopram treatment response in depressed Chinese Han patients. Eur. J. Clin. Pharm. 2018, 74, 1011–1020. [Google Scholar] [CrossRef]
- Januar, V.; Ancelin, M.L.; Ritchie, K.; Saffery, R.; Ryan, J. BDNF promoter methylation and genetic variation in late-life depression. Transl. Psychiatry 2015, 5, e619. [Google Scholar] [CrossRef]
- Kang, H.J.; Kim, J.M.; Lee, J.Y.; Kim, S.Y.; Bae, K.Y.; Kim, S.W.; Shin, I.S.; Kim, H.R.; Shin, M.G.; Yoon, J.S. BDNF promoter methylation and suicidal behavior in depressive patients. J. Affect. Disord. 2013, 151, 679–685. [Google Scholar] [CrossRef]
- Tadić, A.; Müller-Engling, L.; Schlicht, K.F.; Kotsiari, A.; Dreimüller, N.; Kleimann, A.; Bleich, S.; Lieb, K.; Frieling, H. Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol. Psychiatry 2014, 19, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Dreimüller, N.; Schlicht, K.F.; Wagner, S.; Peetz, D.; Borysenko, L.; Hiemke, C.; Lieb, K.; Tadić, A. Early reactions of brain-derived neurotrophic factor in plasma (pBDNF) and outcome to acute antidepressant treatment in patients with Major Depression. Neuropharmacology 2012, 62, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Stewart, R.; Kang, H.J.; Bae, K.Y.; Kim, S.W.; Shin, I.S.; Hong, Y.J.; Ahn, Y.; Jeong, M.H.; Yoon, J.S. BDNF methylation and depressive disorder in acute coronary syndrome: The K-DEPACS and EsDEPACS studies. Psychoneuroendocrinology 2015, 62, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.S.; Ernst, C.; Turecki, G. The epigenetic effects of antidepressant treatment on human prefrontal cortex BDNF expression. Int. J. Neuropsychopharmacol. 2011, 14, 427–429. [Google Scholar] [CrossRef]
- Lopez, J.P.; Mamdani, F.; Beaulieu, M.M.; Yang, J.P.; Berlim, M.T.; Ernst, C.; Turecki, G. Epigenetic regulation of BDNF expression according to antidepressant response. Mol. Psychiatry 2013, 18, 398–399. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.H.; Ginovart, N.; Boovariwala, A.; Sagrati, S.; Hussey, D.; Garcia, A.; Young, T.; Praschak-Rieder, N.; Wilson, A.A.; Houle, S. Elevated monoamine oxidase a levels in the brain: An explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry 2006, 63, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- McDermott, R.; Tingley, D.; Cowden, J.; Frazzetto, G.; Johnson, D.D.P. Monoamine oxidase A gene (MAOA) predicts behavioral aggression following provocation. Proc. Natl. Acad. Sci. USA 2009, 106, 2118–2123. [Google Scholar] [CrossRef] [Green Version]
- Godar, S.C.; Bortolato, M.; Richards, S.E.; Li, F.G.; Chen, K.; Wellman, C.L.; Shih, J.C. Monoamine Oxidase A is Required for Rapid Dendritic Remodeling in Response to Stress. Int. J. Neuropsychopharmacol. 2015, 18. [Google Scholar] [CrossRef]
- Wang, C.C.; Borchert, A.; Ugun-Klusek, A.; Tang, L.Y.; Lui, W.T.; Chu, C.Y.; Billett, E.; Kuhn, H.; Ufer, C. Monoamine Oxidase A Expression Is Vital For Embryonic Brain Development By Modulating Developmental Apoptosis. J. Biol. Chem. 2011, 286, 28322–28330. [Google Scholar] [CrossRef] [Green Version]
- Checknita, D.; Ekström, T.J.; Comasco, E.; Nilsson, K.W.; Tiihonen, J.; Hodgins, S. Associations of monoamine oxidase A gene first exon methylation with sexual abuse and current depression in women. J. Neural. Transm. (Vienna) 2018, 125, 1053–1064. [Google Scholar] [CrossRef] [Green Version]
- Domschke, K.; Tidow, N.; Schwarte, K.; Ziegler, C.; Lesch, K.P.; Deckert, J.; Arolt, V.; Zwanzger, P.; Baune, B.T. Pharmacoepigenetics of depression: No major influence of MAO-A DNA methylation on treatment response. J. Neural. Transm. 2015, 122, 99–108. [Google Scholar] [CrossRef]
