Rapid and Effective Generation of Nanobody Based CARs using PCR and Gibson Assembly

 , , , , , , ,
, , , , , , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
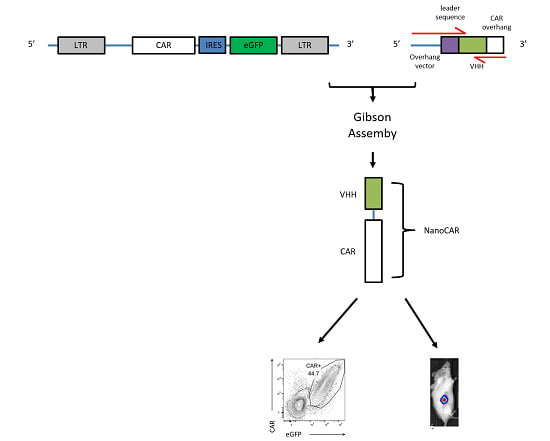
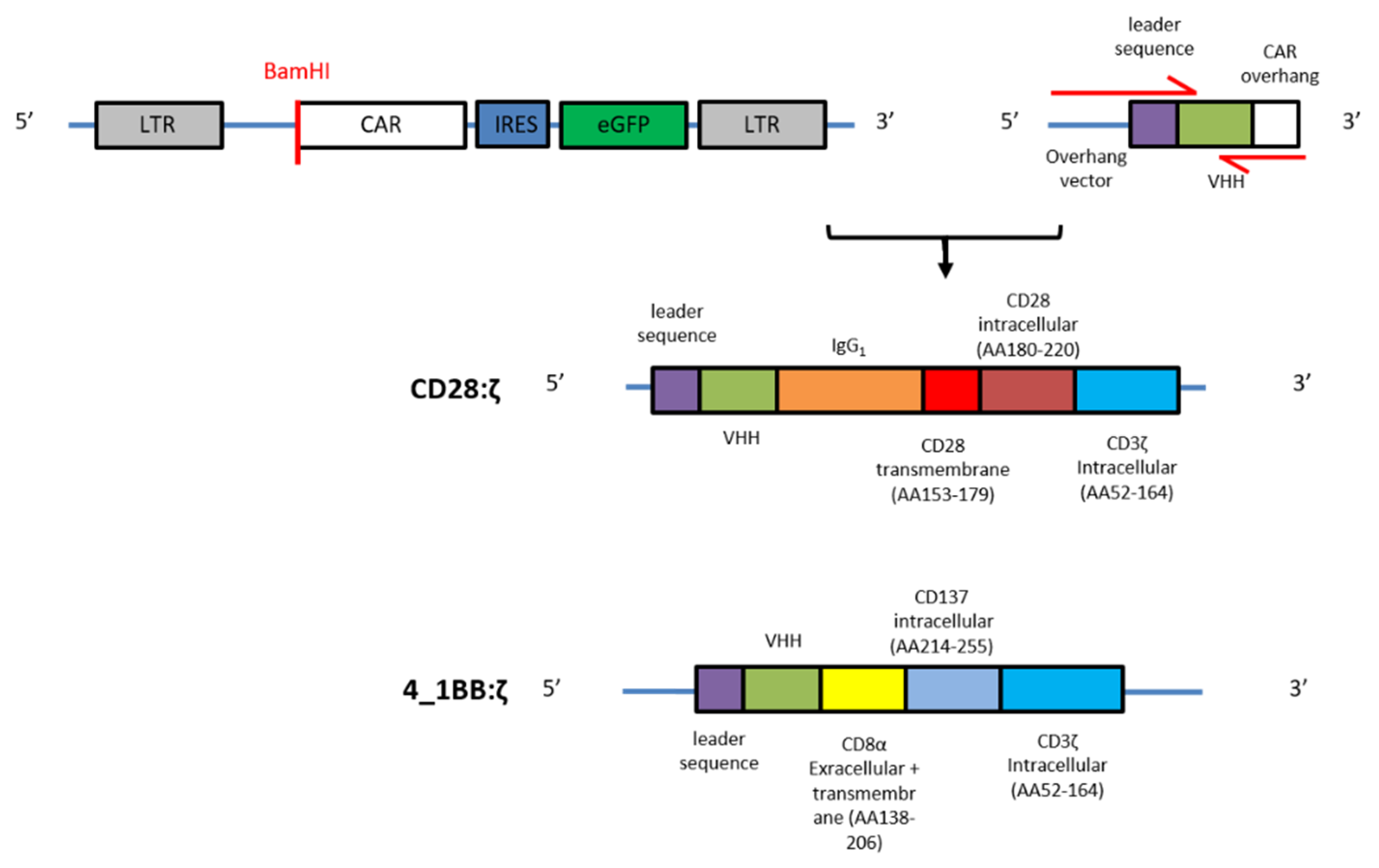
2.1. CAR Construction
2.2. NanoCARs Targeting CD33 are Functional
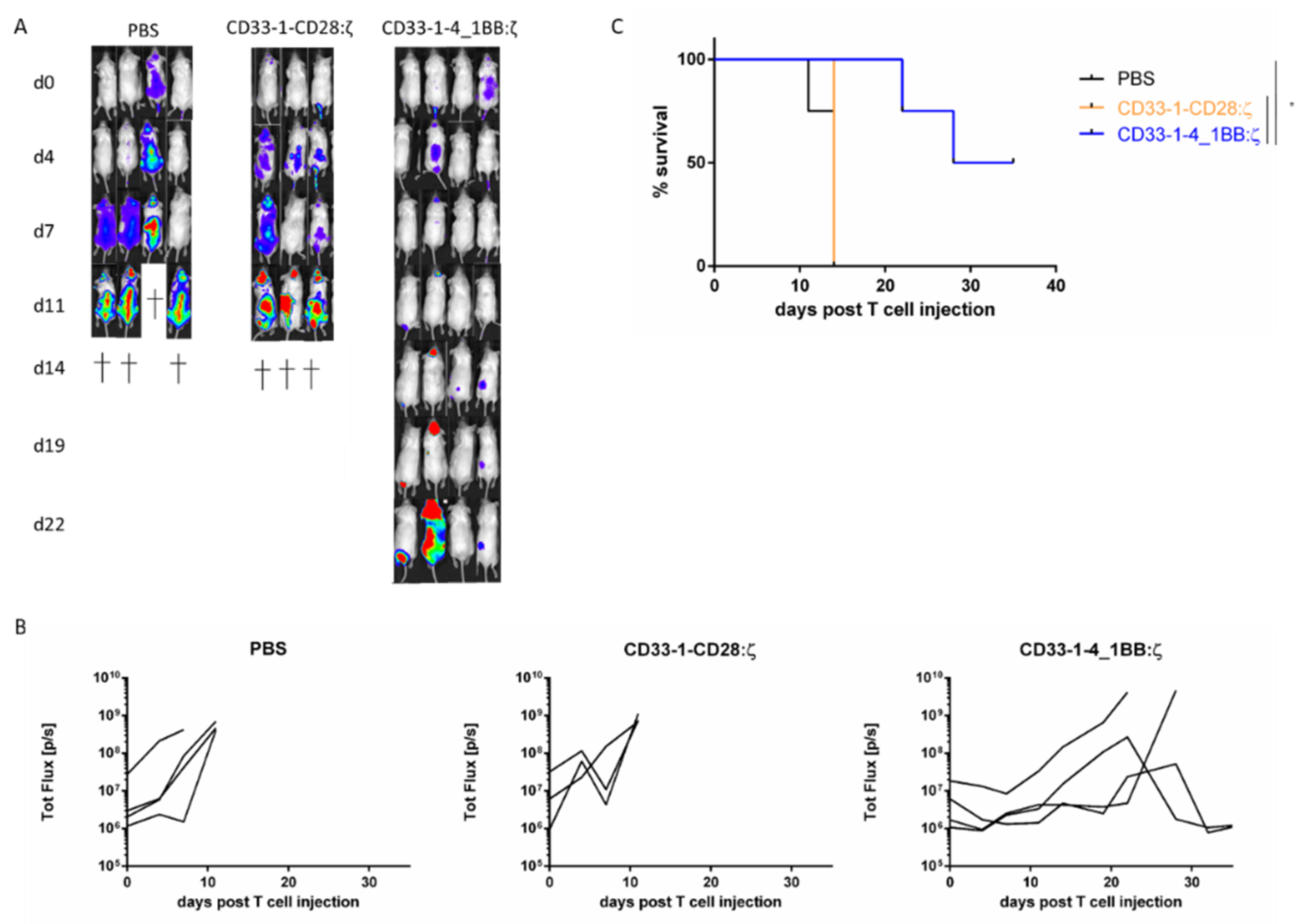
2.3. NanoCAR T Cells Expressing 4_1BB:ζ Outpreform CD28:ζ NanoCAR T Cells in Vivo
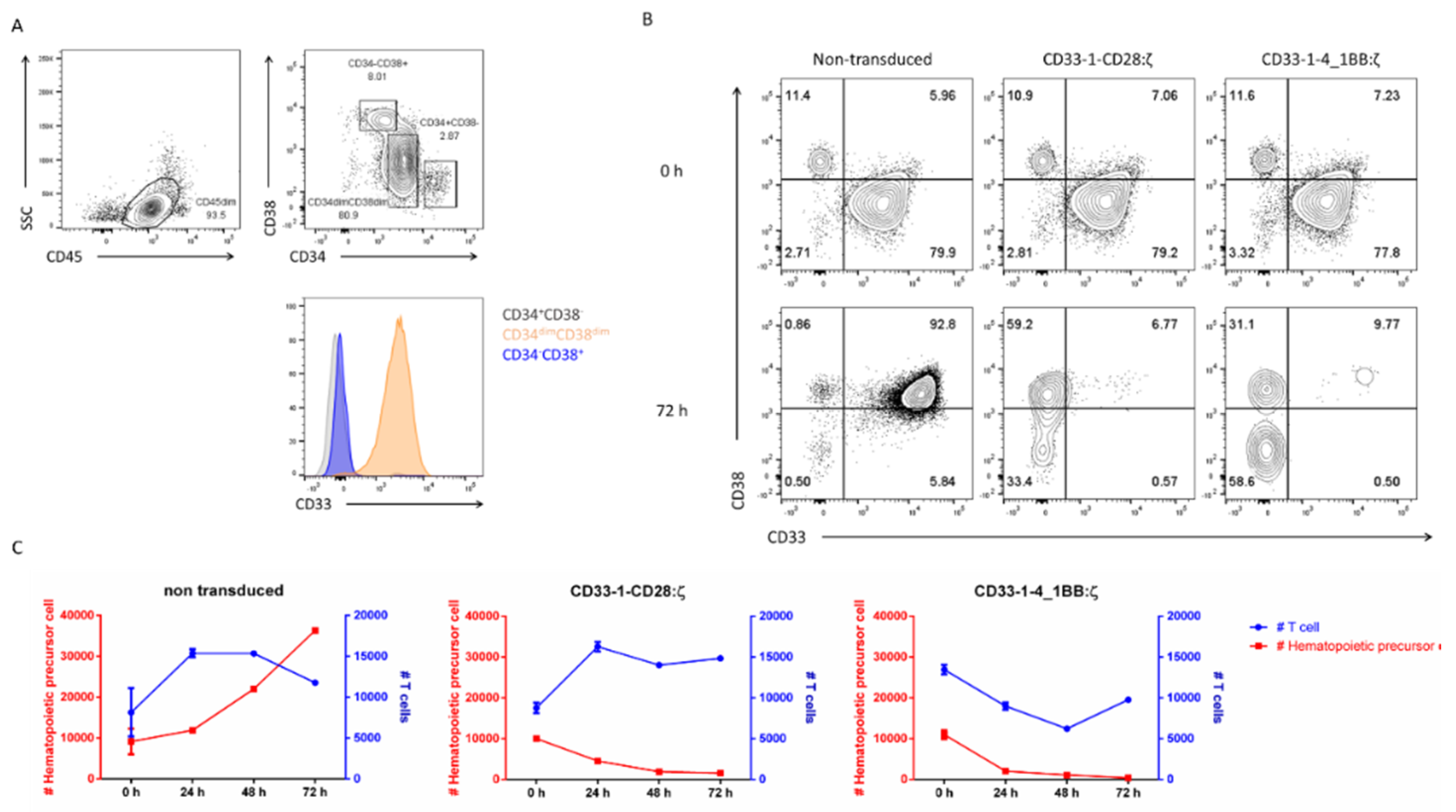
2.4. Targeting of CD33 Results in Hematopoietic Toxicity
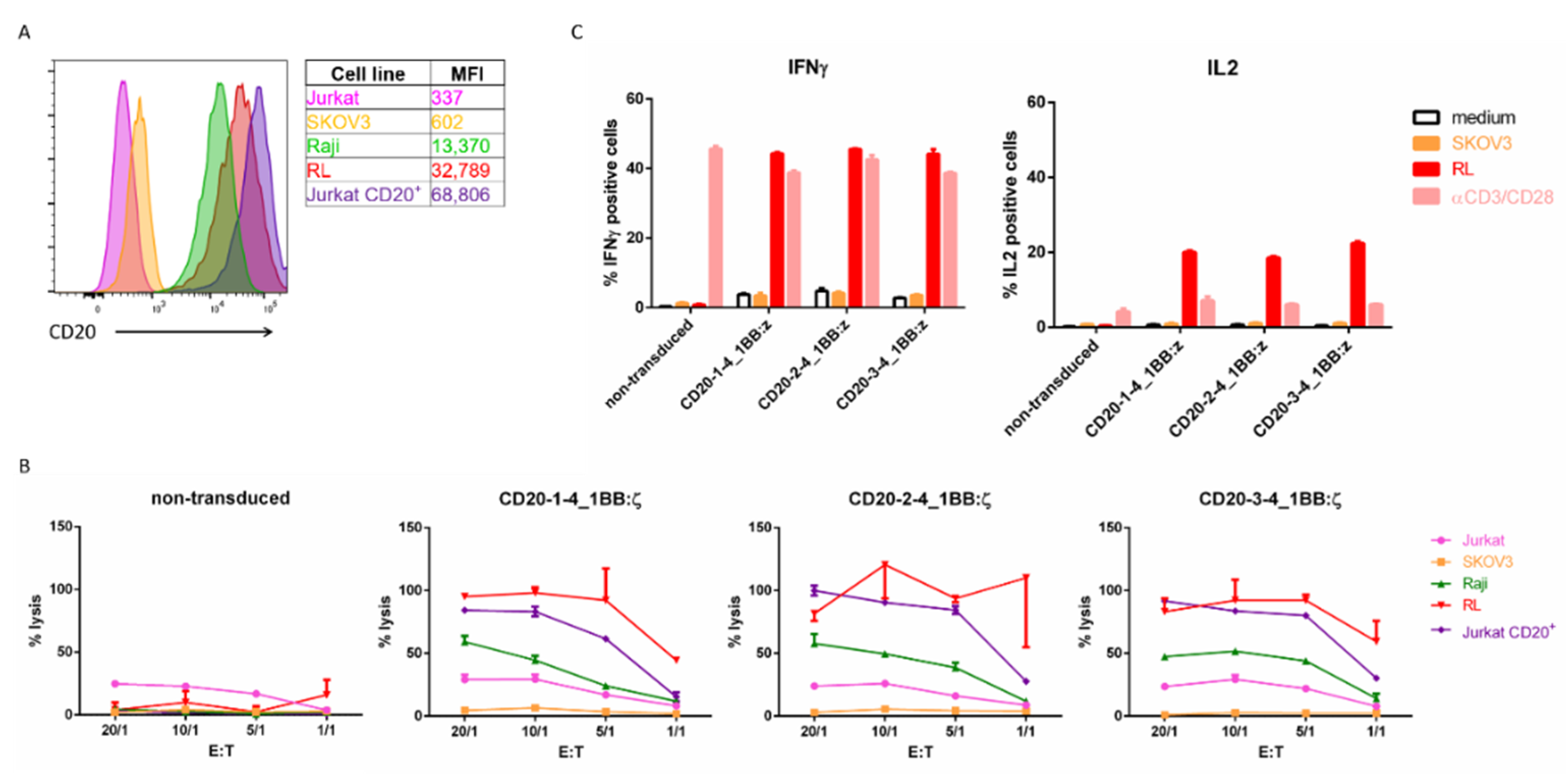
2.5. In vitro Evaluation of CD20 NanoCAR T Cells
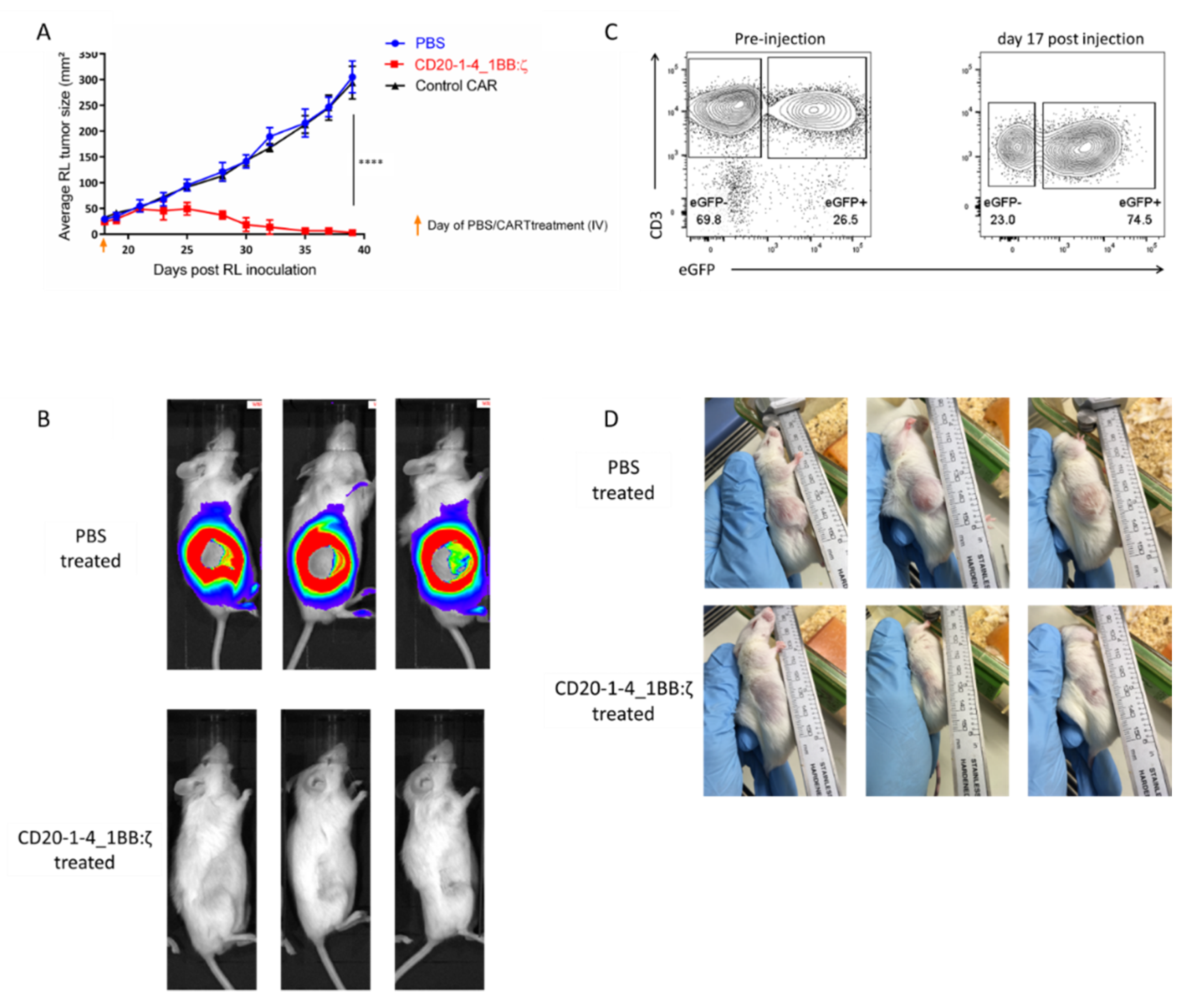
2.6. NanoCARs Targeting CD20 are Functional in Vivo
3. Discussion
4. Materials and Methods
4.1. Generation of the CD33 and CD20 Library
4.2. Culture of Cell Lines
4.3. Generation of NanoCAR Plasmids
4.4. Generation of Retroviral Particles
4.5. Generation of NanoCAR-expressing Human T Cells
4.6. Flow Cytometry and Antibodies
4.7. 51Chromium Release Assay
4.8. Flowcytometric Determination of Cytokine Production
4.9. In Vitro Stress Test
4.10. Tumor Mouse Model
4.11. In vitro Hematopoietic Cytotoxicity
4.12. Sequences and Primers
4.13. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| APC | Allophycocyanin |
| BCMA | B cell maturation antigen |
| CAR | Chimeric antigen receptor |
| CD | Cluster of Differentiation |
| CH | Constant Heavy |
| eGFP | enhanced Green Fluorescent Protein |
