Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction


Abstract
:1. Introduction
2. Results
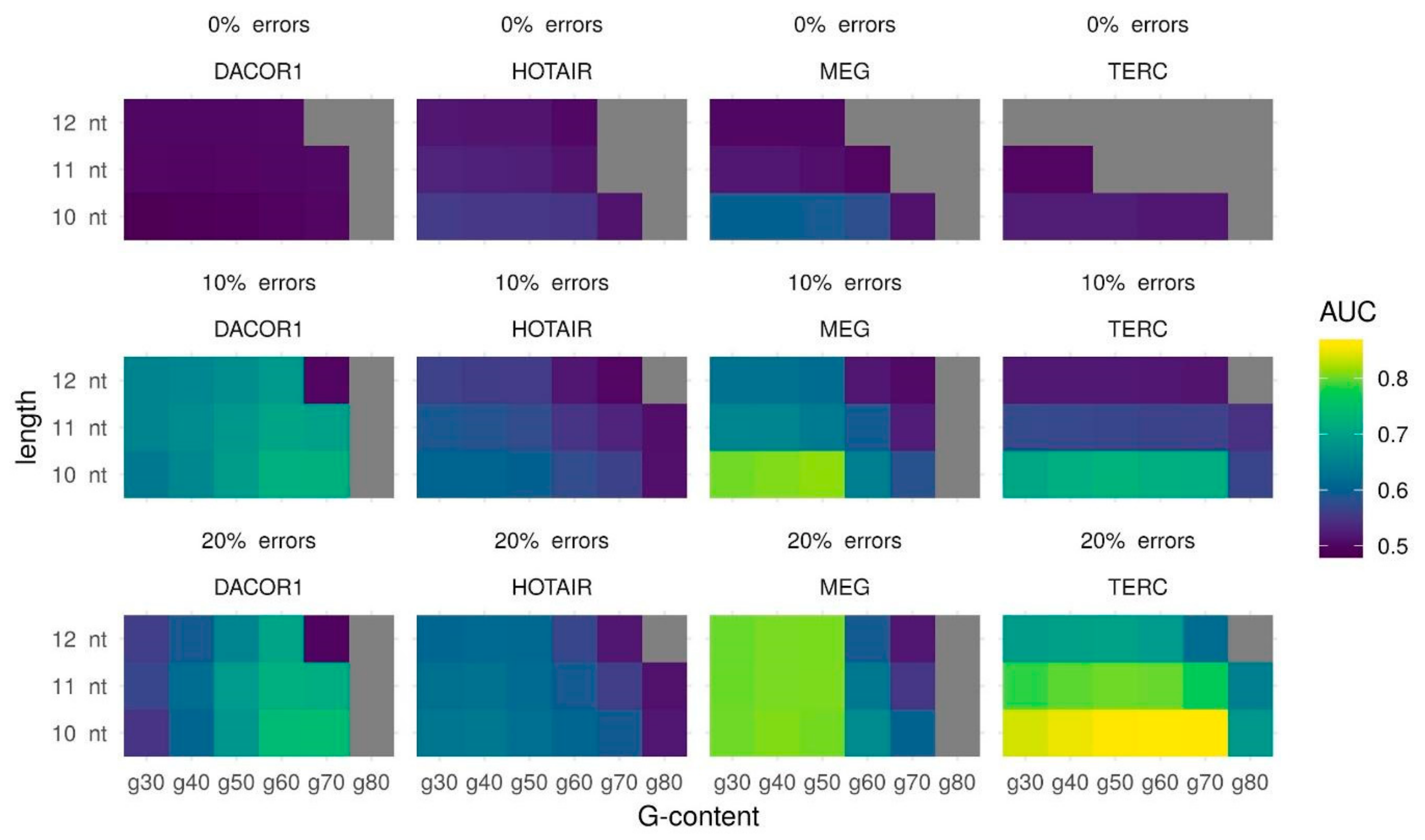
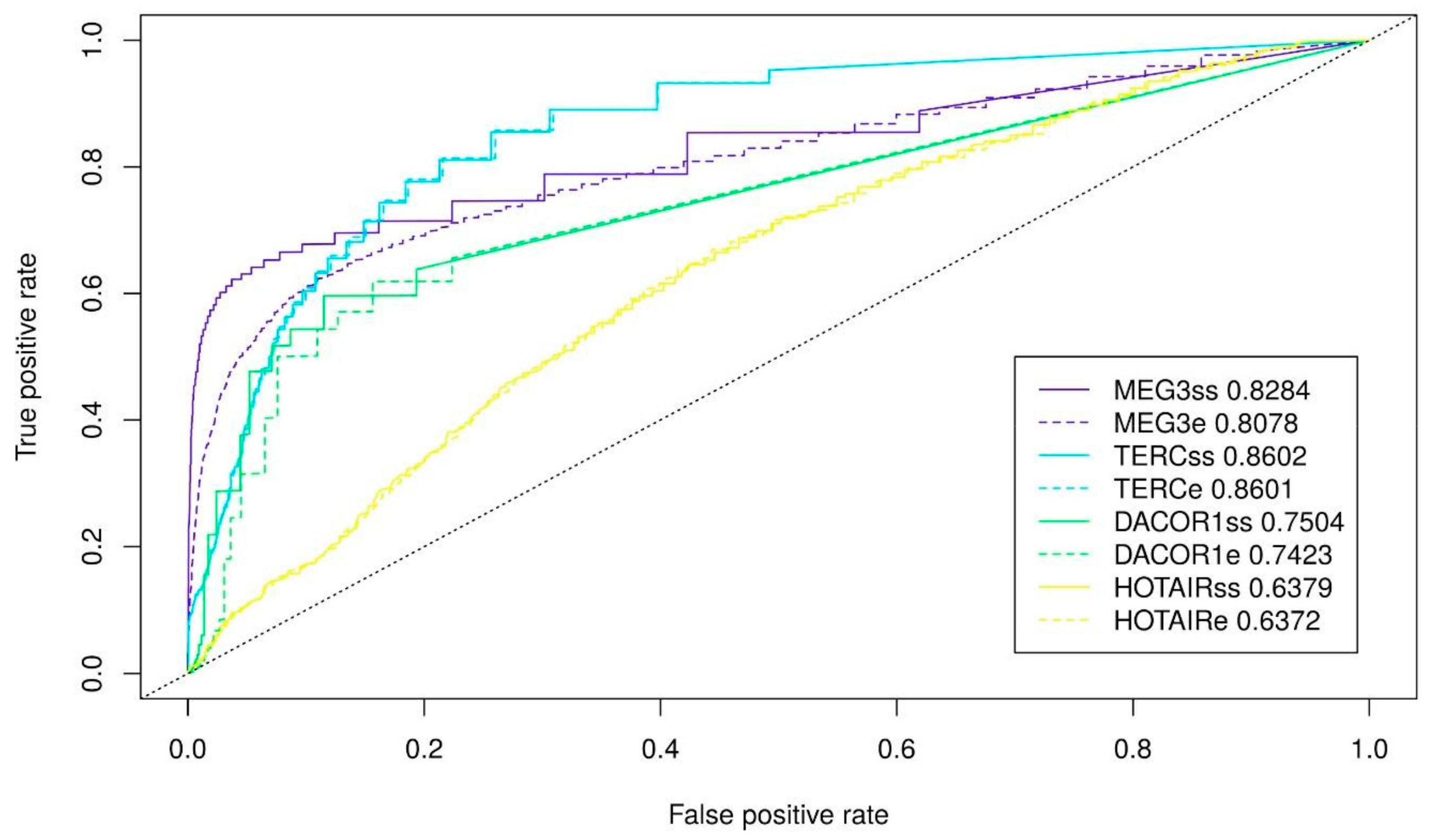
2.1. Fitting Triplex Parameters
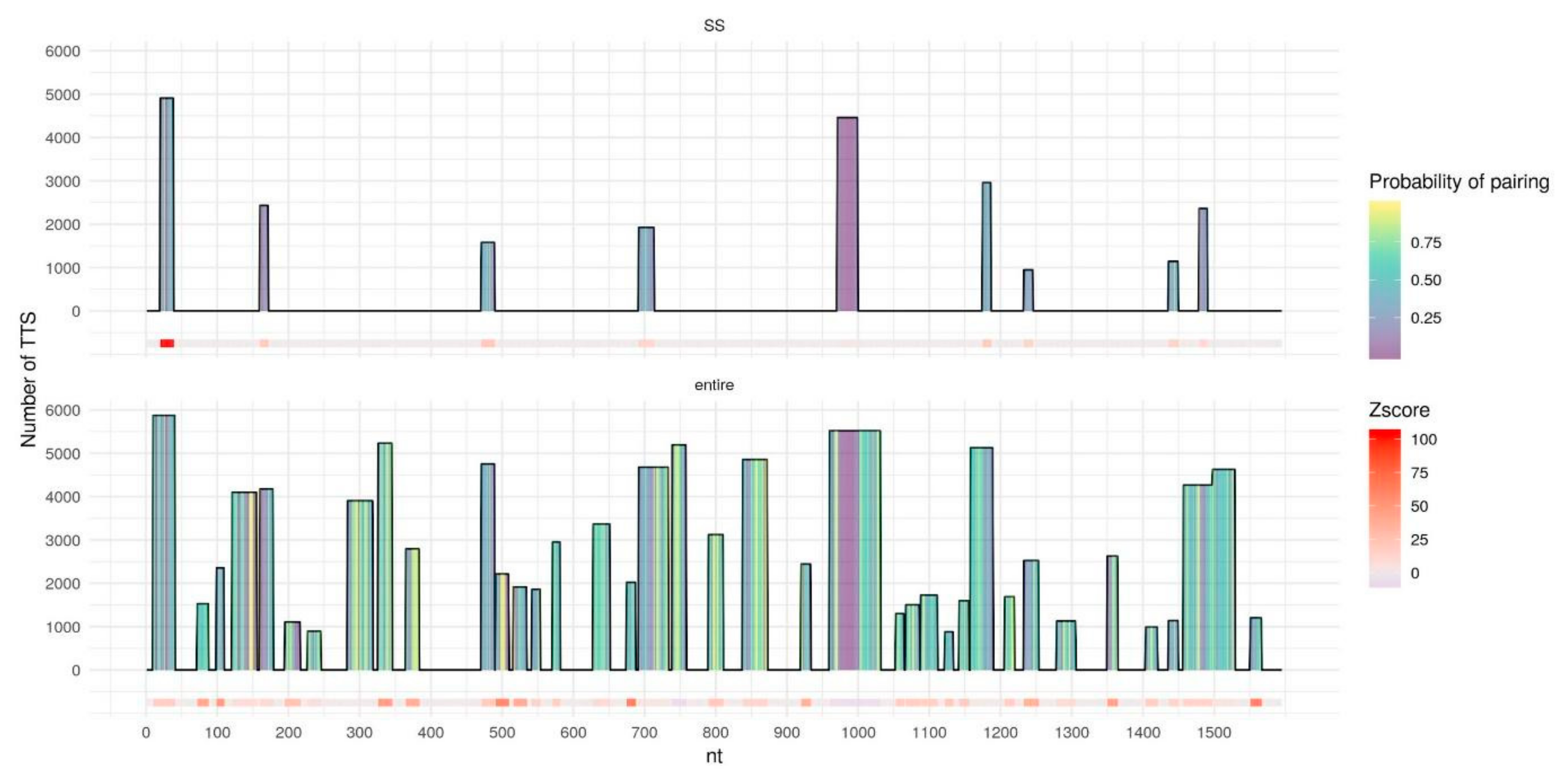
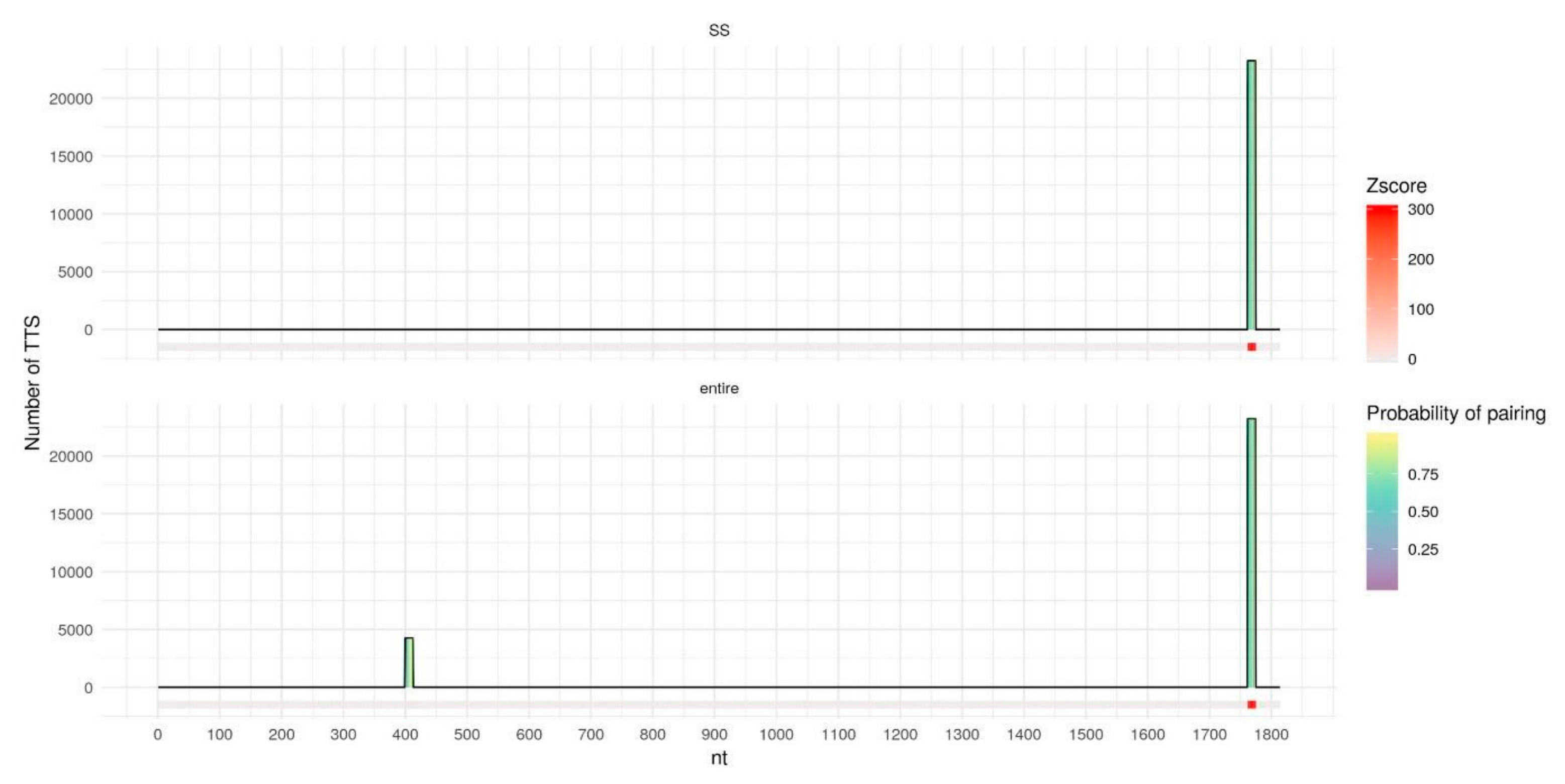
2.2. Secondary Structure Predictions
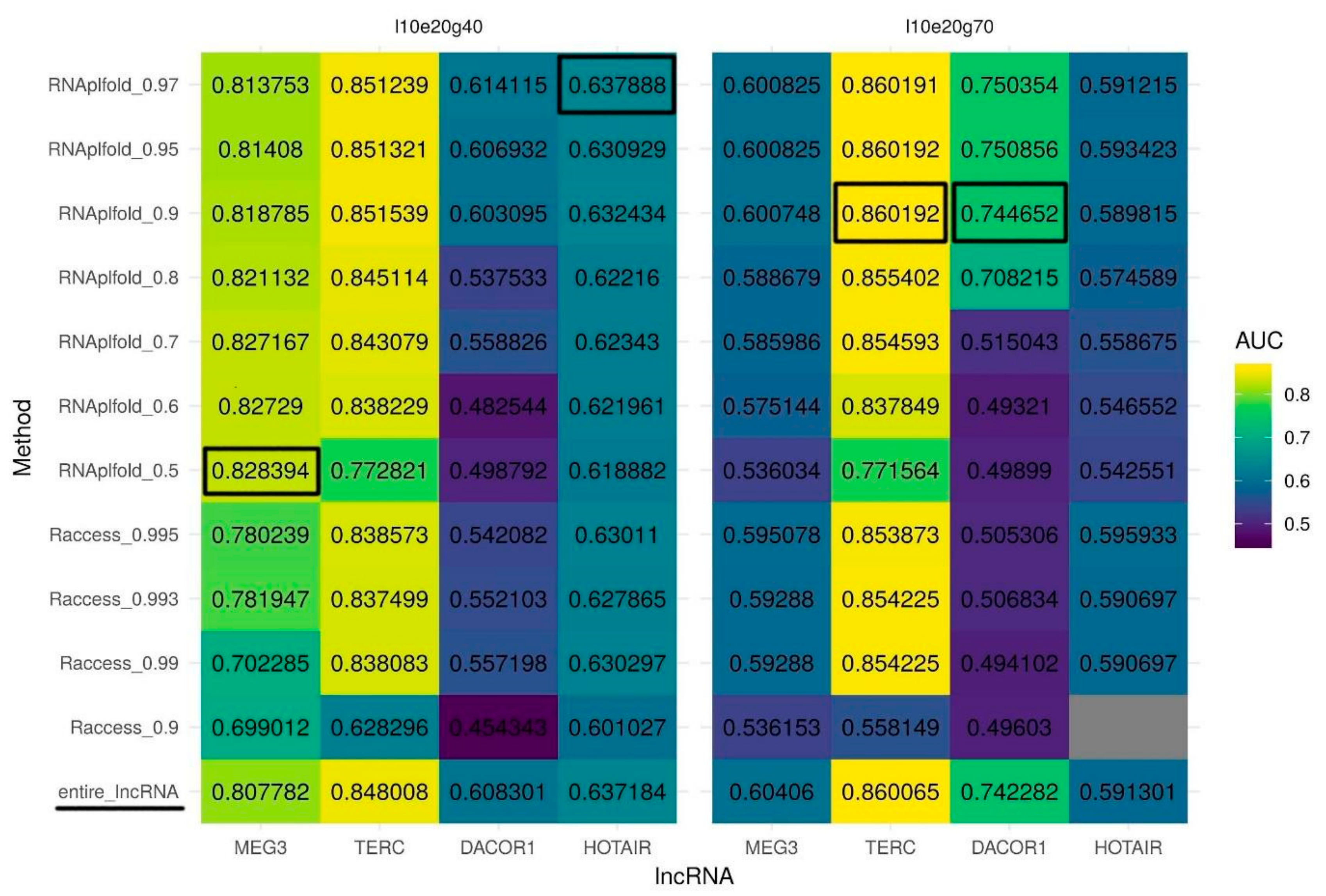
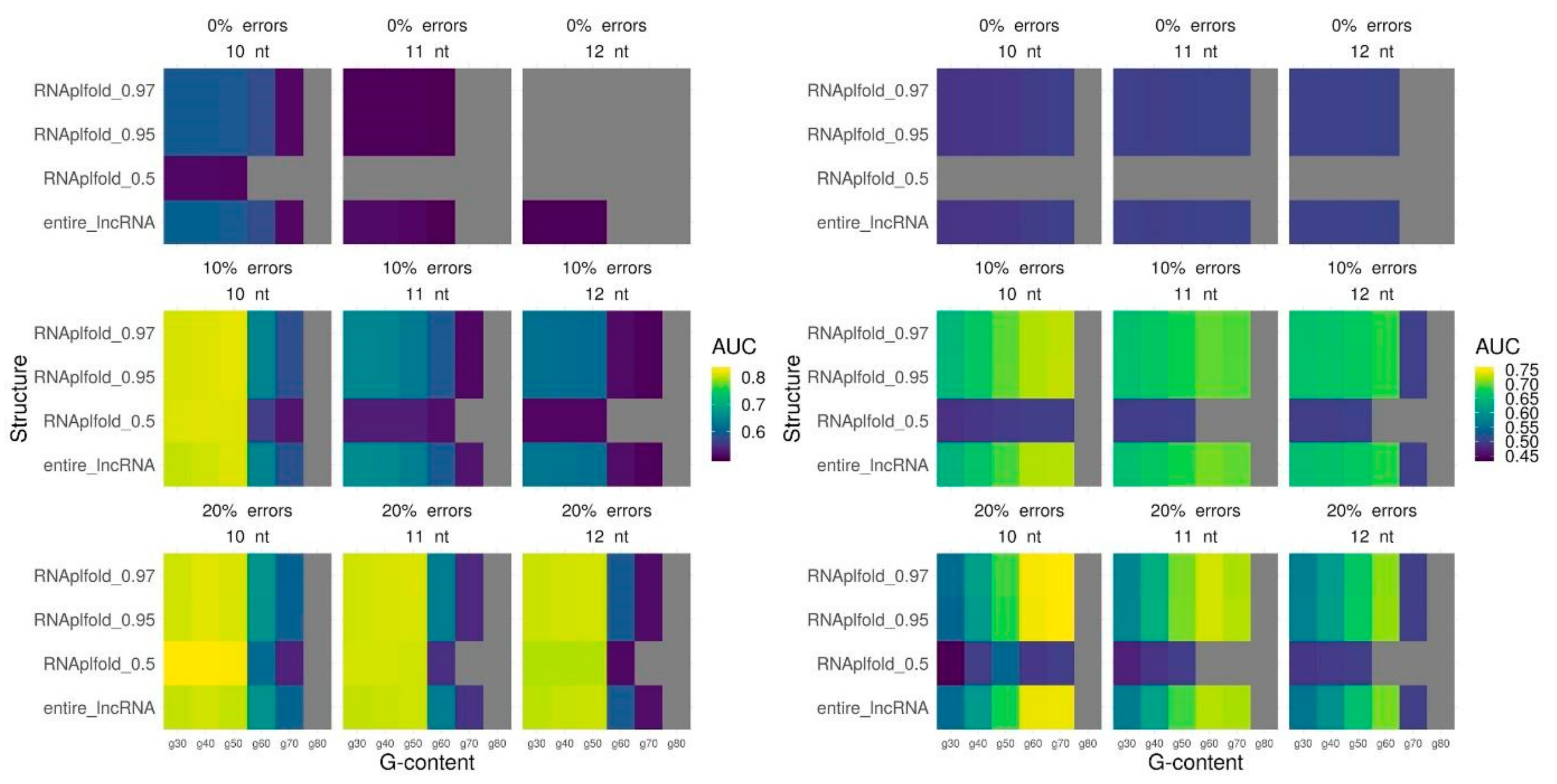
2.3. Triplex Prediction Using Secondary Structure
2.4. DNA Binding Domains (DBDs) and Single-Stranded Fragments
3. Discussion
4. Materials and Methods
4.1. lncRNA Selection and Experimental Data on lncRNA Binding
4.2. LncRNA Secondary Structure Prediction
4.3. Triple Helix Prediction
4.4. ROC-Curves and AUC
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genome Res. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, R.; Andersen, P.R.; Valen, E.; Core, L.J.; Bornholdt, J.; Boyd, M.; Jensen, T.H.; Sandelin, A. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat. Commun. 2014, 5, 5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hon, C.-C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagarde, J.; Uszczynska-Ratajczak, B.; Santoyo-Lopez, J.; Gonzalez, J.M.; Tapanari, E.; Mudge, J.M.; Steward, C.A.; Wilming, L.; Tanzer, A.; Howald, C.; et al. Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat. Commun. 2016, 7, 12339. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.M.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; De Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar]