- Luddington, N.S.; Mandadapu, A.; Husk, M.; El-Mallakh, R.S. Clinical Implications of Genetic Variation in the Serotonin Transporter Promoter Region: A Review. Prim. Care Companion J. Clin. Psychiatry 2009, 11, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Booij, L.; Szyf, M.; Carballedo, A.; Frey, E.-M.; Morris, D.; Dymov, S.; Vaisheva, F.; Ly, V.; Fahey, C.; Meaney, J.; et al. DNA Methylation of the Serotonin Transporter Gene in Peripheral Cells and Stress-Related Changes in Hippocampal Volume: A Study in Depressed Patients and Healthy Controls. PLoS ONE 2015, 10, e0119061. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kim, J.M.; Stewart, R.; Kim, S.Y.; Bae, K.Y.; Kim, S.W.; Shin, I.S.; Shin, M.G.; Yoon, J.S. Association of SLC6A4 methylation with early adversity, characteristics and outcomes in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 44, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Domschke, K.; Tidow, N.; Schwarte, K.; Deckert, J.; Lesch, K.P.; Arolt, V.; Zwanzger, P.; Baune, B.T. Serotonin transporter gene hypomethylation predicts impaired antidepressant treatment response. Int. J. Neuropsychopharmacol. 2014, 17, 1167–1176. [Google Scholar] [CrossRef] [Green Version]
- Iga, J.; Watanabe, S.Y.; Numata, S.; Umehara, H.; Nishi, A.; Kinoshita, M.; Inoshita, M.; Shimodera, S.; Fujita, H.; Ohmori, T. Association study of polymorphism in the serotonin transporter gene promoter, methylation profiles, and expression in patients with major depressive disorder. Hum. Psychopharmacol. 2016, 31, 193–199. [Google Scholar] [CrossRef]
- Okada, S.; Morinobu, S.; Fuchikami, M.; Segawa, M.; Yokomaku, K.; Kataoka, T.; Okamoto, Y.; Yamawaki, S.; Inoue, T.; Kusumi, I.; et al. The potential of SLC6A4 gene methylation analysis for the diagnosis and treatment of major depression. J. Psychiatr. Res. 2014, 53, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Bayles, R.; Baker, E.K.; Jowett, J.B.M.; Barton, D.; Esler, M.; El-Osta, A.; Lambert, G. Methylation of the SLC6a2 Gene Promoter in Major Depression and Panic Disorder. PLoS ONE 2013, 8, e83223. [Google Scholar] [CrossRef]
- Kato, M.; Fukuda, T.; Wakeno, M.; Okugawa, G.; Takekita, Y.; Watanabe, S.; Yamashita, M.; Hosoi, Y.; Azuma, J.; Kinoshita, T.; et al. Effect of 5-HT1A Gene Polymorphisms on Antidepressant Response in Major Depressive Disorder. Am. J. Med. Genet. B Neuopsychiatr. Genet. 2009, 150B, 115–123. [Google Scholar] [CrossRef]
- Yohn, C.N.; Gergues, M.M.; Samuels, B.A. The role of 5-HT receptors in depression. Mol. Brain 2017, 10, 28. [Google Scholar] [CrossRef]
- Lerer, B.; Gelfin, Y.; Gorfine, M.; Allolio, B.; Lesch, K.P.; Newman, M.E. 5-HT1A Receptor Function in Normal Subjects on Clinical Doses of Fluoxetine: Blunted Temperature and Hormone Responses to Ipsapirone Challenge. Neuropsychopharmacology 1999, 20, 628–639. [Google Scholar] [CrossRef] [Green Version]
- Lotrich, F.E.; Pollock, B.G. Candidate genes for antidepressant response to selective serotonin reuptake inhibitors. Neuropsychiatr. Dis. Treat. 2005, 1, 17–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Lv, Q.; Mao, Y.; Zhang, C.; Bao, C.; Sun, H.; Chen, H.; Yi, Z.; Cai, W.; Fang, Y. HTR1A/1B DNA methylation may predict escitalopram treatment response in depressed Chinese Han patients. J. Affect. Disord. 2018, 228, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Gassó, P.; Rodríguez, N.; Blázquez, A.; Monteagudo, A.; Boloc, D.; Plana, M.T.; Lafuente, A.; Lázaro, L.; Arnaiz, J.A.; Mas, S. Epigenetic and genetic variants in the HTR1B gene and clinical improvement in children and adolescents treated with fluoxetine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.; Herane Vives, A.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol 2015, 25, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.; Mondelli, V.; Pariante, C.M. Genetic Contributions of Inflammation to Depression. Neuropsychopharmacology 2017, 42, 81–98. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wu, Z.; Zhao, G.; Wang, F.; Fang, Y. Identification of IL6 as a susceptibility gene for major depressive disorder. Sci. Rep. 2016, 6, 31264. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.; Pilkington, L.; Neuhaus, K.; Ritchie, K.; Ancelin, M.L.; Saffery, R. Investigating the epigenetic profile of the inflammatory gene IL-6 in late-life depression. BMC Psychiatry 2017, 17, 354. [Google Scholar] [CrossRef] [Green Version]