| EMA | European Medicines Agency |
| FDA | Federal and Drug Administration |
| Flt3L | FMS-like tyrosine kinase 3 ligand |
| HBSHcAbs | HEPES-buffered salineHeavy-chain antibodies |
| HPC | Hematopoietic precursor cell |
| VHH | Variable domain of the heavy-chain of HcAbs |
| IFN-γ | Interferon gamma |
| IgG | Immunoglobulin G |
| IL-2 | Interleukin 2 |
| IRES | Internal Ribosomal Entry Site |
| NSG | Nonobese diabetic scid gamma |
| PBMC | Peripheral blood mononuclear cells |
| PBS | Phosphate buffered saline |
| PCR | Polymerase chain reaction |
| PE | Phycoerythrin |
| PerCpCy5.5 | Peridnin-chlorophyll Cy 5.5 |
| PHA | Phytohemagglutinin |
| SCF | Stem cell factor |
| scFv | Single-chain variable fragment |
| SEM | Standard error of the mean |
| TPO | Thrombopoietin |
| VH | Variable heavy |
| VL | Variable light |
| VHH | Variable heavy homodimers |
References
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8 + and CD4 + CD19-specific chimeric antigen receptor–modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Garfall, A.L.; Maus, M.V.; Hwang, W.-T.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 2017, 130, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Weatherill, E.E.; Cain, K.L.; Heywood, S.P.; Compson, J.E.; Heads, J.T.; Adams, R.; Humphreys, D.P. Towards a universal disulphide stabilised single chain Fv format: Importance of interchain disulphide bond location and vL-vH orientation. Protein Eng. Des. Sel. 2012, 25, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.M.; Van der Jeugd, A.; Leinenga, G.; Evans, H.T.; Janowicz, P.W.; Götz, J. Combined effects of scanning ultrasound and a tau-specific single chain antibody in a tau transgenic mouse model. Brain 2017, 140, 1220–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Geddie, M.L.; Kohli, N.; Kornaga, T.; Kirpotin, D.B.; Jiao, Y.; Rennard, R.; Drummond, D.C.; Nielsen, U.B.; Xu, L.; et al. Comprehensive optimization of a single-chain variable domain antibody fragment as a targeting ligand for a cytotoxic nanoparticle. MAbs 2015, 7, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Nieba, L.; Honegger, A.; Krebber, C.; Pluckthun, A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: Improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng. Des. Sel. 1997, 10, 435–444. [Google Scholar] [CrossRef]
- Sun, W.; Xie, J.; Lin, H.; Mi, S.; Li, Z.; Hua, F.; Hu, Z. A combined strategy improves the solubility of aggregation-prone single-chain variable fragment antibodies. Protein Expr. Purif. 2012, 83, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 639–641. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T cells targeting HER2 and IL13R α 2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, M.C.; Riddell, S.R. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol. Rev. 2014, 257, 127–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Muyldermans, S.; Lauwereys, M. Unique single-domain antigen binding fragments derived from naturally occurring camel heavy-chain antibodies. J. Mol. Recognit. 