- Alam, T.; Medvedeva, Y.A.; Jia, H.; Brown, J.B.; Lipovich, L.; Bajic, V.B. Promoter Analysis Reveals Globally Differential Regulation of Human Long Non-Coding RNA and Protein-Coding Genes. PLoS ONE 2014, 9, e109443. [Google Scholar] [CrossRef] [Green Version]
- Böhmdorfer, G.; Wierzbicki, A.T. Control of Chromatin Structure by Long Noncoding RNA. Trends Cell Boil. 2015, 25, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Jandura, A.; Krause, H.M. The New RNA World: Growing Evidence for Long Noncoding RNA Functionality. Trends Genet. 2017, 33, 665–676. [Google Scholar] [CrossRef]
- Qian, X.; Zhao, J.; Yeung, P.Y.; Zhang, Q.C.; Kwok, C.K. Revealing lncRNA Structures and Interactions by Sequencing-Based Approaches. Trends Biochem. Sci. 2019, 44, 33–52. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, D.R.; Thomas, K.; Presser, A.; Bernstein, B.E.; Van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [Green Version]
- Grote, P.; Herrmann, B.G. The long non-coding RNA Fendrr links epigenetic control mechanisms to gene regulatory networks in mammalian embryogenesis. RNA Boil. 2013, 10, 1579–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA–DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.-Y.; Bogu, G.K.; Soh, B.S.; Stanton, L.W. The Long Noncoding RNA RMST Interacts with SOX2 to Regulate Neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.A.; Westhof, E. The Dynamic Landscapes of RNA Architecture. Cell 2009, 136, 604–609. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Meredith, E.K.; Balas, M.M.; Sindy, K.; Haislop, K.; Johnson, A.M. An RNA matchmaker protein regulates the activity of the long noncoding RNA HOTAIR. RNA 2016, 22, 995–1010. [Google Scholar] [CrossRef] [Green Version]
- Postepska-Igielska, A.; Giwojna, A.; Gasri-Plotnitsky, L.; Schmitt, N.; Dold, A.; Ginsberg, D.; Grummt, I. LncRNA Khps1 Regulates Expression of the Proto-oncogene SPHK1 via Triplex-Mediated Changes in Chromatin Structure. Mol. Cell 2015, 60, 626–636. [Google Scholar] [CrossRef]
- O’Leary, V.B.; Ovsepian, S.V.; Carrascosa, L.G.; Buske, F.A.; Radulović, V.; Niyazi, M.; Moertl, S.; Trau, M.; Atkinson, M.J.; Anastasov, N. PARTICLE, a Triplex-Forming Long ncRNA, Regulates Locus-Specific Methylation in Response to Low-Dose Irradiation. Cell Rep. 2015, 11, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA Triplex Formation by Long Noncoding RNAs. Cell Chem. Boil. 2016, 23, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Angrand, P.-O.; Vennin, C.; Le Bourhis, X.; Adriaenssens, E. The role of long non-coding RNAs in genome formatting and expression. Front. Genet. 2015, 6, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Lu, X. Decreased expression of MEG3 contributes to retinoblastoma progression and affects retinoblastoma cell growth by regulating the activity of Wnt/β-catenin pathway. Tumor Boil. 2015, 37, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Gordon, F.E.; Nutt, C.L.; Cheunsuchon, P.; Nakayama, Y.; Provencher, K.A.; Rice, K.A.; Zhou, Y.; Zhang, X.; Klibanski, A. Increased