- Dahl, J.; Ormstad, H.; Aass, H.C.; Malt, U.F.; Bendz, L.T.; Sandvik, L.; Brundin, L.; Andreassen, O.A. The plasma levels of various cytokines are increased during ongoing depression and are reduced to normal levels after recovery. Psychoneuroendocrinology 2014, 45, 77–86. [Google Scholar] [CrossRef]
- Uher, R.; Perroud, N.; Ng, M.Y.; Hauser, J.; Henigsberg, N.; Maier, W.; Mors, O.; Placentino, A.; Rietschel, M.; Souery, D.; et al. Genome-Wide Pharmacogenetics of Antidepressant Response in the GENDEP Project. Am. J. Psychiatry 2010, 167, 555–564. [Google Scholar] [CrossRef]
- Powell, T.R.; Smith, R.G.; Hackinger, S.; Schalkwyk, L.C.; Uher, R.; McGuffin, P.; Mill, J.; Tansey, K.E. DNA methylation in interleukin-11 predicts clinical response to antidepressants in GENDEP. Transl. Psychiatry 2013, 3, e300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, N.; Nonen, S.; Kato, M.; Wakeno, M.; Takekita, Y.; Kinoshita, T.; Kugawa, F. Therapeutic Response to Paroxetine in Major Depressive Disorder Predicted by DNA Methylation. Neuropsychobiology 2017, 75, 81–88. [Google Scholar] [CrossRef]
- Bell, J.T.; Tsai, P.C.; Yang, T.P.; Pidsley, R.; Nisbet, J.; Glass, D.; Mangino, M.; Zhai, G.; Zhang, F.; Valdes, A.; et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012, 8, e1002629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeven, J.E.; Yang, R.; Wolkowitz, O.M.; Bersani, F.S.; Lindqvist, D.; Mellon, S.H.; Yehuda, R.; Flory, J.D.; Lin, J.; Abu-Amara, D.; et al. Epigenetic Age in Male Combat-Exposed War Veterans: Associations with Posttraumatic Stress Disorder Status. Mol. Neuropsychiatry 2018, 4, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Bus, B.A.A.; Molendijk, M.L. The neurotrophic hypothesis of depression. Tijdschr. Psychiatr. 2016, 58, 215–222. [Google Scholar] [PubMed]
- Tadić, A.; Wagner, S.; Schlicht, K.F.; Peetz, D.; Borysenko, L.; Dreimüller, N.; Hiemke, C.; Lieb, K. The early non-increase of serum BDNF predicts failure of antidepressant treatment in patients with major depression: A pilot study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; D’Arcy, C.; Li, X.; Zhang, T.; Joober, R.; Meng, X. What do DNA methylation studies tell us about depression? A systematic review. Transl. Psychiatry 2019, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef] [Green Version]
- Braun, P.R.; Han, S.; Hing, B.; Nagahama, Y.; Gaul, L.N.; Heinzman, J.T.; Grossbach, A.J.; Close, L.; Dlouhy, B.J.; Howard, M.A.; et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiat. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Rudge, J.S.; Eaton, M.J.; Mather, P.; Lindsay, R.M.; Whittemore, S.R. CNTF Induces Raphe Neuronal Precursors to Switch from a Serotonergic to a Cholinergic Phenotype in Vitro. Mol. Cell Neurosci. 1996, 7, 204–221. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; West, J.; Beck, S. Age-associated epigenetic drift: Implications, and a case of epigenetic thrift? Hum. Mol. Genet. 2013, 22, R7–R15. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Gentile, S.; Fusco, M.L. Placental and fetal effects of antenatal exposure to antidepressants or untreated maternal depression. J. Matern. Fetal Neonatal. Med. 2017, 30, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Du Prel, J.B.; Röhrig, B.; Blettner, M. Critical Appraisal of Scientific Articles Part 1 of a Series on Evaluation of Scientific Publications. Dtsch. Arztebl. Int. 2009, 106, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; The, P.G. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Reference | Genomic Region Studied | Study Description | Tissue Examined | Findings |