1999, 12, 131–140. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Coppieters, K.; Dreier, T.; Silence, K.; Haard, H.D.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Wiele, C.V.D.; Staelens, L.; et al. Formatted anti–tumor necrosis factor α VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006, 54, 1856–1866. [Google Scholar] [CrossRef]
- Baral, T.N.; Magez, S.; Stijlemans, B.; Conrath, K.; Vanhollebeke, B.; Pays, E.; Muyldermans, S.; De Baetselier, P. Experimental therapy of African trypanosomiasis with a nanobody-conjugated human trypanolytic factor. Nat. Med. 2006, 12, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.M.; De Bruyn, S.; Duby, C.; Verschueren, K.; Baumeister, J.; Sargentini-Maier, L.; Ververken, C.; Holz, J.-B. A novel individualized treatment approach in open-label extension study of ozoralizumab (ATN-103) in subjects with rheumatoid arthritis on a background of methotrexate. Arthritis Rheum. 2012, 64, S563. [Google Scholar]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Cataland, S.; Knöbl, P.; Wu, H.; Artoni, A.; Westwood, J.-P.; Mansouri Taleghani, M.; Jilma, B.; et al. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrath, K.E.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel Single-domain Antibodies as Modular Building Units in Bispecific and Bivalent Antibody Constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C. Nanobody approval gives domain antibodies a boost. Nat. Rev. Drug Discov. 