expression of angiogenic genes in the brains of mouse meg3-null embryos. Endocrinology 2010, 151, 2443–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonov, I.V.; Mazurov, E.; Borodovsky, M.; A Medvedeva, Y. Prediction of lncRNAs and their interactions with nucleic acids: Benchmarking bioinformatics tools. Briefings Bioinform. 2018, 20, 551–564. [Google Scholar] [CrossRef]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Boil. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Sherpa, C.; Rausch, J.W.; Le Grice, S.F.J. Structural characterization of maternally expressed gene 3 RNA reveals conserved motifs and potential sites of interaction with polycomb repressive complex 2. Nucleic Acids Res. 2018, 46, 10432–10447. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-C.; Hänzelmann, S.; Cetin, N.S.; Frank, S.; Zajzon, B.; Derks, J.-P.; Akhade, V.S.; Ahuja, G.; Kanduri, C.; Grummt, I.; et al. Detection of RNA–DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019, 47, e32. [Google Scholar] [CrossRef] [Green Version]
- Buske, F.A.; Bauer, D.C.; Mattick, J.S.; Bailey, T.L. Triplexator: Detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012, 22, 1372–1381. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Zhang, H.; Liu, H.; Zhu, H. LongTarget: A tool to predict lncRNA DNA-binding motifs and binding sites via Hoogsteen base-pairing analysis. Bioinformatics 2015, 31, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Chainani, P.; White, T.; Yang, J.; Liu, Y.; Soibam, B. Deep learning identifies genome-wide DNA binding sites of long noncoding RNAs. RNA Boil. 2018, 15, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Antonov, I.; Medvedeva, Y.A. Purine-rich low complexity regions are potential RNA binding hubs in the human genome. F1000Research 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Sentürk, C.N.; Cetin, N.S.; Kuo, C.C.; Ribarska, T.; Li, R.; Costa, I.G. Isolation and genome-wide characterization of cellular DNA:RNA triplex structures. Nucleic Acids Res. 2019, 47, 2306–2321. [Google Scholar] [CrossRef]
- Kunkler, C.N.; Hulewicz, J.P.; Hickman, S.C.; Wang, M.C.; McCown, P.J.; A Brown, J. Stability of an RNA•DNA-DNA triple helix depends on base triplet composition and length of the RNA third strand. Nucleic Acids Res. 2019, 47, 7213–7222. [Google Scholar] [CrossRef] [PubMed]
- Maenner, S.; Blaud, M.; Fouillen, L.; Savoye, A.; Marchand, V.; Dubois, A.; Sanglier-Cianférani, S.; Van Dorsselaer, A.; Clerc, P.; Avner, P.; et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Boil. 2010, 8, e1000276. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Boil. 2011, 6, 26. [Google Scholar] [CrossRef]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.-T.; Li, H.; Fu, X.