|---|---|---|---|---|
| D’Addario et al., 2012 [49] | Exon I promoter (chr 11: 27 743 605–27 744 379) | Milanese study of BDNF methylation using fluorescence-based RT-PCR1 in bipolar disorder (BD) patients on mood stabilizers, BD patients on antidepressants + mood stabilizers, and healthy controls. | Peripheral blood mononuclear cells (PBMC) | ● BDNF promoter methylation was increased in BD2 compared to controls, but not in BD1 compared to controls. ● BDNF promoter methylation was increased in patients using antidepressants compared to controls and patients taking mood stabilizers alone. |
| D’Addario et al., 2013 [50] | Exon I promoter | Milanese study of BDNF methylation using fluorescence-based RT-PCR in major depressive disorder (MDD) on antidepressants, MDD on antidepressants + mood stabilizer, and healthy controls. | PBMC | ● MDD patients treated with antidepressants (serotonin or norepinephrine reuptake inhibitors (SSRIs or SNRIs)) alone had higher BDNF promoter methylation compared with patients receiving antidepressant + mood stabilizer. |
| Carlberg et al., 2014 [51] | Exon I promoter | Austrian study of BDNF methylation using PCR on bisulfite-converted genomic DNA from white, European MDD, BD, and unaffected controls. Subgroup of MDD patients analyzed for effects of antidepressants on DNA methylation. | PBMC | ● MDD subgroup treated with antidepressants had significantly increased BDNF promoter methylation compared to controls and MDD patients not treated with antidepressants. ● No increase in % of methylated reference values in MDD without antidepressant therapy compared to control subjects. |
| Wang et al., 2018 [52] | Five CpG islands within the promoter. | Measured methylation of BDNF via PCR amplification of bisulfate-converted DNA in Han Chinese MDD patients before and after 8 weeks of 10–20mg escitalopram daily. | Whole blood genomic DNA isolate | ● Methylation of 4 amplicons in BDNF (1, 3, 4, and 5) was significantly associated with response to escitalopram after 8 weeks, with higher methylation status associated with better response to escitalopram ● Patients with lower LES2 and higher DNA methylation responded better to escitalopram than those with higher LES and lower methylation. ● After 8 weeks of escitalopram, average BDNF and any BDNF amplicon methylation were significantly increased compared to baseline. ● In the remitter group, escitalopram treatment significantly increased DNA methylation, while in the non-remitter group there was no significant increase in BDNF methylation. |
| Januar et al., 2015 [53] | Exon I and IV promoters | Bisulfite conversion and PCR measurement of BDNF methylation in >65-year-old French patients with or without depression. | Buccal swabs | ● After adjustment for age, sex, and antidepressant use, methylation of CpG unit 3.4.5. of exon I and CpG-3 of promoter IV remained significantly higher in depression. |
| Kang et al., 2013 [54] | CpG-rich region of the promoter between -694 and -577 relative to transcriptional start site, including 7 CpG sites. | Measured and averaged methylation at 7 CpG sites of BDNF promoter before 12 week antidepressant treatment (SSRIs, bupropion, mirtazapine, venlafaxine, amitriptyline, or imipramine) in Korean MDD patients. Assessed suicidal ideation during the treatment period. | PBMC | ● Patients with lower BDNF promoter methylation showed greater improvement in BSS3 over the treatment period than patients with higher BDNF promoter methylation before antidepressant treatment. |