2019, 18, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Iri-Sofla, F.J.; Rahbarizadeh, F.; Ahmadvand, D.; Rasaee, M.J. Nanobody-based chimeric receptor gene integration in Jurkat cells mediated by PhiC31 integrase. Exp. Cell Res. 2011, 317, 2630–2641. [Google Scholar] [CrossRef]
- Sharifzadeh, Z.; Rahbarizadeh, F.; Shokrgozar, M.A.; Ahmadvand, D.; Mahboudi, F.; Jamnani, F.R.; Moghimi, S.M. Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Lett. 2013, 334, 237–244. [Google Scholar] [CrossRef]
- Jamnani, F.R.; Rahbarizadeh, F.; Shokrgozar, M.A.; Mahboudi, F.; Ahmadvand, D.; Sharifzadeh, Z.; Parhamifar, L.; Moghimi, S.M. T cells expressing VHH-directed oligoclonal chimeric HER2 antigen receptors: Towards tumor-directed oligoclonal T cell therapy. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 378–386. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, L.; Cui, H.; Wang, X.; Zhang, G.; Ma, J.; Han, H.; He, W.; Wang, W.; Zhao, Y.; et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci. Rep. 2014, 4, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody based dual specific CARs. Int. J. Mol. Sci. 2018, 19, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Chen, L.-J.; Yang, S.-S.; Sun, Y.; Wu, W.; Liu, Y.-F.; Xu, J.; Zhuang, Y.; Zhang, W.; Weng, X.-Q.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.D.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef]