-D. GRID-seq reveals the global RNA–chromatin interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr. Biol. 2017, 27, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, A.; Agostini, F.; Suzuki, A.M.; Hashimoto, K.; Pascarella, G.; Gimenez, J.; Roos, L.; Nash, A.J.; Ghilotti, M.; Cameron, C.J.; et al. RADICL-seq identifies general and cell type-specific principles of genome-wide RNA-chromatin interactions. BioRxiv 2019, 681924. [Google Scholar]
- Li, T.; Mo, X.; Fu, L.; Xiao, B.; Guo, J. Molecular mechanisms of long noncoding RNAs on gastric cancer. Oncotarget 2016, 7, 8601–8612. [Google Scholar] [CrossRef] [Green Version]
- Abbastabar, M.; Sarfi, M.; Golestani, A.; Khalili, E. lncRNA involvement in hepatocellular carcinoma metastasis and prognosis. EXCLI J. 2018, 17, 900–913. [Google Scholar] [PubMed]
- Peng, L.; Yuan, X.; Jiang, B.; Tang, Z.; Li, G.-C. LncRNAs: Key players and novel insights into cervical cancer. Tumor Boil. 2015, 37, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Gupta, S.K.; Schmitz, U.; Marquardt, S.; Knoll, S.; Spitschak, A.; Wolkenhauer, O.; Pützer, B.M.; Vera, J. MiR-205-5p and miR-342-3p cooperate in the repression of the E2F1 transcription factor in the context of anticancer chemotherapy resistance. Theranostics 2018, 8, 1106–1120. [Google Scholar] [CrossRef] [PubMed]
- Paugh, S.W.; Coss, D.R.; Bao, J.; Laudermilk, L.T.; Grace, C.R.; Ferreira, A.M. MicroRNAs Form Triplexes with Double Stranded DNA at Sequence-Specific Binding Sites; a Eukaryotic Mechanism via which microRNAs Could Directly Alter Gene Expression. PLoS Comput. Biol. 2016, 12, e1004744. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wang, G.; Vasquez, K.M. New Perspectives on DNA and RNA Triplexes As Effectors of Biological Activity. PLoS Genet. 2015, 11, e1005696. [Google Scholar] [CrossRef] [Green Version]