| Tadić et al., 2014 [55] | Twelve CpG sites within exon IV promoter | Measured BDNF methylation status in MDD patients before treatment with antidepressants and assessed outcomes at the study endpoint (ranging from 2–6 weeks) with HAM-D-214. | PBMC | ● Baseline methylation at CpG-87 predicted antidepressant response: Non-responders had a significantly lower methylated C-fraction than responders. ● Patients without any methylation at CpG-87 had higher risk of non-response to antidepressant than those with methylation. ● No significant effect of antidepressant class on the association between CpG-87 methylation and antidepressant response. ● DNA methylation of the 12 investigated CpG sites in exon IV did not change significantly during treatment from baseline to end point. |
| Kim et al., 2015 [57] | Nine CpG sites within exon VI promoter | Measured methylation of promoter of BDNF in peripheral blood of Korean ACS patients with and without any depressive disorder. They randomized 127 depressed participants to 24 weeks of escitalopram + ACS treatment and 128 to placebo + ACS treatment. The remaining 123 patients received standard medical treatment for ACS. | Peripheral blood leukocytes | ● In the escitalopram-treated group, significantly higher average methylation % was found in participants with remission compared to those who did not remit. ● Persistence of baseline depressive disorder 1 year later was associated with a higher methylation at CpG-1 and higher average methylation % only in the placebo and the medical treatment-only groups, and not in the escitalopram group. |
| Chen et al., 2011 [58] | Exon IV promoter | Assessed relationship between BDNF methylation, MDD, antidepressant use, exon IV expression, and H3K27 tri-methylation in Caucasian male French Canadians post-mortem. | Prefrontal cortex (post-mortem) | ● Antidepressant use was associated with significantly lower H3K27 methylation levels in BDNF exon IV promoter than the MDD without antidepressant and control groups. |
| Lopez et al., 2013 [59] | Exon IV promoter | Measured BDNF H3K27 trimethylation levels with ChIP5 and peripheral BDNF mRNA6 at baseline and after 8 weeks of citalopram. Subjects were divided into responders and non-responders based on final HAM-D score. | Peripheral blood | ● Trimethylation of H3K27 at BDNF exon IV was significantly decreased after 8 weeks of citalopram in responders to citalopram, but not in non-responders. ● Significant negative correlation between change in depression severity and change in trimethylated H3K27 expression. |
| Reference | Genomic Region Studied | Study Description | Tissue Examined | Findings |
|---|---|---|---|---|
| Checknita et al., 2018 [64] | Exon I promoter | MAOA was genotyped and methylation was measured via bisulfite conversion with PCR in Swedish women with and without substance use disorders, comparing those with and without a history of childhood sexual and/or physical abuse. | Genomic DNA extracted from saliva | ● Among women with current depression, higher methylation was associated with past or current use of any medication (stimulants, hypnotics, anxiolytics, antidepressants, or antipsychotics) at CpG 7/8. ● When antidepressants were considered alone, no differences in methylation were found. |