- Wu, X. Transcription Start Regions in the Human Genome Are Favored Targets for MLV Integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef]
- Re, F. Green fluorescent protein expression in dendritic cells enhances their immunogenicity and elicits specific cytotoxic T-cell responses in humans. Exp. Hematol. 2004, 32, 210–217. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O??Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; Fitzgerald, D.J.; Barrett, D.M.; et al. Anti-CD22—Chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17, 1206–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almåsbak, H.; Walseng, E.; Kristian, A.; Myhre, M.R.; Suso, E.M.; Munthe, L.A.; Andersen, J.T.; Wang, M.Y.; Kvalheim, G.; Gaudernack, G.; et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015, 22, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Jonnalagadda, M.; Mardiros, A.; Urak, R.; Wang, X.; Hoffman, L.J.; Bernanke, A.; Chang, W.-C.; Bretzlaff, W.; Starr, R.; Priceman, S.; et al. Chimeric Antigen Receptors With Mutated IgG4 Fc Spacer Avoid Fc Receptor Binding and Improve T Cell Persistence and Antitumor Efficacy. Mol. Ther. 2015, 23, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [Green Version]
- Zabeau, L.; Verhee, A.; Catteeuw, D.; Faes, L.; Seeuws, S.; Decruy, T.; Elewaut, D.; Peelman, F.; Tavernier, J. Selection of non-competitive leptin antagonists using a random nanobody-based approach. Biochem. J. 2012, 441, 425–434. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Munter, S.; Van Parys, A.; Bral, L.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Billiet, L.; Weening, K.; Verhee, A.; et al. Rapid and Effective Generation of Nanobody Based CARs using PCR and Gibson Assembly. Int. J. Mol. Sci. 2020, 21, 883. https://doi.org/10.3390/ijms21030883
De Munter S, Van Parys A, Bral L, Ingels J, Goetgeluk G, Bonte S, Pille M, Billiet L, Weening K, Verhee A, et al. Rapid and Effective Generation of Nanobody Based CARs using PCR and Gibson Assembly. International Journal of Molecular Sciences. 2020; 21(3):883. https://doi.org/10.3390/ijms21030883
Chicago/Turabian StyleDe Munter, Stijn, Alexander Van Parys, Layla Bral, Joline Ingels, Glenn Goetgeluk, Sarah Bonte, Melissa Pille, Lore Billiet, Karin Weening, Annick Verhee, and et al. 2020. "Rapid and Effective Generation of Nanobody Based CARs using PCR and Gibson Assembly" International Journal of Molecular Sciences 21, no. 3: 883. https://doi.org/10.3390/ijms21030883
APA StyleDe Munter, S., Van Parys, A., Bral, L., Ingels, J., Goetgeluk, G., Bonte, S., Pille, M., Billiet, L., Weening, K., Verhee, A., Van der Heyden, J., Taghon, T., Leclercq, G., Kerre, T., Tavernier, J., & Vandekerckhove, B. (2020). Rapid and Effective Generation of Nanobody Based CARs using PCR and Gibson Assembly. International Journal of Molecular Sciences, 21(3), 883. https://doi.org/10.3390/ijms21030883