- Kalwa, M.; Hänzelmann, S.; Otto, S.; Kuo, C.-C.; Franzen, J.; Joussen, S.; Fernandez-Rebollo, E.; Rath, B.; Koch, C.; Hofmann, A.; et al. The lncRNA HOTAIR impacts on mesenchymal stem cells via triple helix formation. Nucleic Acids Res. 2016, 44, 10631–10643. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, Y.; Ge, Y.; Liu, J.; Zhao, Y. TERC promotes cellular inflammatory response independent of telomerase. Nucleic Acids Res. 2019, 47, 8084–8095. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell. 2011, 44, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Merry, C.R.; Forrest, M.E.; Sabers, J.N.; Beard, L.; Gao, X.-H.; Hatzoglou, M.; Jackson, M.W.; Wang, Z.; Markowitz, S.D.; Khalil, A.M. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 2015, 24, 6240–6253. [Google Scholar] [CrossRef]
- Kiryu, H.; Terai, G.; Imamura, O.; Yoneyama, H.; Suzuki, K.; Asai, K. A detailed investigation of accessibilities around target sites of siRNAs and miRNAs. Bioinformatics 2011, 27, 1788–1797. [Google Scholar] [CrossRef]
- Bernhart, S.H.; Hofacker, I.L.; Stadler, P.F. Local RNA base pairing probabilities in large sequences. Bioinformatics 2006, 22, 614–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lncRNA | Min Length (nt) | Max Error-Rate (%) | Min G-Content (%) | AUC | Symbol |
|---|---|---|---|---|---|