| Domschke et al., 2015 [65] | Forty-three CpG sites within exon I promoter. | Methylation of MAOA was measured in peripheral blood of German MDD patients. Clinical response to 6 weeks of escitalopram was assessed by intra-individual changes in HAM-D-21 scores between weeks 1 and 6. | Peripheral blood | ● In females, overall methylation across all 3 amplicons and single CpGs showed no association with intake of medication (SSRI, SSRI + mirtazapine, antipsychotics, or mood stabilizers). ● Average methylation across all CpGs showed no association with response to escitalopram after 6 weeks in females. ● Lower methylation at CpG-1 in Amplicon A and CpG-5 in Amplicon B were nominally associated with worse treatment response after 6 weeks of escitalopram in females. ● In males, neither average methylation across all sites nor methylation status of individual CpG sites showed association with response to escitalopram after 6 weeks. |
| Reference | Genomic Region Studied | Study Description | Tissue Examined | Findings |
|---|---|---|---|---|
| Booij et al., 2015 [67] | SLC6A4 CpG sites 5–15 within the 214–625 bp regulatory region upstream of the promoter. | Analysis of SLC6A4 methylation via pyrosequencing and luciferase reporter vector; compared with SLC6A4 mRNA expression in T cells via RT-PCR; compared between healthy controls, childhood trauma, and MDD. | Peripheral blood T cells and monocytes | ● SSRIs associated with increased methylation at CpG-11 and -12 compared to no antidepressants or dual-acting antidepressants. ● SSRI use predicted increased methylation at CpG-11 and -12. |
| Kang et al., 2013 [68] | SLC6A4 CpG-rich region of the promoter between -479 and -350 relative to the transcriptional start site, including 7 CpG sites. | Measured DNA methylation of SLC6A4 in Korean MDD patients. Patients were treated with a variety of antidepressants for 12 weeks and clinical outcome was measured by scales for depression, anxiety, functioning, disability, and quality of life before and after 12 weeks of antidepressant treatment. | Peripheral blood | ● Higher methylation at CpG-2 and higher average SLC6A4 methylation predicted less HAM-D improvement in depression. ● Higher methylation percentage at CpG-1 was associated with less improvement in HAM-A 1. ● Higher average promoter methylation was associated with decreased SOFAS 2. ● All above findings lost significance after Bonferroni correction. |
| Domschke et al., 2014 [69] | SLC6A4 Nine CpG sites within the transcriptional control region upstream of exon 1A. | Analyzed blood sample DNA methylation status in Caucasian MDD patients. Clinical response to 6 weeks of escitalopram treatment was assessed by intra-individual changes in HAM-D-21 scores. | Peripheral blood | ● Overall SLC6A4 methylation in the analyzed amplicon showed no association with medication intake (SSRI vs. SSRI + mirtazapine), or comedication with antipsychotics or mood stabilizers. ● Average methylation across 9 CpG sites was significantly associated with response to escitalopram after 6 weeks: Lower methylation was associated with impaired treatment response, while higher methylation was associated with better treatment response. ● Average methylation across CpGs post-treatment showed a nominally significant association between lower methylation status and impaired treatment response. ● Methylation status of individual CpG-1 and -2 were significantly associated with treatment response after 6 weeks. CpG-4 methylation was nominally associated with treatment response. |
| Iga, et al., 2016 [70] | SLC6A4 One CpG-rich region in the promoter, including 9 CpG sites. | Measured DNA methylation in peripheral blood of Japanese MDD patients before and after 8 weeks of treatment with various antidepressants. Clinical outcome was assessed with HAM-D. | Peripheral blood | Lower CpG-2 methylation levels were associated with greater clinical improvement as assessed by HAM-D scores. |