| MEG3 | 10 | 20 | 40 | 0.8078 | l10 e20g40 |
| HOTAIR | 10 | 20 | 40 | 0.6372 | l10e20g40 |
| TERC | 10 | 20 | 70 | 0.8601 | l10e20g70 |
| DACOR1 | 10 | 20 | 70 | 0.7423 | l10e20g70 |
| lncRNA | lncRNA ID | lncRNA Length | Number of DNA Peaks | Median Peaks Length | Method | Ref |
|---|---|---|---|---|---|---|
| MEG3 | ENST00000451743.6 | 1595 nt | 6798 | 400 nt | ChOP-seq | [13] |
| TERC | ENST00000363312.1 | 451 nt | 2198 | 756 nt | ChIRP-seq | [48] |
| DACOR1 | TCONS_00023265 | 1814 nt | 40213 | 279 nt | ChIRP-seq | [49] |
| HOTAIR | ENST00000424518.5 | 2421 nt | 832 | 678 nt | ChIRP-seq | [48] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matveishina, E.; Antonov, I.; Medvedeva, Y.A. Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction. Int. J. Mol. Sci. 2020, 21, 830. https://doi.org/10.3390/ijms21030830
Matveishina E, Antonov I, Medvedeva YA. Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction. International Journal of Molecular Sciences. 2020; 21(3):830. https://doi.org/10.3390/ijms21030830
Chicago/Turabian StyleMatveishina, Elena, Ivan Antonov, and Yulia A. Medvedeva. 2020. "Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction" International Journal of Molecular Sciences 21, no. 3: 830. https://doi.org/10.3390/ijms21030830
APA StyleMatveishina, E., Antonov, I., & Medvedeva, Y. A. (2020). Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction. International Journal of Molecular Sciences, 21(3), 830. https://doi.org/10.3390/ijms21030830