| Okada et al., 2014 [71] | SLC6A4 CpG island in exon I promoter —sequence chr 17: 28562388–28563186 | Measured DNA methylation and responses to antidepressant therapy in unmedicated, Japanese MDD patients. | Peripheral blood | ● A significant increase in methylation was found in CpG-3 after 6 weeks of antidepressant treatment in MDD. ● Pre-treatment methylation of CpG-3 showed significant positive correlation with IR 3 in MDD. ● No significant difference in methylation rates between the patients with >50% IR and <50% IR. ● No correlation between IR and methylation change of CpG-3 before and after antidepressant treatment. |
| Bayles et al., 2013 [72] | SLC6A2 Promoter region 1 (−515 bp to −225 bp), promoter region 2 (−180 bp–+167 bp), region A, and region B. | Study of SLC6A2 methylation using EpiTYPER assays in an Australian population of MDD, panic disorder, and healthy controls. Subset comparison of SLC6A2 promoter methylation before and after 3 month treatment with SSRIs in MDD and panic disorder. | Peripheral blood leukocytes | Statistically significant increase in methylation of CpG sites 14 and 15 (Region A) in MDD and panic disorder patients after 3 months of SSRI treatment. |
| Reference | Genomic Region Studied | Study Description | Tissue Examined | Findings |
|---|---|---|---|---|
| Wang et al., 2018 [77] | HTR1A and HTR1B Ninety-six CpG sites within promoter regions. | Measured DNA methylation via PCR amplification of bisulfate-converted DNA, in Han Chinese MDD patients before and after 8 week treatment with escitalopram 10–20 mg daily. | Whole blood genomic DNA isolate | ● Average methylation level of HTR1A or HTR1B was not significantly associated with treatment response to escitalopram. ● 2 CpG sites significantly predicted antidepressant response: CpG-668, amplicon HTR1A_1 and CpG-1401, amplicon HTR1B_4. Lower methylation at those sites was associated with impaired response to escitalopram. ● Patients with lower LES and higher DNA methylation at 4 CpG sites (HTR1A_1 CpG-659, HTR1A_1 CpG-668, HTR1A_1 CpG-706, HTR1B_2 CpG-107) responded better to escitalopram than those with higher LES and lower methylation. ● No significant difference in average DNA methylation of HTR1A and HTR1B between baseline and treatment week 8. ● Methylation at 4 individual CpG sites within HTR1A/1B was significantly increased after 8 weeks of escitalopram (HTR1B_1 CpG-336, HTR1B_2 CpG-105, HTR1B_2 CpG-107, HTR1B_4 CpG-1443). ● Significant differences in 6 CpG sites’ methylation in remitter and non-remitter groups (HTR1A_2 CpG-2793, HTR1A_2 CpG-2834, HTR1A_2 CpG-2927, HTR1A_2 CpG-2937, HTR1B_2 CpG-100, HTR1B_4 CpG-1401). ● DNA methylation was increased after escitalopram treatment in the remitter group, while there was no influence of escitalopram on methylation in the non-remitter group. |
| Gassó et al., 2017 [78] | HTR1B CpG islands in promoter, including 7 CpG sites. Chromosome 6: (77463994 – 77464019) | Measured DNA methylation after Spanish children with MDD, OCD1, or GAD2 completed 12 weeks of fluoxetine treatment for the first time and assessed whether DNA methylation was associated with clinical response to fluoxetine. | Peripheral blood | Negative correlation between average DNA methylation of the 7 CpGs analyzed in the HTR1B promoter and clinical response to fluoxetine as measured by GAF3/CGAS4. |
| Reference | Genomic Region Studied | Study Description | Tissue Examined | Findings |
|---|---|---|---|---|
| Ryan et al., 2017 [82] | IL6 234 bp region of the promoter | Measured DNA methylation of IL6 in peripheral tissue of French patients >65-years-old with and without depression, and with and without antidepressant treatment. | Buccal swabs | ● Depression was associated with a 2.4% decreased overall IL6 methylation compared to controls. ● Antidepressant use was associated with a mean 4.6% increase in methylation of IL6. |
| Powell et al., 2013 [85] | IL11 CpG island in the promoter (chr 19: 55880511–55880989) | Measured baseline DNA methylation the IL11 promoter in peripheral blood of Caucasian European MDD patients randomized to 12 weeks of either escitalopram or nortriptyline. | Whole blood genomic DNA isolate | ● Methylation of CpG-5 predicted response to either antidepressant. ● Lower baseline CpG-5 methylation was associated with better antidepressant response. ● CpG-4 methylation predicted differential response to the two medications: High methylation levels were associated with better response to escitalopram, but with worse response to nortriptyline. ● Methylation at CpG-11 and rs1126757 significantly predicted response to treatment: Homozygous G allele (GG) individuals who had higher levels of CpG-11 methylation responded better to antidepressant treatment than those who were homozygous for the A allele (AA). |
| Reference | Genomic Region Studied | Study Description | Tissue Examined | Findings |
|---|---|---|---|---|
| Takeuchi et al., 2017 [86] | Whole genome | Measured genome-wide methylation in peripheral blood cells of MDD patients before 6 week treatment with paroxetine. Compared the patients who were the best and worst responders to paroxetine. | Whole blood genomic DNA isolate | ● 623 CpG sites had a >10% difference in methylation status between the best (BR) and worst (WR) responders to paroxetine, with 218 sites nominally significantly different and 2 sites significantly different: cg00594917 (PPFIA4 exon I) and cg07260927 (in the 5′ UTR of HS3ST1). ● Methylation difference between WR and BR was greatest at cg00594917 in PPFIA4 exon I. ● Hierarchical cluster analysis of 23 CpG sites in PPFIA4 distinguished BR and WR patients except for 1 patient. ● Methylation of 6 CpG sites within PPFIA4 was significantly different between BR and WR. At all 6 sites, WR had higher methylation than BR. ● Hierarchical cluster analysis of 28 CpG sites in HS3ST1 distinguished BR and WR patients except for 1 patient. ● Methylation of 5 CpG sites within HS3ST1 was significantly different between WR and BR. At 4 of 5 sites, methylation levels of WR were higher than those of BR. |
| Verhoeven et al., 2018 [88] | Whole genome | Applied Horvath’s epigenetic clock algorithm to calculate epigenetic age of leukocyte genomes of combat-exposed veterans with and without PTSD. | PBMC | ● Current antidepressant use was associated with lower epigenetic age in veterans across the sample compared to subjects not taking antidepressants. ● PTSD subjects taking antidepressants had a significantly lower epigenetic age than participants without PTSD. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webb, L.M.; Phillips, K.E.; Ho, M.C.; Veldic, M.; Blacker, C.J. The Relationship between DNA Methylation and Antidepressant Medications: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 826. https://doi.org/10.3390/ijms21030826
Webb LM, Phillips KE, Ho MC, Veldic M, Blacker CJ. The Relationship between DNA Methylation and Antidepressant Medications: A Systematic Review. International Journal of Molecular Sciences. 2020; 21(3):826. https://doi.org/10.3390/ijms21030826
Chicago/Turabian StyleWebb, Lauren M., Kathryn E. Phillips, Man Choi Ho, Marin Veldic, and Caren J. Blacker. 2020. "The Relationship between DNA Methylation and Antidepressant Medications: A Systematic Review" International Journal of Molecular Sciences 21, no. 3: 826. https://doi.org/10.3390/ijms21030826
APA StyleWebb, L. M., Phillips, K. E., Ho, M. C., Veldic, M., & Blacker, C. J. (2020). The Relationship between DNA Methylation and Antidepressant Medications: A Systematic Review. International Journal of Molecular Sciences, 21(3), 826. https://doi.org/10.3390/ijms21030826